
Abstract
Recent studies have demonstrated that Janus kinase (JAK) plays a crucial role in signal transduction by directly affecting various cytokine receptors involved in inflammatory diseases such as atopic dermatitis (AD). Large-scale clinical trials on AD utilizing JAK inhibitors and biologic reagents, such as dupilumab, which targets the IL-4Rα receptor subunit of the Th2 cytokines IL-4 and IL-13, have yielded highly favorable results in comparison to traditional therapies. This indicates that therapeutic strategies based on molecular biology are efficacious in clinical settings. However, in September 2021, the U.S. Food and Drug Administration (FDA) indicated that tofacitinib, a JAK inhibitor, may carry various risks, including severe heart disease. Similar concerns have been raised for other JAK inhibitors, and further safety evaluations are underway. Thus, human biology involving JAKs appeared more complicated than we expected. In this article, we provide an overview of the molecular mechanisms of AD and examine the molecular targeting drugs for AD from the perspective of JAK-related biology.
1. Introduction
In treating atopic dermatitis (AD), topical steroids have been the pivotal treatment for half a century. After a while, the therapeutic options were extended to phototherapy and oral steroids [Citation1]. In Japan, additional immunosuppressive agents such as tacrolimus ointment and oral cyclosporine became clinically available in 1999 and 2008. However, recent breakthroughs in molecular biological research have led to a better understanding of the pathophysiology of AD. The development of drugs targeting the molecules in AD pathogenesis significantly improved treatment options. Dupilumab, a receptor antagonist of interleukin (IL)-4 and IL-13, was approved in 2018 [Citation2]. This drug is an antibody to bind specifically to the IL-4 receptor alpha (IL-4Rα) subunit, which inhibits downstream signal transduction by interfering with the complex formation after IL-4 and IL-13 bind to the receptor. Thereby it blocks the signaling and exerts anti-inflammatory effects. Downstream signaling of various Th2 cytokines is mediated by two groups of molecules: Janus kinase (JAK) and signal transducers and activators of transcription (STAT). Thus, the JAK-STAT pathway was extensively analyzed for many years, and finally, JAK inhibitors recently became commercially available for various diseases, including AD. This article introduces JAK inhibitors to aim attention at molecular biological aspects of detailed conditions of AD and large-scale clinical studies for AD treatments [Citation3].
2. JAK-associated receptors and cytokines
The terminology of ‘cytokine’ encompasses many structurally unrelated proteins which are classified by their binding to receptor superfamilies, such as the tumor necrosis factor (TNF) receptor family, the interleukin (IL)-1 receptor superfamily, interferon (IFN) receptor superfamily [Citation4]. Each family uses a unique mode of signal transduction system. The receptors are mediated by Janus kinases (JAKs), namely, JAK1, JAK2, JAK3, and tyrosine kinase 2 (Tyk2). JAKs share four components; 1) the kinase domain, which contains ATP-binding pocket; 2) the pseudokinase domain; 3) the four-point-one protein, ezrin, radixin, moesin (FERM) domain; 4) and the Src homology 2 (SH2)-like domain, which bind directly to the intracellular domains of receptors for Th2 cytokines and IFNs [Citation5]. It composes a wide range of receptors that are bound by approximately 50 cytokines, including interleukins, IFNs, hormones, and so on [Citation6]. The major receptor families, corresponding binding cytokines, and related JAKs are illustrated in . JAKs regulate gene expressions in cell proliferation, development, and differentiation via the JAK-STAT pathway and play an essential role in immune cells and blood cells [Citation7]. The receptors bound by cytokines trigger dimerization and recruitment of related JAKs. The activated JAKs phosphorylate the tails of receptors. The phosphorylated receptor forms a docking site for STATs (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6) [Citation8]. Afterward, STATs dissociate from the receptor to form homodimers or heterodimers, then enter the nucleus. Subsequently, binding to DNA regulates target gene expression as transcriptional factors [Citation9]. Notably, the inactivation of one side of a pair of JAKs is sufficient to inhibit the dimerization of STATs and downstream signaling [Citation10]. Thus, for example, JAK1- or JAK3-specific inhibition can suppress signal transduction through the common γ-chain cytokines receptor family (). Various combinations of JAKs are activated by multiple cytokines, such as IL- 2, IL- 4, IL-7, IL- 9, IL-13, IL-15, and IL-21 through JAK1-JAK3, IL-6, IL-7, IL-11, IL-13, TSLP, and IFN-γ via JAK1/JAK2, etc., as summarized in .
Figure 1. An overview of Janus kinase (JAK)-related cytokines. From the top, the coupled- receptor families, corresponding cytokines, combinations of JAKs, and coverage of subtypes of JAK inhibitors. INF: interferon, GM-CSF: Granulocyte-macrophage colony-stimulating factor, EPO: erythropoietin, TP: thrombopoietin.
