
Abstract
Background
Fostemsavir is an oral prodrug of temsavir, a first‐in‐class attachment inhibitor that binds HIV‐1 gp120, preventing initial HIV attachment and entry into host immune cells.
Objective
The pharmacokinetic interaction was determined between temsavir and maraviroc, a CCR5 allosteric inhibitor indicated for CCR5-tropic HIV-1 that may be co-administered with fostemsavir as part of combination antiretroviral therapy in heavily treatment-experienced adults with multidrug-resistant HIV-1 infection.
Methods
This was a Phase 1, open-label, single-sequence, 3-period crossover study evaluating the effect of fostemsavir on maraviroc pharmacokinetics and the effect of maraviroc on temsavir pharmacokinetics (ClinicalTrials.gov, NCT02480894). Fourteen healthy participants received fostemsavir 600 mg twice daily (BID) for 4 days in Period 1 (followed by a 3-day washout), maraviroc 300 mg BID for 5 days in Period 2, and fostemsavir 600 mg BID with maraviroc 300 mg BID for 7 days in Period 3. Study drugs were administered orally with a standard meal.
Results
Following fostemsavir and maraviroc co-administration, maraviroc area under the plasma concentration-time curve over the dosing interval (AUCτ) increased 25% (from 1914 to 2382 ng.h/mL) and maraviroc plasma concentration at the end of the dosing interval (Ctrough) increased 37% (from 36.5 to 49.9 ng/mL), but there was no change in maximum observed concentration (Cmax). Following fostemsavir and maraviroc co-administration, temsavir AUCτ and Cmax increased 10-13% and Ctrough decreased 10%.
Conclusions
Co-administration of fostemsavir and maraviroc did not result in clinically relevant changes in maraviroc or temsavir exposure. Fostemsavir and maraviroc may be co-administered without dose adjustment of either antiretroviral agent.
Introduction
Fostemsavir is a highly soluble methyl phosphate prodrug of the membrane-permeable but poorly soluble antiretroviral (ARV) temsavir, approved in combination with other ARVs for heavily treatment-experienced adults with multidrug-resistant HIV-1 infection (Reference ID: 4635448). A wide range of ARV combinations are used in patients with multidrug-resistant HIV-1 infection. Therefore, it is important to understand the drug interaction potential between fostemsavir and other ARV treatments, such as maraviroc.
Fostemsavir is metabolized by alkaline phosphatase at the luminal surface of the small intestine to yield active temsavir.Citation1 Pre-systemic conversion to temsavir is supported by the lack of quantifiable fostemsavir observed in blood after fostemsavir administration.Citation2 Temsavir, the active metabolite of fostemsavir, is a first-in-class attachment inhibitor that binds directly to the viral envelope glycoprotein 120 (gp120). The attachment of temsavir locks gp120 into a closed state and prohibits the conformational change necessary for initial interaction between HIV and the surface receptors on CD4 cells, thereby preventing attachment and subsequent entry into host T cells and other immune cells.Citation3–6
Fostemsavir has no in vitro cross-resistance with other classes of ARV drugs, including entry inhibitors.Citation6,Citation7 In addition, fostemsavir can be used regardless of HIV-1 tropism.Citation6–10 Among clinical isolates of HIV-1, a broad range of in vitro susceptibility to temsavir has been observed, which may be due to the substantial diversity in HIV-1 gp120.Citation6,Citation9
The absolute bioavailability of temsavir following administration of oral fostemsavir is 26.9%. Temsavir is predominantly metabolized by an esterase‐mediated hydrolysis pathway with contributions from a cytochrome P450 (CYP) 3A4‐mediated oxidative pathway.Citation11 Temsavir is a substrate of P‐glycoprotein (P‐gp) and breast cancer resistance protein (BCRP). Drugs that inhibit or induce CYP3A4, P-gp, and/or BCRP have the potential to alter the pharmacokinetic (PK) profile of temsavir. Temsavir does not inhibit or induce major CYP or uridine diphosphate glucuronosyltransferase (UGT) enzymes. Based on in vitro data, temsavir may be a clinically relevant inhibitor of BCRP, organic anion transporter protein (OATP)1B1, and OATP1B3, and this was confirmed in a clinical drug interaction study where co-administration of fostemsavir increased rosuvastatin, a BCRP and OATP1B1/3 substrate, area under the concentration-time curve from zero to infinity (AUC∞) by 69%.Citation12 A temsavir metabolite, BMS‐930644, inhibits CYP3A4, BCRP, multidrug and toxin extrusion 2 (MATE2-K), and organic cation transporter 1 (OCT1) with half maximal inhibitory concentration (IC50) values <10 μM; however, circulating BMS‐930644 concentrations are low (maximum concentration [Cmax] of approximately 458 ng/mL [∼1 μM] with fostemsavir 600 mg twice daily [BID] dosing), such that clinically significant interactions are unlikely.
