
ABSTRACT
Cardiac Arrhythmias (CA) are responsible for syncope and sudden cardiac death (SCD) in several million cases worldwide. The molecular basis of CA pathogenesis is known, but the underlying mechanisms connecting genotype to phenotype changes in the proteins are not yet fully explored. Hence, in this study, three LQT2-linked missense mutations (Thr613Met, Ser641Phe and Gly648Ser) located in exon-7 region of KCNH2 gene identified in CA patients was assessed for their deleteriousness using diverse computational algorithms. The SIFT and PolyPhen-2 methods, which depends on sequence conservation across species, and protein structure-based information have attributed pathogenicity to these variants. Protein behavior studies at 3D level have identified that these three missense mutations induce biochemically severe amino acid changes that may alters the structure and function of KCNH2. Our molecular docking suggested that mutant KCNH2 protein shows a similar binding interactions to KB-R7943 as compared to the wild type model. These findings could be useful in resorting to alternate therapies for subset of CA patients who show limited therapeutic response to KB-R7943. This study supports that in silico approaches could act as a first-line filters to screen the potential impact of CA linked KCNH2 mutations depending upon the perspective of molecular evolution and phenotype of proteins.
1. Introduction
Cardiac arrhythmias or irregular heartbeats are prevalent across all age groups and are responsible for syncope (fainting) and sudden cardiac death (SCD) in several million cases worldwide (Afzal et al. Citation2019). Primary cardiac arrhythmias, which constitute a significant percentage of all forms of arrhythmias are caused by a heritable abnormality of ion channel functions in heart (Fu Citation2015). Hence, exploring the intricate and complex mechanisms underlying cardiac arrhythmias has become the main challenge for scientists and clinicians. Genetic pathology has been described in clinical subclassifications of hereditary arrhythmias e.g. Long QT syndrome, short QT syndrome, Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia, sick sinus syndrome, cardiac conduction defect, atrial fibrillation, etc (Li et al. Citation2016). Long QT syndrome (LQTS), a prolonged ventricular repolarization condition is a genetically heritable cardiac arrhythmic disease which leads to sudden death. Genetic mutations in cardiac ion channel proteins are described to be the underlying causes for different types of LQTS phenotypes (Tester and Ackerman Citation2014). LQTS2 is a frequent form accounting for 35% to 40% of all the different types of LQTS phenotypes (Singh et al. Citation2019). The gene which is known to cause LQTS2 is called Potassium Voltage-Gated Channe; Subfamily H Member 2 (KCNH2), it codes for α-subunit of voltage-gated inwardly rectifying K+ channel (Saprungruang et al. Citation2018). The mutations in KCNH2 lead to a reduction in rectifer K+ current (IKr), which causes prolonged QT intervals and ventricular fibrillation based on the nature of mutation (De Zio et al. Citation2019).
With the advancement in sequence technologies, the number of KCNH2 mutations (missense mutations, indels, frameshift mutations, duplications) in CA patients are continuously increasing (Guillen Sacoto et al. Citation2015; Saprungruang et al. Citation2018; Fan et al. Citation2019). Of these, the mutations which result in substitution of amino acids are presumed to be deleterious as they could potentially alter both biophysical characteristics and functional aspects of the encoded protein. In KCNH2, the structural and functional aspects of most missense mutations are not fully characterized. The technical complexity surrounding the traditional cell line and animal model-based mutation characterization strategies makes them a relatively less attractive option. On the other hand, the ability to study these missense mutations can be improved based on the information, like conservation of sequences across species (Sim et al. Citation2012), structural attributes (Ittisoponpisan et al. Citation2019), stability (Witvliet et al. Citation2016) and physicochemical properties of polypeptides (Li et al. Citation2014). In this context, we studied LQT2- linked KCNH2 missense mutations (T613M, S641P and G648S) using different bioinformatic webservers and assessed their prediction potential in exploring the genetic sequence – protein structure relationships in terms structure deviation, secondary structure and stability parameters by using different computational methods. The three KCNH2 mutations i.e. T613 (pore helix), S641 (upper end of S6) and G648 (hinge region at which S6 kinks to open the channel) fall in potassium voltage gated channel regions, hence they are ideal candidates to study the mutation induced to perturbations in the structure and function of protein.