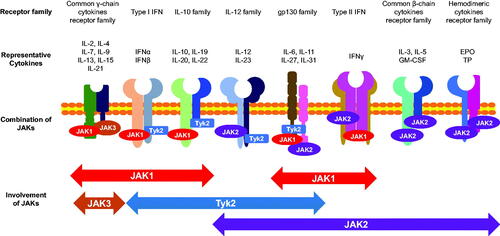
Table 1. Pairs of Janus kinases (JAK) and corresponding cytokines. The table was modified from REF [Citation11], which shows the cytokines expression downstream of a couple of JAKs in a permutational combinatorial manner. Notably, yellow-colored areas indicate homodimeric pairs of JAKs and exist at the table’s diagonal. In contrast, the pink-colored areas, which show a heterodimeric couple of JAKs, mirror the region of the orange-colored space from the diagonal lines. INF: interferon, GM-CSF: Granulocyte-macrophage colony-stimulating factor, EPO: erythropoietin, TP: thrombopoietin.
3. JAK inhibitors and target of cytokines
Some inherited immune deficient diseases, such as severe combined immunodeficiency (SCID), are caused by non-redundant mutations related to JAK-STAT pathways [Citation12]. In addition, overexpression of molecules in JAK-STAT pathways is associated with autoimmune disease and malignancy [Citation13]. Therefore, much effort has been devoted to comprehending the overall picture of the underlying mechanism whereby JAK works in vitro and in vivo [Citation12]. These previous studies indicated the importance of the inhibition of JAK in immune-related diseases [Citation8]. Thus, it was natural to urge pharmaceutical developers to search for small chemicals to block the JAK-STAT pathway as a new immune modulator. JAK inhibitors (also known as Jakinubs) were first approved in rheumatoid arthritis (RA) therapy in 2012 in the USA [Citation14]. Compared to conventional disease-modifying antirheumatic drugs (DMARDs), JAK inhibitors are described as targeted synthetic DMARDs (tsDMARs). JAK inhibitors are grouped into the first and second generations. The first generation comprises tofacitinib, ruxolitinib, baricitinib, and oclacitinib, while the second generation was improved for more JAK isoform selectivity, including abrocitinib and upadacitinib. Other newer JAK inhibitors, such as brepocitinib, cerdulatinib, ifidancitinib, and gusacitinib, are now upcoming to the market [Citation15]. The conformation of JAKs contains an ATP binding pocket essential to supply the phosphate group intrinsic to JAK activity [Citation5]. Each JAK has a unique ATP binding pocket, albeit with structural similarity. The fundamental strategy for inventing JAK inhibitors was to impede ATP binding [Citation5]. JAK inhibitors in newer generation harbors can recognize the slighter conformational difference of ATP binding pockets between JAKs to block a particular JAK subtype selectively [Citation15]. Thus, in contrast with biologics which usually target limited kinds of cytokines, JAK inhibitors can be applied to block spectra of cytokine networks in a theoretically designed way (, ). As mentioned in the previous section, signal transduction through a receptor bound to JAKs is suppressed by one molecule of the paired JAKs. Thus, not only pan-JAK inhibitors but JAK1/2 inhibitors practically suppress all the receptor signals due to combinatory restriction (, ). Of note, JAK2 homodimers mediate the signals from the hematopoietic growth factors, such as erythropoietin (EPO), thrombopoietin (TP), granulocyte macrophage colony-stimulating factor (GM-CSF), IL-3, and IL-5 [Citation16] (, ). IL-5 is known to play critical pathophysiological roles in many eosinophilic dermatoses. However, blocking this pathway theoretically could cause anemia, leukopenia, and thrombocytopenia [Citation17]. Thus, it is also necessary to understand the possible side effects in the targeted spectra.
4. JAK molecules in the pathophysiology of atopic dermatitis
From a clinical viewpoint, three factors are paramount to comprehending the pathophysiology of AD, i.e., the disruption of the epidermal barrier, the formation of allergic inflammation (Th2 inflammation), and the induction of itch. Inflammatory reaction in AD skin is triggered by foreign antigens invasion [Citation18,Citation19]. Subsequently, epidermal keratinocytes and epidermal Langerhans cells recognize them and elicit inflammatory responses by producing alarmins, such as IL-25, IL-33, and thymic stromal lymphopoietin (TSLP). Alarmins activate group 2 innate lymphoid cells (ILC2) that produce Th2 cytokines (IL-4, IL-13, etc.), leading to type 2 allergic inflammation. IL-4 also contributes to the class switch to IgE of B cells in draining lymph nodes [Citation20].
Although allergic inflammation is considered a defensive response against pathogens such as parasites, uncontrollable allergic inflammation due to pathological conditions can lead to allergic diseases such as AD [Citation21]. A complex interplay of multifaceted cytokines forms the pathogenesis of AD, where JAKs are involved in many of these pathways (summarized in ).
Figure 2. Cytokine pathways in atopic dermatitis pathogenesis and molecular-targeted therapies. Note that JAK2-, JAK3-, and Tyk2-specific inhibitors whose coverages are indicated by dashed lines are not approved for AD treatment.