Maraviroc is a selective C-C chemokine receptor type 5 (CCR5) antagonist preventing CCR5-tropic HIV-1 entry into cells. Maraviroc is co-administered with other ARV drugs for the treatment of HIV. The absolute bioavailability of maraviroc is 23%.Citation13 Maraviroc is primarily metabolized by CYP3A with renal clearance accounting for approximately 23% of total clearance.Citation13,Citation14 Maraviroc is considered a sensitive CYP3A substrate because a ≥5-fold increase in maraviroc AUC was observed when strong CYP3A inhibitors, such as ketoconazole and saquinavir/ritonavir, were co-administered.Citation15 Furthermore, co-administration of the strong CYP3A inducer rifampin reduced maraviroc AUC over the dosing interval (AUCτ) by 68%.Citation16 Maraviroc is also a substrate of P-gp and OATP1B1, and inhibition of these transporters contributes to the overall magnitude of drug interaction.Citation17–19 Drugs that inhibit or induce CYP3A, P-gp, and/or OATP1B1 have the potential to alter the PK profile of maraviroc. Maraviroc is not an inhibitor or inducer of CYP enzymes and is not an inhibitor of P-gp, BCRP, OATP1B1, MRP2, or renal transporters.Citation20,Citation21
Because temsavir and its metabolite, BMS‐930644, have the potential to inhibit OATP1B1 and CYP3A4 pathways involved in maraviroc disposition, respectively, it is possible that co-administration of fostemsavir with maraviroc will increase plasma maraviroc exposure. Co-administration of fostemsavir with maraviroc is not expected to alter the PK profile of temsavir because maraviroc does not inhibit or induce drug-metabolizing enzymes or transporters.
The objective of the current study was to evaluate the potential for drug-drug interaction when co-administering fostemsavir 600 mg BID and maraviroc 300 mg BID in healthy adults. The primary PK parameters were AUCτ, Cmax, and concentration at the end of the dosing interval (Ctrough) for each drug when administered alone and in combination. Safety and tolerability were assessed by collection of adverse events (AEs) and clinical safety laboratory results.
Materials and methods
Ethics and study participants
The study was conducted at ICON Early Phase Services, LLC, San Antonio, TX, USA. The study protocol, protocol amendments, and participant consent forms were reviewed and approved by an independent ethics committee (IntegReview, Austin, TX, USA). All participants provided written informed consent.
Healthy men and women 18-55 years, with a body mass index of 18.0-32.0 kg/m2, were eligible for the study. Eligible participants had no clinically significant deviations from normal in medical history, physical examinations, 12-lead electrocardiograms (ECGs), or clinical laboratory test results. Women of childbearing potential (WOCBP) who were not nursing or pregnant, were using acceptable non-hormonal methods of contraception, and had a negative serum or urine pregnancy test within 24 hours prior to the start of study drug were eligible for inclusion in the study. Investigators advised WOCBP and male participants who were sexually active with WOCBP on the use of highly effective methods of contraception.