2. Methods
2.1. KCNH2 mutation data collection
The molecular genetic data on KCNH2 mutations were retrieved from databases (ex: pubmed, and HGMD) and research publications (Tester et al. Citation2005; Kapplinger et al. Citation2009; Saprungruang et al. Citation2018). The information about KCNH2 mutations including transcript ID, nucleotide and amino acid sequences of KCNH2 gene were collected from KEGG gene database (https://www.genome.jp/kegg/genes.html) and ENSEMBLE (https://www.ensembl.org/) (Table ).
Table 1. Basic characteristics of KCNH2 missense variants analyzed in this study.
2.2. Predicting the deleterious potential of KCNH2 missense mutations by computational methods
We measured the deleterious potential of missense mutations using different computational methods that were trained to discriminate the mutations into deleterious or non-deleterious based on the support vector algorithmic programs. In brief, we submitted the mutation details like chromosome number and base-pair position followed by allele change to an empirical rule-based tool viz. SIFT (Sorting Intolerant from Tolerant) BLink Beta version for homology searching (Ng and Henikoff Citation2003). SIFT provides prediction score for each mutation and classify them as borderline (0.101–0.20) or tolerant (0.201–1.00), potentially damaging (0.051–0.10) and damaging (0.00–0.05). The PolyPhen-2 (Polymorphism Phenotyping version 2) method predicts the probable deleterious effects of amino acid changes based on the structure and function of human proteins (phylogenetic and structure). Input options constitute protein sequence, database ID/accession number and position of the amino acid variants. The PolyPhen-2 score classify the mutations as probably damaging (1.00–0.80), possibly damaging (0.80–0.50) or benign (0.49–0.0) (Adzhubei et al. Citation2010).
2.3. 3-dimensional modeling, structural differences, stability, solvent accessibility and secondary structural analysis of KCNH2 protein models
The Protein Databank (PDB) has a 544 aa (from 333th to 877th aa) long 3-dimensional crystal structure of KCNH2 protein with 3.8 Å resolution (PDB; reference ID is 5VA2). The crystal structure KCNH2 model (of the original 1159 aa) was used as a coordinate to construct the remaining 615 aa long polypeptide chain following the I-Tasser ab-initio and Modeller 9v2, Swiss Model homology modeling approaches, which is basically used when the identical structure is not available, or the target sequence has less than 30% identity. The I-Tasser built protein models were energy minimized, by applying the steepest decent algorithm available in Gromacs software (Roy et al. Citation2010). The energy minimized protein models, structural quality assessment was carried out by PROCHECK (Laskowski et al. Citation1993). The superimposition, secondary structure and solvent accessibility tests were carried out using Pymol software (Rigsby and Parker Citation2016), PDBSUM (Laskowski et al. Citation2018) and DUET (Pires et al. Citation2014) webservers.
2.4. KCNH2 protein structure alignment analysis
The deleterious missense mutations not only change the sequence of amino acids, but they will also alter the physical configuration of proteins by inducing physical constraints both at residue and whole structure levels. In this study, we performed structural alignment of mutant and wild type KCNH2 proteins by superimposing their Cα traces and backbone atoms to calculate the root mean squared deviation (RMSD). A RMSD score of <2.0 Å for proteins and <0.2 Å for amino acids represents that the given amino acid substitution is deleterious to the structure of protein (Zemla Citation2003; Kufareva and Abagyan Citation2012).