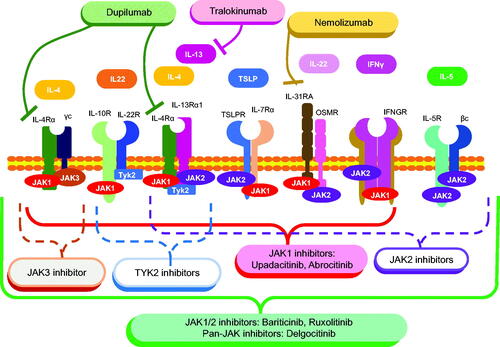
This review will discuss Th2 cytokines and other related molecules, such as IL-22, involved in epidermal thickening (acanthosis). IL-4 receptors are subclassified into type I and type II IL-4 receptors. Type I IL-4 receptors consist of a heterodimer of the IL-4Rα subunit and γC subunit to which IL-4 binds, whereas type II IL-4 receptors consist of IL-4Rα subunit and IL-13Rα1 [Citation22]. As the signal transduction mechanisms, the binding of IL-4 to type I IL-4 receptor induces phosphorylation of JAK1 and JAK3, while the binding of IL-4 and IL-13 to type II IL-4 receptor induces phosphorylation of JAK1, JAK2, and Tyk2 [Citation23]. IL-4 suppresses the production of filaggrin and antimicrobial peptides from epidermal keratinocytes, which disrupts the skin barrier and increases the risk of skin infections, such as infectious impetigo [Citation24]. IL-4 also promotes IgE production, which triggers itch-inducing histamine release from mast cells and basophils upon antigen stimulation. However, itch in AD is resistant to antihistamines [Citation25]. This observation suggests the presence of itch-inducing factors other than histamine. Accordingly, the presence of IL-4/IL-13 receptors on neurons has tempted many researchers to investigate the involvement of the JAK-STAT pathway in itch in AD [Citation26]. Though many Th2 cytokines induce Th2 inflammation via activation of STAT6 in the JAK-STAT pathway, IL-13 additionally activates STAT3, which inhibits epidermal keratinocyte differentiation and reduces barrier function. Correspondingly, IL-13 inhibits epidermal keratinocyte differentiation, leading to the impairment of barrier function [Citation27]. Recent studies showed that the expression of IL-13 is significantly higher than that of IL-4, suggesting that the physiological activity of IL-13 may be more deeply involved in the pathophysiology of AD [Citation28].
In addition to IL-4 and IL-13, various signal cascades are involved with JAK in AD pathophysiology. TSLP is secreted by epidermal keratinocytes upon stimulation and acts on ILC2 and dendritic cells expressing TSLP receptors to induce Th2 inflammation via phosphorylation of JAK1 and JAK2. TSLP has also been shown to cause pruritus via neurons in mice [Citation20].
IL-5 is a Th2 cytokine that selectively affects eosinophil precursor cells in the bone marrow to induce eosinophils’ production, maturation, and inflammatory localization. IL-5 acts as a proliferative activator of eosinophils through phosphorylation of the IL-5 receptor and its downstream JAK2 and induces inflammatory cytokine production by eosinophils. Although eosinophils are associated with many allergic diseases, the contribution of IL-5 in AD pathogenesis is unclear.
IL-31 binds to IL-31 receptors on neurons and activates the JAK-STAT pathway via phosphorylation of JAK1 and JAK2. Transgenic mice overexpressing IL-31 exhibit dermatitis development and scratching behavior. In humans, the severity of AD was shown to correlate with serum IL-31 levels [Citation29].
IFN-γ, Th1 cytokine, induces apoptosis in keratinocytes and further promotes the breakdown of the stratum corneum barrier. IFN-γ is a tetramer composed of IFNGR-1 and IFNGR-2, and it signals by activating STAT1 through phosphorylation of intracellular JAK1 and JAK2.
IL-22 is a cytokine produced by Th22 cells, not Th2 cytokine, but activates the JAK-STAT pathway via phosphorylation of JAK1 and Tyk2 after receptor binding [Citation30,Citation31]. IL-22 induces keratinocyte proliferation and epidermal thickening in AD and psoriasis vulgaris. At the same time, IL-22 inhibits differentiation and maturation and downregulates the expression of barrier function-related factors, contributing to lichenification [Citation30]. Consistent with this, epidermal thickness and proliferation are known to be significantly increased in chronic lesions than in the acute phase [Citation32].
