
Abstract
Kabuki syndrome (KS) is a genetic disorder caused by gene mutations in either lysine-specific methyltransferase 2D (KMT2D) or lysine demethylase 6A (KDM6A). This congenital disorder exhibits characteristic facial features, developmental delays in psychomotor skills, and skeletal abnormalities. Moreover, it is classified as a congenital immunodeficient disorder under the category of combined immunodeficiency, leading to hypogammaglobulinemia and the onset of autoimmune diseases. Here, we present the first case of KS complicated by idiopathic pulmonary hemosiderosis (IPH). The KS patient, a 2-year-old Japanese girl with a history of hypoplastic left heart syndrome and recurrent bacterial infection, developed severe respiratory distress and anemia. She had autoimmune hemolytic anemia and gouty nephropathy. Hemophagocytic macrophages with hemosiderin ingestion were identified in bronchoalveolar lavage fluid, excluding differential diagnoses and leading to the diagnosis of idiopathic pulmonary hemosiderosis. Intravenous prednisolone (2 mg/kg/day) was administered, but symptoms did not improve. However, pulmonary hemorrhage disappeared with methylprednisolone pulse therapy. IPH warrants consideration in cases where individuals with KS manifest idiopathic pneumonia and concurrent anemia.
Graphical Abstract
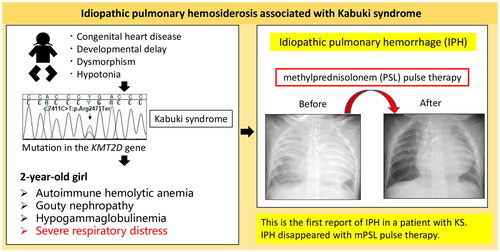
Kabuki syndrome (KS), a genetic disorder characterized by distinctive facial features, musculoskeletal abnormalities, and developmental delays, is commonly caused by mutations in the lysine-specific methyltransferase 2D (KMT2D) gene or lysine demethylase 6A (KDM6A). According to the International Union of Immunological Societies (IUIS) 2022 Update, KS is classified as a combined immunodeficiency. KS may manifest as recurrent otitis media, pneumonia, or other infections due to hypogammaglobulinemia, possibly leading to the development of various autoimmune diseases [Citation1]. Here, we present the first case of KS diagnosed with idiopathic pulmonary hemosiderosis (IPH) associated with severe respiratory distress and anemia that improved with methylprednisolone pulse therapy.
A 2-year 3-month-old Japanese girl with severe respiratory distress was admitted to our hospital. Her past medical history was as follows: at 6 weeks of age, she underwent the Norwood procedure for hypoplastic left heart syndrome. Whole-exome sequencing at 7 months of age revealed a recurrent pathogenic loss-of-function mutation, specifically the c.7411C > T (p.Arg2471Ter) mutation, in KMT2D (); she was diagnosed with KS. She had recurrent pneumonia and urinary tract infections and underwent a tracheotomy at 2 years of age. However, she developed bloody secretions and severe anemia requiring frequent erythrocyte transfusions. She was referred to our department for severe respiratory distress evaluation.
Figure 1. (A) The heterozygous KMT2D mutation c.7411C > T (p.Arg2471Ter) was identified in the proband. (B) Chest radiograph showing severe diffuse infiltration of bilateral lungs. (C) Chest computed tomography showing multiple ground glass opacities and consolidation in both lung fields. (D) Chest radiograph after methylprednisolone pulse therapy showing markedly decreased lung infiltration.
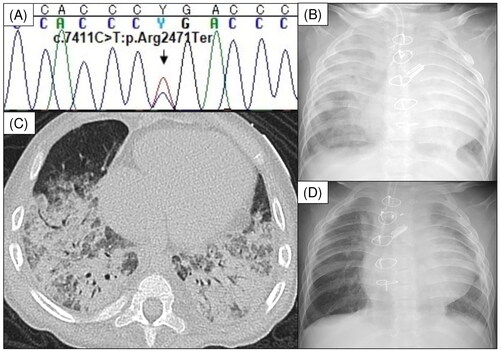
On admission, the patient was afebrile but had a SpO2 of 80% on 70% inspired oxygen fraction. Laboratory findings showed a white blood cell count of 12,290/μL, with differential neutrophil counts (83.8%). Her hemoglobin level was 11.8 g/dL, with a reticulocyte count of 30.6‰. Her direct antiglobulin test results were positive and haptoglobin level was 3 mg/dL. Her serum urea nitrogen, creatinine, and uric acid levels were elevated to 31, 0.53, and 11.7 mg/dL, respectively. She had hypogammaglobulinemia, with an immunoglobulin G (IgG) level of 334 mg/dL. There was no evidence of hypocomplementemia; test results for antinuclear, anti-neutrophil cytoplasmic, and anti-glomerular basement membrane antibodies were negative. Allergen-specific IgE antibodies against cow milk and wheat were not detected. Chest radiography and computed tomography revealed diffuse alveolar hemorrhage (DAH) in both lung fields (), while bronchoalveolar lavage (BAL) fluid staining with Prussian blue revealed hemosiderin-laden macrophage infiltration. The patient was diagnosed with IPH, autoimmune hemolytic anemia, and gouty nephropathy. Treatment with 2 mg/kg/d prednisolone did not improve her pulmonary hemorrhage; therefore, we administered 30 mg/kg methylprednisolone intravenously daily for 3 d, which resolved the pulmonary hemorrhage (). IgG level was maintained above 700 mg/dL, and intravenous immunoglobulin was administered monthly at a dose of 600 mg/kg. At the age of 2 years and 11 months, daily prednisolone was discontinued, and she received maintenance therapy with dexamethasone palmitate (1 mg/dose every 2 weeks). No further infections, recurrence of severe anemia, or respiratory distress were reported. Whole-exome sequencing did not reveal any pathogenic mutation related to inborn errors of immunity reported by IUIS 2022 besides that in KMT2D.