2.5. Molecular docking for protein-inhibitor complex
AutoDock program was used to execute a docking simulation between the known sodium–calcium exchange inhibitor and KCNH2 protein. The binding conformation of inhibitor to the protein was analyzed by Lamarckian Genetic Algorithm (LGA). The protein structure was charged by adding charged ions and Histidine molecules to neutralize the whole protein. Finally, gigester charges were applied to neutralize the charges on the whole protein. Each atom type of the ligand was placed in a grid point, and interaction energy with all atoms of the protein was calculated. The default parameters used in constructing grid parameter file are 60 × 60 × 60 in X, Y, Z directions with an 0.367 Å spacing in the grid. Autodock MGLtools were used to prepare the docking parameter file. Lamarckian genetic algorithm with default parameters like the genetic algorithm settings set to 150 runs, 150 possible conformations; population size to 50 individuals; and the number of energy evaluations to 2,500,000 were used. In the RMSD based on positional all atoms, the results with a difference of less than 1 Å were gathered. At the end of docking, ligands with the maximum promising binding energy were selected. The resultant complex structures were explored using Pymol-0.98 program.
3. Results
3.1 KCNH2 mutation details and pathogenicity prediction
SIFT depends on nucleotide sequence homology across species and amino acid properties to classify any amino acid substitution as neutral or pathogenic to the protein functionality. In the present study, SIFT analysis predicted that T613M, S641P, and G648S variants are deleterious because all of them possessed ‘0’ prediction score. Any variant which is predicted to have >0.1 score by SIFT is deleterious. PolyPhen-2, which takes into account variant effects on nucleotide sequence and structural conservation changes, has also predicted T613M, S641P and G648S variants as damaging. PolyPhen-2 considers any variant as damaging if its prediction score is above 0.9 (Table ).
Table 2. Shows pathogenic predictions scores and amino acid changes of all three CA linked KCNH2 mutaions.
3.2 Stability analysis
The location and type of a substituted amino acid residue determine the stability of protein. In particular, amino acid substitutions that lead to changes in their orientation i.e. from exposed to buried state or vice versa may affect the secondary properties (ex. solvent accessibility) and also structural stability of proteins. In this context, we examined the protein stability (in terms of free energy values) and solvent accessibility of Thr613Met, Ser641Phe and Gly648Ser mutations using DUET web server. The negative free energy values suggest the query mutation could destabilize the protein, hence deleterious (Table ).
3.3 KCNH2 protein modeling
3.3.1 3-dimensional (3D) protein modeling.
The KCHN2 sequence (1159 aa) to protein structure alignment in Blastp program identified that experimentally solved structures available from Protein Data Bank (PDB) have a maximum sequence coverage of 72%. The structure of this covered protein structure spans over a NH2-terminal (1–140 aa), transmembrane segment (337–877 aa) and COOH terminal (1006–1159 aa) regions. The structure of missing regions lying in between 141 aa to 332 aa & 878 aa to 1005 aa is generated through I-Tasser Tool. From the top 5 predicted models, the best KCNH2 protein structure was selected based on its confidence score, template model and root mean square deviation score (Supplementary Table 1). High-quality polypeptides showed a C-score of −3.44 (Chain 1) and −3.77 (Chain 2). Using Modeller and Swiss Model tools we joined all the chains (experimentally solved structures and ab-initio models) and built 100 different confirmations of KCNH2 full-length model with open state pore. The built model was energy minimized and further used as a template to build KCNH2 mutated models (T613M, S641P and G648S). The stereochemical quality of the proteins are validated using the Ramachandran plot, which revealed that 97% amino acids are in core region and only 3% of amino acids were in disallowed regions (Figure ).
Figure 1. Molecular view of built KCNH2 model (a) cytosolic N-terminal PAS domain (b) voltage gated transmembrane domain (VAS) (c) cytosolic C-terminal domain.