5. Types and characteristics of JAK inhibitors for atopic dermatitis
The oral JAK inhibitors baricitinib, upadacitinib, and abrocitinib apply to AD treatment in Japan. Baricitinib inhibits JAK1/2. On the other hand, the other two are JAK1-specific inhibitors and can be prescribed in double doses depending on the patient’s symptoms. JAK1-specific and JAK1/2 inhibitors affect the majority of cytokine pathways in AD pathogenesis, and the significant spectral difference between these drugs is the coverage of IL-5 and IL-12 family (). The recent reports indicated that 59.8% of the dupilumab treatment group (300 mg/2 weeks) achieved EASI 75 after 16 weeks [Citation33]. In contrast, 47.7% of baricitinib (4 mg/day) completed EASI 75 in the clinical trial BREEZE AD7, although this is not a direct comparison [Citation34]. In addition, the phase 3b Heads Up study compared upadacitinib (30 mg/day) with dupilumab (300 mg/2 weeks). 71% of the former and 61% of the latter achieved EASI 75 at 16 weeks. This result indicated the superiority of upadacitinib over dupilumab [Citation35]. The usual dose of upadacitinib for RA treatment is 15 mg/day, whereas the Heads Up study administered a double dose of 30 mg/day. In the JADE COMPARE study, abrocitinib was compared with dupilumab on the primary endpoint of EASI75 at 12 weeks. 58.7% of abrocitinib (100 mg/day) and 58.1% of dupilumab (300 mg/day) were achieved, respectively. In addition, 70.3% of the double-dose abrocitinib (200 mg/day) group achieved EASI 75 at 12 weeks, respectively. Furthermore, abrocitinib showed a higher percentage of pruritus improvement than dupilumab at two weeks of treatment, although this difference was not statistically significant [Citation36].
Tralokinumab, a monoclonal antibody that neutralizes IL-13, became available for AD treatment. In ECZTRA 1 (NCT03131648) and ECZTRA 2 (NCT03160885, identical phase 3 randomized, double-blind trials, the more AD patients who were treated with tralokinumab achieved IGA reduction to 1 or less (ECZTRA 1: 15.8% and ECZTRA 2: 22.2%) than placebo-treated patients (ECZTRA 1: 7.1% and ECZTRA 2: 10.9%) at 16 weeks. EASI75 was also more achieved by the tralokinumab-treated group (ECZTRA 1: 25.0% and ECZTRA 2: 33.2%) than placebo-treated group (ECZTRA 1: 12.7% and ECZTRA 2: 11.4%) at 16 weeks [Citation37]. Previously, IL-4 was considered a pivotal cytokine for AD pathogenesis. However, recent data from experimental settings indicate that IL-13 is a primary cytokine to play a role in AD [Citation38]. On the contrary, clinical data implies that dupilumab seems more efficient in achieving EASI75 than tralokinumab. Nonetheless, a direct comparison of tralokinumab with dupilumab has not been conducted.
Nemolizumab, a humanized monoclonal antibody against IL-31 receptor A, is a recently approved biological reagent [Citation39]. In a double-blind, phase 3 study, the mean percent change in visual analog scale score at week 16 was −42.8% in the nemolizumab group, compared to −21.4% in the placebo group. The mean percent change in EASI score was −45.9% for nemolizumab and −33.2% for placebo. Improvement of DLQI and ISI was also observed more in the nemolizumab-treated group than in the placebo-treated group [Citation39]. As shown in , IL-31 is mediated by a heterodimer of JAK1/JAK2 (). Thus, JAK inhibitors, currently approved for AD, affect the IL-31 signaling pathway. Therefore, the high efficacy of JAK inhibitors for AD treatment might partly be attributable to inhibitory effects on IL-31 signaling.
In terms of topical appreciation, ruxolitinib, which is a JAK1/2 inhibitor, is available in the USA [Citation40]. Since JAK2 inhibitor suppresses EPO, TP, and GM-CSF, ruxolitinib was approved for hematologic disorders such as polycythemia vera and myelofibrosis [Citation41]. In the dermatological aspect, TRuE-AD1 (NCT03745638) and TruE-AD2 (NCT03745651), phase III clinical trials evaluated the efficacy and safety of ruxolitinib cream in patients with AD. In the trials, IGA treatment success was assessed as an IGA score of 0 or 1 with a ≥ 2-point improvement from baseline by week 8. ruxolitinib 1.5% group showed better results of IGA treatment success (TruE-AD1: 53.8%, TruE-AD2: 51.3%) compared to the vehicle group (TruE-AD1: 15.1%, TruE-AD2: 7.6%) [Citation37]. In addition, no significant adverse events have been identified in connection to topical ruxolitinib, albeit with minor treatment-emergent adverse events, including burning sensation (ruxolitinib; 0.8% and vehicle; 4.4%).
Delgocitinib, 0.5% ointment, was also approved for topical use for AD in 2020 in Japan but has yet to be in the USA. Delgocitinib is a pan-JAK inhibitor because it inhibits the phosphorylation of all JAK family members [Citation42]. Thus, it is expected to comprehensively suppress Th2 inflammation (). Regarding side effects, delgocitinib is unlikely to cause skin atrophy and thinning seen with topical steroids or the burning sensation and other skin irritations seen with tacrolimus. In addition, folliculitis (3.6%), acne (3.2%), Kaposi’s varicelliform eruption (3.4%), herpes simplex (3.0%), and contact dermatitis (4.5%) have also been reported at the site of delgocitinib application. However, these were not reported to be more common than with steroids or tacrolimus ointments [Citation43].