Exclusion criteria were related to medical history and concurrent diseases, physical examination findings and clinical laboratory test results, allergies (for example, history of allergy to fostemsavir, HIV attachment inhibitors, CCR5 antagonists or related compounds), and adverse drug reactions. Participants were excluded if they had a positive blood screen for hepatitis C antibody, hepatitis B surface antigen, or HIV-1 and HIV-2 antibodies and HIV-1 RNA. Prohibited and/or restricted medications included prior exposure to fostemsavir or maraviroc within 12 weeks of study drug administration, exposure to any investigational drug or placebo within 4 weeks of study drug administration, and use of any prescription drugs or over-the-counter acid-reducing agents within 4 weeks prior to study drug administration except those medications cleared by the medical monitor. No concomitant medications (prescription, over-the-counter, or herbal) were to be administered during the study unless prescribed for treatment of specific clinical events.
Study design and treatments
This was a Phase 1, non-randomized, open-label, 3-period, single-sequence, crossover study to evaluate the drug-drug interaction between fostemsavir 600 mg BID and maraviroc 300 mg BID in 14 healthy participants (). Eligible participants were admitted to the clinic the day before dosing (Day −1) and remained confined to the clinic until study discharge on Day 19. Fostemsavir and maraviroc were administered as oral tablets. Morning doses were administered with standard meals (400-500 calories; approximately 30% fat) and evening doses were administered with snacks (300-350 calories). Identical meals and snacks were administered on intensive PK sampling days (Days 4, 11, 18). Participants consumed the meal or snack within 25 minutes and study drug was administered within 5 minutes of completion of the meal or snack.
Figure 1. Study design. BID, twice daily.
Assessments and bioanalytical methods
Blood samples for analysis of temsavir in plasma were collected in di-potassium ethylenediaminetetraacetic acid (K2EDTA)-containing tubes pre-dose and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, and 12 hours after dosing on Day 4 (Period 1) and Day 18 (Period 3). Blood samples for analysis of maraviroc in plasma were collected in K2EDTA-containing tubes pre-dose and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, and 12 hours after dosing on Day 11 (Period 2) and Day 18 (Period 3). Plasma samples were assayed for temsavir and maraviroc concentrations using validated methods at Covance, West Trenton, NJ, USA. Both analytical methods used protein precipitation to extract drug from plasma, and extracts were analyzed by high performance liquid chromatography with tandem mass spectrometry. The lower and upper limits of quantification were 5 and 5000 ng/mL for temsavir and 1 and 1000 ng/mL for maraviroc. Based on analytical quality control samples, between-run precision (%CV), within-run precision (%CV), and accuracy (% bias) were ≤3.3%, ≤5.5%, and −1.3% to 2.0% for temsavir and ≤5.0%, ≤4.1%, and 7.8% to 10.5% for maraviroc.
Temsavir and maraviroc PK parameters, Cmax, time to Cmax (tmax), AUCτ, and Ctrough were derived from the plasma concentration-time data using noncompartmental methods with Phoenix WinNonlin Professional Network Edition, Version 6.3 (Certara, Princeton, NJ, USA); the linear up-log down method was used for the AUCτ calculations.
Safety was assessed by AEs, physical examinations, vital signs, ECGs, and clinical laboratory tests. AEs were collected throughout the study. Physical examination was completed at Days −1 and 19, and at the follow-up visit; vital signs were collected at Days −1, 4, 10, 15, 18, and at the follow-up visit; and ECGs were collected at Days −1 and 18. Clinical laboratory tests were collected on Days −1, 5, 11, and 18, with additional liver function tests on Days 8, 13, and 15.