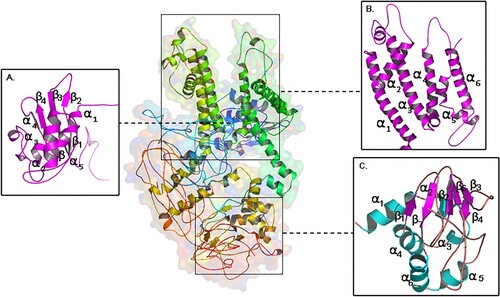
3.3.2 Structure analysis
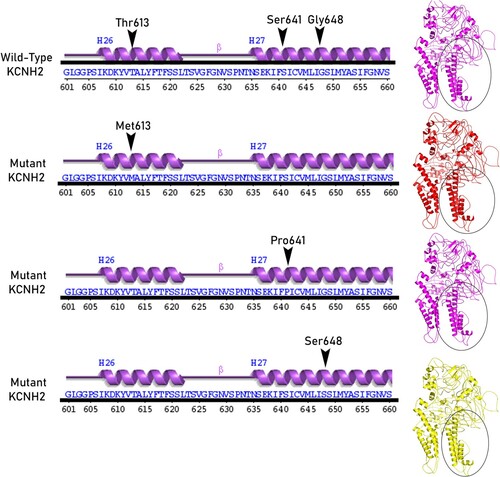
The core Per-Arnt-Sim (PAS) domain (cytosolic N-terminal region) of KCNH2 protein consists of five α-helices and four β-sheets. The transmembrane segment (VSD) consists of six α-helices, where the helices 1 to 4 make up voltage sensor domain and the helices 5,6 cytosolic pore domain. The nucleotide-binding domain (cytosolic C-terminal region) consists of five α-helices, and seven β-sheets. The mutated residue T613 is located in the short helix (connecting the S5 and S6 segments), and residues S641 and G648 are located in the S6 helix which lines the inner channel pore (Figure ). In structure-based superposition method, the differences of the Cα-atom coordinates, between the structures or subset of atoms are estimated in form of RMSD by rotating them in 3D space. All the mutant KCNH2 models (T613M, S641P and G648S) demonstrated 99.68% sequence identity in their amino acid sequence excluding the mutated residue. No significant difference in the main structure of KCNH2 protein in mutant conditions, as reflected by the RMSD scores of individual missense mutations (RMSD scores are <0.2 Å), is observed. However, all 3 KCNH2 variants portrayed structural differences at amino acid residue level, as reflected by their RMSD score (>2 Å). All the KCNH2 amino acid variants T613M (2.48 Å), S641P (2.33 Å) and G648S (1.86 Å) showed significant structural deviations compared to the wild type amino acid residues. The secondary structure of KCNH2 protein molecule is characterized by 4 β-sheets, 4 β-hairpins,12 β-strands, 38 α-helices, 44 helix-helix interactions, 117 β-turns and 29 γ-turns. The native threonine residue at 613 amino acid position is localized in helix 26 of KCNH2 molecule and the mutant Methionine residue does not induce any change in its helix position. Similarly, both Ser641Phe and Gly648Ser, localized in helix 27 position and does not induce any change in its orientation. None of the T613M, S641P and G648S variants have shown any changes in any of the secondary structure properties (α-sheets, β-hairpins, β-strands, helices, helix-helix interactions, β-turns and γ-turns) of KCNH2 protein (Figures and ).
Figure 2. Shows the superpose simulation of both wild type KCNH2 against its mutated protein models, with description of the amino acid changes and their RMSD values at the position of the all mutations.
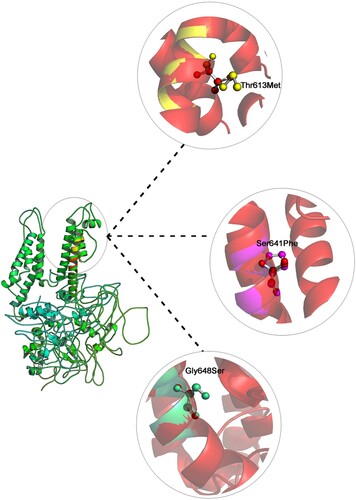
Figure 3. KCNH2 mutations site secondary structural elements at KCNH2 Wild and mutant models.