Although the specificity of JAK inhibitors clarifies the target in the molecular mechanism, the pharmacokinetics of drugs is also the paramount determinant of drug efficacy. Absorption rates of the drugs depend on the solubility and the membrane permeability for enteral administration [Citation44]. Distribution behavior is affected by plasma protein bindings such as drug transporters and carrier proteins. The unbound forms of JAK inhibitors are required to enter the cytoplasm, while albumin-bound forms (for example, ruxolitinib) cannot pass through the blood-brain barrier [Citation45]. Other paramount factors for the pharmacokinetics of JAK inhibitors are their metabolism and elimination. The biotransformation processes of JAK inhibitor molecules are composed of two differential phases [Citation44]. The first phase is to modify the functional group by chemical modification, such as oxidation, reduction, and hydrolysis [Citation46]. In this phase, the cytochrome P450 system plays pivotal roles. The second phase is the conjugation reaction, where functional groups are linked to water-soluble molecules, which eliminates metabolites through feces or urine [Citation46]. These processes also influence the half-life of the drugs. Thus, the precise prediction of the drug efficacy in in-vivo situations in humans is complex, albeit with well-studied JAK-related biology. In effect, recent studies demonstrated that the plasma concentration of tofacitinib, initially reported as an inhibitor of JAK1/3 and JAK2/2, was marginally lower than the JAK2/2 IC50 values, while that of upadacitinib, a JAK1-specific inhibitor, is now known to be higher than JAK 2/2 IC50 values for several hours at their approved dose [Citation47]. The topics about pharmacokinetics are well-summarized in the review [Citation44,Citation48].
6. Adverse effects of JAK inhibitors and a cautionary note
Based on the nature of JAKs involved in the inflammatory cytokine signalings, the expected side effects of inhibition of JAKs are infectious diseases. The reported side effects include nasopharyngitis, upper respiratory tract infection, urinary tract infection, and herpes virus infection, such as reactivation of varicella-zoster virus [Citation49,Citation50]. Notably, previous studies revealed that the treatment by various JAK inhibitors poses a higher risk of herpes zosters (HZ) in the Asian population. On the other hand, recent studies indicated that AD patients treated with JAK inhibitors do not pose increased risks of HZ [Citation50]. Furthermore, epidemiologic research provided arguable results of ethnic differences in HZ morbidity in the Asian population [Citation51,Citation52]. Thus, Asian ethnicity does not necessarily associate with a higher incidence rate than other ethnicities. Notably, a study with genome-wide association study meta-analyses revealed genetic factors for risk/faster onset of HZ in the use of tofacitinib, which includes single-nucleotide polymorphisms near CD83 and IL17RB attributable to the more rapid onset of HZ for subjects of the European and the Asian, respectively [Citation53]. However, the molecular mechanisms of how these genetic background modifications contribute to the susceptibility to HZ in the use of tofacitinib are yet to be clarified.
Further current research demonstrated that upadacitinib and abrocitinib correlate with a dose-dependent correlation to acne. Particularly, upadacitinib caused more acne in AD patients than in RA patients [Citation54]. One explanation of why acne occurs more in AD than RA is the age of the patients. The mean age of patients who developed acne after JAK inhibitors was 29.6 years, and that of the oldest was 37 years old [Citation55]. The observations correspond to the previous research that acne was most frequently seen at 18 to 29 years old in the general population and AD patients [Citation56]. On the other hand, the RA patients who were enrolled in the JAK inhibitor study were older than 50 years [Citation57]. Thus, the age of subjects who are treated with JAK inhibitors could lead to the difference between RA and AD [Citation55]. The consistent report was the case where acne exacerbation was seen in a 25-year-old patient of alopecia areata after tofacitinib treatment [Citation58].
Another possible explanation has been suggested by epidemiological studies indicating that obesity and morbidity of acne are inversely correlated [Citation59]. Thus, some speculated that the activated adipose tissue might play a protective role in acne. Moreover, a recent study showed that AD patients with higher body mass indexes are prone to manifest acne using JAK inhibitors [Citation55]. The phenomena tempted the researchers to assume that the pathophysiology of JAK inhibitor-related acne might be due to the disruption of the protective functions of the adipose tissue [Citation55]. Therefore, some speculated that the mammalian target of rapamycin complex 1(mTORC1) might play a role in the acne pathophysiology since mTORC1 is transcriptionally activated by STATs and regulate adipose tissue function [Citation55,Citation60]. Along the same line, TLSP, which JAK1 and JAK2 bind, comes forward as a key factor for lipid metabolism. A seminal study by the mice engineered to produce elevated TSLP recently demonstrated that TSLP induces the migration of conventional T cells to sebaceous glands, leading to hypersecretion of sebum that augments the skin barrier function [Citation61]. Although the mechanistic insight is one of the complexities of the JAK-related molecular network, the molecules in lipid metabolism of the sebaceous gland could be therapeutic targets for acne as an adverse effect due to JAK inhibitors.