Statistical analyses
SAS® software Version 9.3 (SAS Institute, Inc., Cary, NC, USA) was used for statistical analyses. To assess the effect of fostemsavir on the PK of maraviroc and the effect of maraviroc on the PK of temsavir, for each analyte separately, a general linear mixed effect model with treatment as fixed effect and measurements within each participant as repeated measures were fitted to the log-transformed Cmax, AUCτ, and Ctrough for use in the estimation of effects and construction of 90% confidence intervals (CIs). Kenward-Rogers degrees of freedom were specified in the model. Point estimates and 90% CIs for treatment differences on the log scale were exponentiated to obtain estimates for geometric mean ratios (GMRs) and 90% CIs on the original scale.
Results from a food effect study demonstrated that co-administration of maraviroc with a high-fat breakfast reduced the Cmax and AUCτ by 33%, but maraviroc can be administered with or without food based on the prescribing information.Citation21 Results from a hepatic impairment study showed that AUCτ was ∼50% higher in patients with moderate impairment compared with healthy volunteers; however, no dose adjustment is recommended in this population.Citation20,Citation21 Therefore, pre-specified no-clinically-significant-effect bounds were 0.67 to 1.50 for maraviroc Cmax and AUCτ based on the food effect study for the lower bound and the hepatic study for the upper bound.Citation20,Citation21 Assuming 12 participants completed the study, the GMRs were close to 1.00, log(Cmax) and log(AUCτ) were normally distributed, and within-participant variability was ≤27% for Cmax and ≤19% for AUCτ, the power that the 90% CI for the GMRs of Cmax and AUCτ of maraviroc were both within 0.67 to 1.50 was 92%. Default no effect bounds of 0.80 to 1.25 were pre-specified for temsavir Cmax and AUCτ. Assuming 12 participants completed the study, log(Cmax) and log(AUCτ) were normally distributed, and within-participant variability was ≤27% for Cmax and ≤19% for AUCτ, there was an 80% probability for the 90% CI to be within 80% and 125% of the point estimate of the GMR for Cmax and within 85% and 117% of the point estimate of the GMR for AUCτ.
Results
Participant disposition and characteristics
In total, 14 participants were enrolled. All 14 participants completed Period 1, 13 participants completed Period 2, and 12 participants (86%) completed the study. Two participants discontinued due to AEs (1 participant discontinued due to nausea, dizziness, and cold sweat and 1 participant discontinued due to gastritis). Participants in this study were predominantly male (71%) and white (64%) or Black/African American (36%). The mean (range) age was 33 (21-47) years and weight was 77.4 (61.1-101) kg.
Pharmacokinetics
Effect of fostemsavir on maraviroc PK
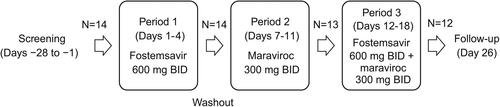
Mean maraviroc plasma concentration-time profiles with and without co-administration of fostemsavir are shown in . Statistical analysis of maraviroc PK parameters following administration with and without fostemsavir is shown in . Co-administration of maraviroc 300 mg BID with fostemsavir 600 mg BID increased geometric mean plasma maraviroc AUCτ 25% and Ctrough 37%, but there was no change in Cmax. The 90% CI for plasma maraviroc Cmax and AUCτ GMRs were contained entirely within the pre-specified limits of 0.67 to 1.50 to conclude no clinically relevant effect of fostemsavir on maraviroc PK. Individual treatment ratios of maraviroc PK parameters are presented in .
Figure 2. Arithmetic mean (standard deviation) plasma concentration-time profiles of maraviroc after administration of maraviroc 300 mg BID alone and in combination with fostemsavir 600 mg BID. BID, twice daily.