The cause of IPH remains unclear, but risk factors such as immunological associations, allergic reactions, genetic predisposition, and environmental exposure are considered for its development [Citation2]. Gürbüz et al. [Citation3] reported a case of Kabuki syndrome with a de novo heterozygous c.7411C > T (p.Arg2471Ter) mutation in the KMT2D gene, which was complicated by autoimmune thyroiditis. However, the patient in this study did not have IPH. To date, three cases of DAH complicating Kabuki syndrome presenting with IgA vasculitis [Citation4], immune thrombocytopenia [Citation5], and Goodpasture’s syndrome with bronchial artery abnormality [Citation6], respectively, have been reported. In each case report, the DAH improved after treatment of the underlying disease. In our case, the patient had no allergies to cow milk or wheat, and tests for various autoantibodies yielded negative results. Hemosiderin-laden macrophages were confirmed in BAL. It was difficult to rule out the possibility of pulmonary hemorrhage due to the patient’s right-to-left shunt and severe pulmonary hypertension that precluded Glenn surgery. The patient’s respiratory condition was poor, and considering the risks and benefits, a lung biopsy was not performed. However, based on the patient’s clinical course, which showed resolution of pulmonary hemorrhage with steroid pulse therapy without changing circulatory agonists, IPH was diagnosed.
There are currently no established guidelines for IPH treatment. Although steroids are commonly used, various immunosuppressants are also used [Citation2]. It has been reported that the administration of liposome-incorporated dexamethasone, specifically dexamethasone palmitate, is effective in treating both the acute and chronic phases of idiopathic pulmonary hemosiderosis (IPH). Dexamethasone palmitate at a dose of 0.05–0.08 mg/kg/day for three consecutive days is effective in arresting hemorrhage during the acute phase of pulmonary hemorrhage in IPH [Citation7, Citation8]. Furthermore, for maintenance therapy, it has been reported that extending the dosing interval to from weekly to biweekly to a monthly is effective [Citation7, Citation9]. In our case, we administered steroid pulse therapy to induce remission, switched to a dexamethasone palmitate once every two weeks for maintenance therapy, and discontinued daily oral prednisolone. Immunosuppressants, such as methotrexate and azathioprine, were not selected in view of renal impairment and contraindications to concomitant medications.
To our knowledge, this is the first report of IPH in a patient with KS. IPH should be considered when idiopathic pneumonia and anemia occur in patients with KS.
Author’s contributions
Y.U. was responsible for writing the manuscript and drafted the manuscript. K.Y. and H.O. revised the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was performed in line with the principles of the Declaration of Helsinki. Informed consent for participation and publication of this report and accompanying images was obtained from the patient’s parents. A copy of the written consent is available for review by the editor of this journal. This study was granted an exemption from requiring ethics approval by the research ethics committee of Saitama Children’s Medical Center.
Acknowledgments
We appreciate the support provided by the patient and their parents.
Disclosure statement
The authors declare that they have no conflict of interest.
Additional information
Funding
References
- Margot H, Boursier G, Duflos C, et al. Immunopathological manifestations in Kabuki syndrome: a registry study of 177 individuals. Genet Med. 2020;22(1):181–188. doi: 10.1038/s41436-019-0623-x.
- Saha BK. Idiopathic pulmonary hemosiderosis: a state of the art review. Respir Med. 2021;176:106234. doi: 10.1016/j.rmed.2020.106234.
- Gürbüz F, Özalp Yüreğir Ö, Ceylaner S, et al. Coexistence of Kabuki Syndrome and autoimmune thyroiditis. Jcrpe. 2016;8(1):105–106. doi: 10.4274/jcrpe.2686.
- Oto J, Mano A, Nakataki E, et al. An adult patient with Kabuki syndrome presenting with Henoch-Schönlein purpura complicated with pulmonary hemorrhage. J Anesth. 2008;22(4):460–463. doi: 10.1007/s00540-008-0656-9.
- Das S, Cherian S, Hamarneh W, et al. Diffuse alveolar hemorrhage secondary to idiopathic thrombocytopenic purpura; an extremely rare presentation. Chest. 2012;142(4):469A. doi: 10.1378/chest.1390830.
- Li S, Liu J, Yuan Y, et al. Case report: a study on the de novo KMT2D variant of Kabuki syndrome with Goodpasture’s syndrome by whole exome sequencing. Front Pediatr. 2022;10:933693. doi: 10.3389/fped.2022.933693.
- Doi T, Ohga S, Ishimura M, et al. Long-term liposteroid therapy for idiopathic pulmonary hemosiderosis. Eur J Pediatr. 2013;172(11):1475–1481. doi: 10.1007/s00431-013-2065-9.
- Doi T, Ohga S, Ishimura M, et al. Erratum to: long-term liposteroid therapy for idiopathic pulmonary hemosiderosis. Eur J Pediatr. 2015;174(12):1701–1701. doi: 10.1007/s00431-015-2638-x.
- Tobai H, Yano J, Sato N, et al. Successful liposteroid therapy for a recurrent idiopathic pulmonary hemosiderosis with down syndrome. Case Rep Pediatr. 2020;2020:5292947.