Except for infectious diseases, JAK inhibitors are highly effective in treating AD. However, a cautionary note has recently emerged. ORAL-Surveillance study which assessed the risk of tofacitinib, a pan-JAK inhibitor, was conducted in RA patients aged 50 years or older with cardiovascular risk, using a TNF inhibitor as the target drug [Citation57]. The results showed an increased risk of severe heart disease, malignancy, deep venous thrombosis (DVT), and death, which the U.S. Food and Drug Administration (FDA) concluded in September 2021. The study also noted the possibility of similar risks with other oral JAK inhibitors. As mentioned above, JAK inhibitors pose a risk of thrombocytopenia since JAK2 inhibition causes suppression of hematopoietic growth factors, such as EPO. Thus, the predisposition of DVT is not fully explained by current knowledge of the pathophysiology related to the JAK-STAT pathway. Thus, it is unknown how JAK inhibitors induce DVT. However, it needs to be careful to understand the report since the patients in ORAL-Surveillance were at high risk of cardiovascular events, and some of them were administered 10 mg twice daily of tofacitinib which is a double dose of the current clinical use [Citation57]. Therefore, although it is reasonable to pay attention to the possible risks suggested by the ORAL-Surveillance study for our clinical practice, further risk assessments will need to conclude the risk of JAK inhibitors. As we introduce the differential adverse effects of JAK inhibitors between RA and AD, the age of patients might be significant. Of note, a cautionary note about tofacitinib from FDA is based on the research about RA patients with age > 50 years. It is yet to be clarified if the spectra of adverse effects in using JAK inhibitors for RA patients are identical to that of AD patients whose affected age tends to be much younger than RA.
7. Concluding remarks
JAK is a vital signaling pathway regulator in the pathophysiology of AD, and JAK inhibitors have shown promising results in treating AD. On the other hand, risks of adverse events have begun to be reported, and further information is needed to determine the long-term safety and cost-effectiveness of these drugs.
8. Statement of all funding sources that supported the work
KOSE Cosmetology Research Foundation, Grants-in-aid for Scientific Research (21K16227, 20K08649, and 21K16227)
Acknowledgments
None
Disclosure statement
Kenji Kabashima has received consulting fees or advisory board honoraria from Japan Tobacco Inc., Kao, LEO Pharma, Torii, Chugai Pharmaceutical, Maruho, Pola Pharma, Abbvie, Eli Lilly, Sanofi, and Pfizer, and has received research grants from LEO Pharma, Japan Tobacco Inc., P&G Japan, Eli Lilly Japan, Tanabe Mitsubishi, Ono Pharmaceutical, Kyowa Kirin, Pola Pharma, AbbVie, Sanofi, Kose, Maruho, and Kyorin Pharmaceutical.
Additional information
Funding
References
- Eichenfield LF, DiBonaventura M, Xenakis J, et al. Costs and treatment patterns among patients with atopic dermatitis using advanced therapies in the United States: analysis of a retrospective claims database. Dermatol Ther . 2020; 10(4):791–806.
- Akinlade B, Guttman‐Yassky E, Bruin‐Weller M, et al. Conjunctivitis in dupilumab clinical trials. Br J Dermatol. 2019; 181(3):459–473.
- Šteňová E, Tarabčáková L, Babál P, et al. Hypereosinophilic syndrome—a rare adverse event of anti-cytokine treatment in rheumatoid arthritis resolved after Janus kinase inhibitor therapy. Clin Rheumatol. 2020; 39(11):3507–3510.
- Freidin M, Kessler JA. Cytokine regulation of substance P expression in sympathetic neurons. Proc Natl Acad Sci U S A. 1991; 88(8):3200–3203.
- Silvennoinen O, Haikarainen T, Virtanen A. Aiming for the pocket. Nat Chem Biol. 2022;18(12):1296–1297.
- Darnell JE. STATs and gene regulation. Science. 1997; 277(5332):1630–1635.
- Koride S, Nayak S, Banfield C, et al. Evaluating the role of Janus kinase pathways in platelet homeostasis using a systems modeling approach. CPT Pharmacometrics Syst Pharmacol. 2019;8(7):478–488.
- Schwartz DM, Kanno Y, Villarino A, et al. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. 2017; 16(12):843–862.
- Hu X, Li J, Fu M, et al. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. 2021; 66(1):402.
- Chen M, Dai SM. A novel treatment for psoriatic arthritis: Janus kinase inhibitors. Chin Med J. 2020; 133(8):959–967.
- Garcia-Melendo C, Cubiró X, Puig L. Janus kinase inhibitors in dermatology: part 1—general considerations and applications in vitiligo and alopecia areata. Actas Dermo-Sifiliográficas (English Edition). 2021; 112(6):503–515.
- Philips RL, Wang Y, Cheon H, et al. The JAK-STAT pathway at 30: much learned, much more to do. Cell. 2022; Oct185(21):3857–3876.