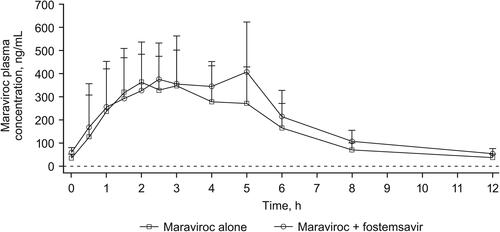
Figure 3. Individual treatment ratios of PK parameters of maraviroc (evaluable PK analysis set). Open circles represent individual ratios and closed circles represent adjusted geometric mean ratios, and connected bars represent 90% confidence intervals of the geometric mean ratios. AUCτ, area under the concentration-time curve over the dosing interval; BID, twice daily; Ctrough, concentration at the end of the dosing interval; Cmax, maximum concentration; PK, pharmacokinetic.
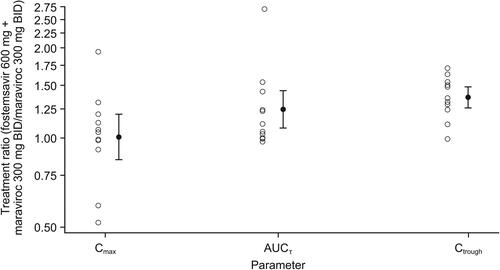
Table 1. Effect of fostemsavir 600 mg BID on maraviroc PK.
Effect of maraviroc on temsavir PK
Mean temsavir plasma concentration-time profiles following administration of fostemsavir with and without maraviroc are shown in . Statistical analysis of temsavir PK parameters following administration of fostemsavir with and without maraviroc is shown in . Co-administration of maraviroc 300 mg BID with fostemsavir 600 mg BID increased geometric mean temsavir Cmax 13% and AUCτ 10% and decreased Ctrough 10%; these changes are not clinically relevant. Individual treatment ratios of temsavir PK parameters are presented in .
Figure 4. Arithmetic mean (standard deviation) plasma concentration-time profiles of temsavir after administration of fostemsavir 600 mg BID alone and in combination with maraviroc 300 mg BID. BID, twice daily.
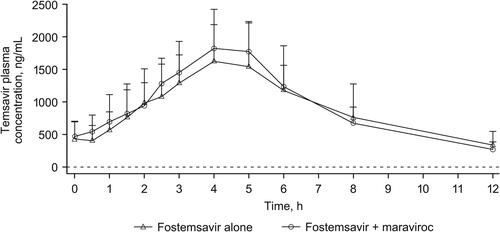
Figure 5. Individual treatment ratios of PK parameters of temsavir (evaluable PK analysis set). Open circles represent individual ratios and closed circles represent adjusted geometric mean ratios, and connected bars represent 90% confidence intervals of the geometric mean ratios. AUCτ, area under the concentration-time curve over the dosing interval; BID, twice daily; Ctrough, concentration at the end of the dosing interval; Cmax, maximum concentration; PK, pharmacokinetic.
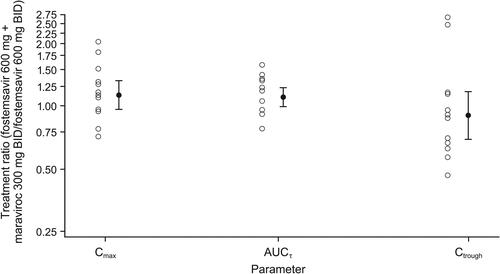
Table 2. Effect of maraviroc 300 mg BID on temsavir PK.
Safety
Administration of fostemsavir 600 mg BID alone, maraviroc 300 mg BID alone, and the combination of fostemsavir 600 mg BID with maraviroc 300 mg BID for up to 7 days were well tolerated in healthy adult participants. There were no deaths or serious AEs. There were no clinically significant findings in clinical laboratory test results, physical examinations, ECGs, or vital signs.
Overall, 8 participants (57%) reported at least 1 AE, 3 participants (21-23%) in each period. AEs reported in at least 2 participants overall were dizziness, headache, and nausea. All AEs were mild, except for the moderate AEs experienced by one participant who discontinued from the study. Two AEs, mild abdominal distention and mild decreased appetite (each reported by 1 participant), were considered related to study drug.
Two participants discontinued due to AEs. One participant discontinued due to nausea, dizziness, and cold sweat on Day 18 (fostemsavir + maraviroc); symptoms started prior to dosing on Day 18, the participant was given fluids, and symptoms resolved after approximately 2 hours. One participant discontinued due to gastroenteritis; this participant developed moderate nausea, moderate gastroenteritis, mild diarrhea, and mild vomiting on Day 6 (during the washout between and 2 days after the last dose of fostemsavir alone). Ondansetron was administered on Day 6, but the nausea, gastroenteritis, diarrhea, and vomiting continued on Day 7, and the participant also reported myalgia. Ondansetron, loperamide, intravenous fluids, and ibuprofen were given on Day 7, and the AEs resolved on Day 7 (nausea, diarrhea, emesis, myalgia) or Day 8 (gastroenteritis).
Discussion
Fostemsavir, a first-in-class attachment inhibitor, is approved in combination with other ARVs for heavily treatment-experienced adults with multidrug-resistant HIV-1 infection. A wide range of ARV combinations are used in patients with multidrug-resistant HIV-1 infection, and selection of ARV agents for individual patients is based on factors such as history of prior ARV use, viral susceptibility data, comorbidities, and potential for drug interactions. To support the use of fostemsavir in various ARV combinations, it is important to understand the drug interaction potential of fostemsavir. This study evaluated the PK interaction between fostemsavir and maraviroc, a CCR5 antagonist for the treatment of CCR5-tropic HIV-1 infection, to inform dosing recommendations in anticipation of their possible co-administration in clinical trials (eg, the BRIGHTE study) and ultimately in the broader patient population as fostemsavir has no in vitro cross-resistance with other classes of ARV drugs, including entry inhibitors.
Administration of fostemsavir with standard meals had no impact on temsavir Cmax or AUCCitation22 and a high-fat meal reduced maraviroc Cmax and AUC by 33%, which is not considered clinically meaningful.Citation21 The conduct of this study in the fed state (standard meals; 400-500 calories; approximately 30% fat) is not expected to influence the drug interaction results of this study because both treatment periods were in the fed state and given the minimal impact of food on fostemsavir and maraviroc absorption.
It was hypothesized that co-administration of fostemsavir with maraviroc may increase maraviroc exposure as a result of OATP1B1 inhibition by temsavir and/or CYP3A4 inhibition by its metabolite BMS‐930644. Co-administration of fostemsavir 600 mg BID with maraviroc 300 mg BID increased geometric mean plasma maraviroc AUCτ and Ctrough by 25% and 37%, respectively, with no change in Cmax. Importantly, the 90% CIs for the treatment ratio (fostemsavir + maraviroc vs. maraviroc alone) were contained within the protocol‐defined boundaries for no clinically significant effect. It was also hypothesized that co-administration of fostemsavir with maraviroc would not alter the PK profile of temsavir because maraviroc does not inhibit or induce CYPs or transporters relevant to temsavir. Results were consistent with this hypothesis because geometric mean changes in plasma temsavir Cmax, AUCτ, and Ctrough were ≤13%. Furthermore, co-administration of fostemsavir 600 mg BID with maraviroc 300 mg BID was well tolerated and produced no new safety signals.
Results of this drug interaction study determined that fostemsavir and maraviroc may be co-administered without dose adjustment of either drug. Maraviroc can be an important treatment option for heavily treatment-experienced patients with multidrug-resistant HIV-1-infection. In fact, 16% (60/371) of all participants in the Phase 3 BRIGHTE trial included maraviroc as part of the initial optimized background regimen to be paired with fostemsavir. The ongoing BRIGHTE study (NCT02362503), being conducted in patients with multidrug resistance and limited therapeutic options, has demonstrated good efficacy (virologic and immunologic response) and a favorable safety and tolerability profile through 96 weeks of treatment with fostemsavir plus optimized background therapy.Citation23–25
Conclusion
Co-administration of fostemsavir and maraviroc did not result in clinically relevant changes in maraviroc or temsavir exposure. Fostemsavir and maraviroc may be co-administered without dose adjustment of either ARV agent.