- Tzeng HT, Chyuan IT, Lai JH. Targeting the JAK-STAT pathway in autoimmune diseases and cancers: a focus on molecular mechanisms and therapeutic potential. Biochem Pharmacol. 2021; Nov193:114760.
- Cutolo M, Meroni M. Clinical utility of the oral JAK inhibitor tofacitinib in the treatment of rheumatoid arthritis. JIR. 2013; 6:129–137. DOI:10.2147/JIR.S35901
- Shawky AM, Almalki FA, Abdalla AN, et al. A comprehensive overview of globally approved JAK inhibitors. Pharmaceutics. 2022; 14(5):1001.
- Parganas E, Wang D, Stravopodis D, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998 ;93(3):385–395.
- Guglielmelli P, Barosi G, Specchia G, et al. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood. 2009; Aug114(8):1477–1483.
- Kabashima K. New concept of the pathogenesis of atopic dermatitis: interplay among the barrier, allergy, and pruritus as a Trinity. J Dermatol Sci. 2013; 70(1):3–11.
- Otsuka A, Nomura T, Rerknimitr P, et al. The interplay between genetic and environmental factors in the pathogenesis of atopic dermatitis. Immunol Rev. 2017; 278(1):246–262.
- Weidinger S, Beck LA, Bieber T, et al. Atopic dermatitis. Nat Rev Dis Primers. 2018; 4(1):1.
- Sousa-Santos ACAF, Moreno AS, Santos ABR, et al. Parasite infections, allergy and asthma: a role for tropomyosin in promoting type 2 immune responses. Int Arch Allergy Immunol. 2020;181(3):221–227.
- Harb H, Chatila TA. Mechanisms of dupilumab. Clin Exp Allergy. 2020; 50(1):5–14.
- Furue M. Regulation of skin barrier function via competition between AHR axis versus IL-13/IL-4–JAK–STAT6/STAT3 axis: pathogenic and therapeutic implications in atopic dermatitis. JCM. 2020; Nov 209(11):3741.
- Chieosilapatham P, Kiatsurayanon C, Umehara Y, et al. Keratinocytes: innate immune cells in atopic dermatitis. Clin Exp Immunol. 2021 May 13;204(3):296–309.
- Genois A, Haig M, Roches AD, et al. Case report of atopic dermatitis with refractory pruritus markedly improved with the novel use of clonidine and trimeprazine: dramatic relief of pruritus with clonidine. Pediatr Dermatol. 2014;31(1):76–79.
- Nakashima C, Ishida Y, Kaku Y, et al. Dupilumab improved atypical fibrotic skin plaques in atopic dermatitis. Br J Dermatol. 2020;182(2):487–488.
- Furue K, Ito T, Tsuji G, et al. The IL-13–OVOL1–FLG axis in atopic dermatitis. Immunology. 2019;158(4):281–286.
- Bieber T. Interleukin‐13: targeting an underestimated cytokine in atopic dermatitis. Allergy. 2020; 75(1):54–62.
- Erickson S, Heul AV, Kim BS. New and emerging treatments for inflammatory itch. Ann Allergy Asthma Immunol. 2021; Jan126(1):13–20.
- Wollenberg A, Fölster-Holst R, Saint Aroman M, et al. Effects of a protein-free oat plantlet extract on microinflammation and skin barrier function in atopic dermatitis patients. J Eur Acad Dermatol Venereol. 2018; Apr32:1–15.
- Nomura T, Honda T, Kabashima K. Multipolarity of cytokine axes in the pathogenesis of atopic dermatitis in terms of age, race, species, disease stage and biomarkers. Int Immunol. 2018; Aug 3030(9):419–428.
- Gittler JK, Shemer A, Suárez-Fariñas M, et al. Progressive activation of TH2/TH22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol. 2012; 130(6):1344–1354.
- Halling AS, Loft N, Silverberg JI, et al. Real-world evidence of dupilumab efficacy and risk of adverse events: a systematic review and meta-analysis. J Am Acad Dermatol. 2021; 84(1):139–147.
- Radi G, Simonetti O, Rizzetto G, et al. Baricitinib: the first jak inhibitor approved in Europe for the treatment of moderate to severe atopic dermatitis in adult patients. Healthcare. 2021; 9(11):1575.
- Blauvelt A, Teixeira HD, Simpson EL, et al. Efficacy and safety of upadacitinib vs dupilumab in adults with moderate-to-evere atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2021;157(9):1047–1055.
- Bieber T, Simpson EL, Silverberg JI, et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N Engl J Med. 2021;384(12):1101–1112.
- Mohney LA, Singh R, Feldman SR. Review of ruxolitinib in the treatment of atopic dermatitis. Ann Pharmacother. 2023; 57(2):207–216.
- Tsoi LC, Rodriguez E, Degenhardt F, et al. Atopic dermatitis is an IL-13–dominant disease with greater molecular heterogeneity compared to psoriasis. J Invest Dermatol. 2019; 139(7):1480–1489.
- Di Salvo E, Ventura-Spagnolo E, Casciaro M, et al. IL-33/IL-31 axis: a potential inflammatory pathway. Mediators Inflamm. 2018;2018:1–8.
- Owji S, Caldas SA, Ungar B. Management of atopic dermatitis: clinical utility of ruxolitinib. J Asthma Allergy. 2022;15:1527–1537.
- Kirito K. Recent progress of JAK inhibitors for hematological disorders. Immunol Med. 2022:1–12. DOI:10.1080/25785826.2022.2139317
- Tanimoto A, Shinozaki Y, Nozawa K, et al. Improvement of spontaneous locomotor activity with JAK inhibition by JTE-052 in rat adjuvant-induced arthritis. BMC Musculoskelet Disord. 2015;16(1):339.
- Nakagawa H, Nemoto O, Igarashi A, et al. Long‐term safety and efficacy of delgocitinib ointment, a topical janus kinase inhibitor, in adult patients with atopic dermatitis. J Dermatol. 2020;47(2):114–120.
- Eichner A, Wohlrab J. Pharmacology of inhibitors of janus kinases – part 1: pharmacokinetics. J Dtsch Dermatol Ges. 2022; Nov20(11):1485–1499.
- Mohamed MEF, Klünder B, Othman AA. Clinical pharmacokinetics of upadacitinib: review of data relevant to the rheumatoid arthritis indication. Clin Pharmacokinet. 2020 May;59(5):531–544.
- Shi JG, Chen X, Emm T, et al. The effect of CYP3A4 inhibition or induction on the pharmacokinetics and pharmacodynamics of orally administered ruxolitinib (INCB018424 phosphate) in healthy volunteers. J Clin Pharmacol. 2012; 52(6):809–818.
- Chimalakonda A, Burke J, Cheng L, et al. Selectivity profile of the tyrosine kinase 2 inhibitor deucravacitinib compared with janus kinase 1/2/3 inhibitors. Dermatol Ther. 2021;11(5):1763–1776.
- Eichner A, Wohlrab J. Pharmacology of inhibitors of janus kinases – part 2: pharmacodynamics. J Dtsch Dermatol Ges. 2022; 20(12):1621–1631.
- Winthrop KL, Nash P, Yamaoka K, et al. Incidence and risk factors for herpes zoster in patients with rheumatoid arthritis receiving upadacitinib: a pooled analysis of six phase III clinical trials. Ann Rheum Dis. 2022; 81(2):206–213.
- Alves C, Penedones A, Mendes D, et al. The safety of systemic janus kinase inhibitors in atopic dermatitis: a systematic review and network meta-analysis. Eur J Clin Pharmacol. 2022; 78(12):1923–1933.
- Toyama N, Shiraki K. and Epidemiology of herpes zoster and its relationship to varicella in Japan: a 10-year survey of 48,388 herpes zoster cases in Miyazaki prefecture. J Med Virol. 2009; 81(12):2053–2058.
- Thompson RR, Kong CL, Porco TC, et al. Herpes zoster and postherpetic neuralgia: changing incidence rates from 1994 to 2018 in the United States. Clin Infect Dis. 2021;73(9):e3210–7–e3217.
- Bing N, Zhou H, Chen X, et al. Contribution of a european‐prevalent variant near CD83 and an east asian–prevalent variant near IL17RB to herpes zoster risk in tofacitinib treatment: results of genome‐wide association study meta‐analyses. Arthritis Rheumatol. 2021; Jul73(7):1155–1166.
- Chang P, Huang S, Chang P, et al. Safety considerations of systemic Janus kinase inhibitors in atopic dermatitis applications. J Dermatol. 2021; 48(11):1631–1639.
- Lee SD, Ahn HJ, Shin MK. A case series of acne following Janus kinase inhibitors in patients with atopic dermatitis. JAAD Case Rep. 2022; 30:11–16.
- Thyssen JP, Yosipovitch G, Paul C, et al. Patient‐reported outcomes from the JADE COMPARE randomized phase 3 study of abrocitinib in adults with moderate‐to‐severe atopic dermatitis. J Eur Acad Dermatol Venereol. 2022;36(3):434–443.
- Ytterberg SR, Bhatt DL, Mikuls TR, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N Engl J Med. 2022; 386(4):316–326.
- Alharthi S, Turkmani M, AlJasser MI. Acne exacerbation after tofacitinib treatment for alopecia areata. Dermatol Reports. 2022; [cited 14(2):9396. Available fromhttps://www.pagepress.org/journals/index.php/dr/article/view/9396
- Snast I, Dalal A, Twig G, et al. Acne and obesity: a nationwide study of 600,404 adolescents. J Am Acad Dermatol. 2019;81(3):723–729.
- Saleiro D, Platanias LC. Intersection of mTOR and STAT signaling in immunity. Trends Immunol. 2015; 36(1):21–29.
- Choa R, Tohyama J, Wada S, et al. Thymic stromal lymphopoietin induces adipose loss through sebum hypersecretion. Science. 2021; 373(6554):eabd2893.