Disclosure of interest
K.M. and P.A. are employees of ViiV Healthcare. M.W. and C.L. were employees of ViiV Healthcare at the time the manuscript was developed. M.M. is an employee of GlaxoSmithKline.
Acknowledgments
We thank all study participants and their families, and ICON Early Phase Services, LLC, San Antonio, TX, USA. ViiV Healthcare acquired fostemsavir from Bristol‐Myers Squibb and we thank all Bristol‐Myers Squibb scientists for their contributions.
Data availability statement
Anonymized individual participant data and study documents can be requested for further research from www.clinicalstudydatarequest.com.
Additional information
Funding
References
- Brown J, Chien C, Timmins P, et al. Compartmental absorption modeling and site of absorption studies to determine feasibility of an extended-release formulation of an HIV-1 attachment inhibitor phosphate ester prodrug. J Pharm Sci. 2013;102(6):1742–1751.
- Nettles RE, Chien C, Elefant E, et al. Single and multiple dose pharmacokinetics and safety in non‐HIV‐infected healthy subjects dosed with BMS‐663068, an oral HIV attachment inhibitor. Paper presented at: 12th International Workshop on Clinical Pharmacology of HIV Therapy; April 13–15, 2011; Miami, FL. Abstract O_04.
- Langley DR, Kimura SR, Sivaprakasam P, et al. Homology models of the HIV-1 attachment inhibitor BMS-626529 bound to gp120 suggest a unique mechanism of action. Proteins. 2015;83(2):331–350.
- Pancera M, Lai Y-T, Bylund T, et al. Crystal structures of trimeric HIV envelope with entry inhibitors BMS-378806 and BMS-626529. Nat Chem Biol. 2017;13(10):1115–1122.
- Lataillade M, Krystal M, Ackerman P. Conceptualization and design of mechanism of action for temsavir. Paper presented at: 16th European AIDS Conference; October 25–27, 2017; Milan, Italy. Abstract.
- Nowicka‐Sans B, Gong Y-F, McAuliffe B, et al. In vitro antiviral characteristics of HIV‐1 attachment inhibitor BMS‐626529, the active component of the prodrug BMS‐663068. Antimicrob Agents Chemother. 2012;56(7):3498–3507.
- Li Z, Zhou N, Sun Y, et al. Activity of the HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068, against CD4-independent viruses and HIV-1 envelopes resistant to other entry inhibitors. Antimicrob Agents Chemother. 2013;57(9):4172–4180.
- Ray N, Hwang C, Healy MD, et al. Prediction of virological response and assessment of resistance emergence to the HIV-1 attachment inhibitor BMS-626529 during 8-day monotherapy with its prodrug BMS-663068. J Acquir Immune Defic Syndr. 2013;64(1):7–15.
- Zhou N, Nowicka‐Sans B, McAuliffe B, et al. Genotypic correlates of susceptibility to HIV-1 attachment inhibitor BMS-626529, the active agent of the prodrug BMS-663068. J Antimicrob Chemother. 2014;69(3):573–581.
- Lalezari JP, Latiff GH, Brinson C, et al. Safety and efficacy of the HIV-1 attachment inhibitor prodrug BMS-663068 in treatment-experienced individuals: 24 week results of AI438011, a phase 2b, randomised controlled trial. Lancet HIV. 2015;2(10):e427–e437.
- Gorycki P, Magee M, Ackerman P, et al. Pharmacokinetics, metabolism and excretion of radiolabeled fostemsavir administered with or without ritonavir in healthy male subjects. Paper presented at: 19th International Workshop on Clinical Pharmacology of Antiviral Therapy; May 22–24, 2018; Baltimore, MD. Abstract 42.
- Landry I, Vakkalagadda B, Lubin S, et al. HIV-1 attachment inhibitor prodrug BMS-663068: PK assessment with rosuvastatin. Paper presented at: Conference on Retroviruses and Opportunistic Infections; February 22–25, 2016; Boston, MA. Abstract 460.
- Abel S, Russell D, Whitlock LA, Ridgway CE, Nedderman ANR, Walker DK. Assessment of the absorption, metabolism and absolute bioavailability of maraviroc in healthy male subjects. Br J Clin Pharmacol. 2008;65(s1):60–67.
- Hyland R, Dickins M, Collins C, Jones H, Jones B. Maraviroc: in vitro assessment of drug-drug interaction potential. Br J Clin Pharmacol. 2008;66(4):498–507.
- Abel S, Russell D, Taylor-Worth RJ, Ridgway CE, Muirhead GJ. Effects of CYP3A4 inhibitors on the pharmacokinetics of maraviroc in healthy volunteers. Br J Clin Pharmacol. 2008;65(s1):27–37.
- Abel S, Jenkins TM, Whitlock LA, Ridgway CE, Muirhead GJ. Effects of CYP3A4 inducers with and without CYP3A4 inhibitors on the pharmacokinetics of maraviroc in healthy volunteers. Br J Clin Pharmacol. 2008;65(s1):38–46.
- Kimoto E, Vourvahis M, Scialis RJ, Eng H, Rodrigues AD, Varma MVS. Mechanistic evaluation of the complex drug-drug interactions of maraviroc: contribution of cytochrome P450 3A, P-glycoprotein and organic anion transporting polypeptide 1B1. Drug Metab Dispos. 2019;47(5):493–503.
- Siccardi M, D'Avolio A, Nozza S, et al. Maraviroc is a substrate for OATP1B1 in vitro and maraviroc plasma concentrations are influenced by SLCO1B1 521 T > C polymorphism. Pharmacogenet Genomics. 2010;20(12):759–765.
- Walker DK, Abel S, Comby P, Muirhead GJ, Nedderman ANR, Smith DA. Species differences in the disposition of the CCR5 antagonist, UK-427,857, a new potential treatment for HIV. Drug Metab Dispos. 2005;33(4):587–595.
- Abel S, Back DJ, Vourvahis M. Maraviroc: pharmacokinetics and drug interactions. Antivir Ther. 2009;14(5):607–618.
- SELZENTRY® (maraviroc) [package insert]. Research Triangle Park, NC: ViiV Healthcare; October, 2020.
- Sevinsky H, Magee M, Ackerman P, et al. The effect of food on the pharmacokinetics of the HIV-1 attachment inhibitor temsavir, the active moiety of the prodrug fostemsavir. Paper presented at: 18th International Workshop on Clinical Pharmacology of Antiviral Therapy; June 14–17, 2017; Chicago, IL. Poster 23.
- Kozal M, Aberg J, Pialoux G, et al. Fostemsavir in adults with multidrug-resistant HIV-1 infection. N Engl J Med. 2020;382(13):1232–1243.
- Lataillade M, Lalezari JP, Kozal M, et al. Safety and efficacy of the HIV-1 attachment inhibitor prodrug fostemsavir in heavily treatment-experienced individuals: week 96 results of the phase 3 BRIGHTE study. Lancet HIV. 2020;7(11):e740–e751.
- Ackerman P, Wilkin T, Pierce A, et al. Clinical impact of antiretroviral agents used in optimized background therapy with fostemsavir in heavily treatment-experienced adults with HIV-1: exploratory analyses of the phase 3 BRIGHTE study. Paper presented at: 11th IAS Conference on HIV Science; July 18‒21, 2021; Virtual. Poster PEB155.