3.4 Docking study of KCNH2 and ligand molecule
KB-R7943 is a well-known inhibitor of the reverse Na+/Ca2+ exchange and it plays a pivotal role in regulating contractility and electrical activity in the heart (Antoons et al. Citation2012). From docking analysis, it is revealed that the KB-R7943 inhibitor strongly binds at the transmembrane channel (S4 and S5) of wildtype KCNH2. The nitrobenzyl and methyl sulfanyl methane diamine groups of KB-R7943 binds to Ala558 and Thr623 residues (S4 and S5 pores) of wildtype KCNH2, with the lowest binding energy of 6.1 Kcal/Mol. The KCNH2 in two mutant forms (Ser641 and Gly648), binds to the inhibitor at S4 and S5 voltage transmembrane regions of the protein (Asp609, Thr623, Asn629 and Thr613) by forming more stable bonds with inhibitor showing highest binding energy of 7.8 and 7.7 Kcal/Mol (). However, the Thr613 mutant protein binds to the inhibitor in a different pose, involving Glu575 and Ser636 residues of S5 and S6 pores of KCNH2 voltage channels, with an binding energy of 7.1 K.Cal/Mol. Interestingly, in all the docking interactions nitrobenzyl group forms strong H-bond interactions with Thr623, which eventually alters the confirmations of pore or cytoplasmic tail of the KCNH2. So, from our analysis, we predict that KB-R7943, a Na+/Ca2+ exchange inhibitor can effectively bind to KCNH2 even under mutant conditions (Figure ).
Figure 4. Illustration of the docking simulation combining all KB-R7943 with KCNH2 with (a) wild type protein model (b) mutant Thr613Meth protein model (c) mutant Ser641Phe protein model (d) mutant Gly648Ser protein model.
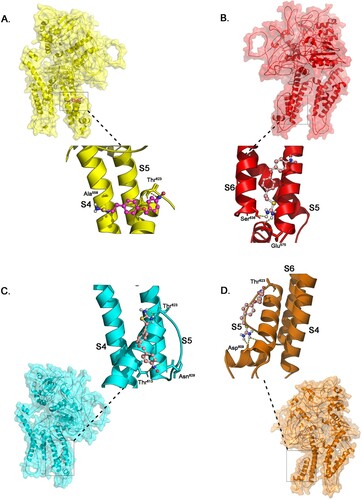
Table 3. Auto dock binding energies of KB-R7943 with KCNH2 wild and mutant forms.
4. Discussion
The recent molecular genetic reports have implicated several nucleotide sequence alterations with many inherited arrhythmic diseases, more particularly, ‘ion channel diseases,’ where genetic mutations affect functional units of ion channels and/or their transporter-associated proteins in patients without primary cardiac structural abnormalities (Lieve Citation2015; Imberti et al. Citation2016). KCNH2 (previously known as hERG) gene encodes subunits of potassium channels, which transports potassium ions to the cells’ exterior and determinate the generation and transmission of electric signals. Potassium channels made KCNH2 actively recharge the cardiac muscle in order to maintain the regular rhythm after each heartbeat (Gianulis and Trudeau Citation2011). KCNH2 gene is localized at 7q35–7q36, and it consists of 15 exons, reads in reverse direction and encodes a protein of 1159 amino acids that forms a VGK (rectifier voltage gated K+) channel. Genetic mutations in KCNH2 can reduce IKr (a repolarizing current in the heart) slow action potential (AP) repolarization and can trigger cardiac arrhythmias. Mutations in KCNH2 cause different forms of CAs. For example, approximately 38% of patients with CA possess mutations in KCNH2. According to HGMD, Ensemble and NCBI data bases, a total of 364 genetic mutations in KCNH2 gene has been described in Cardiac Arrhythmias [356 genetic mutations in long QT syndrome (LQT) and 8 genetic mutations in Short QT syndrome (SQT)]. Interestingly, all the 364 variants are non-synonymous variants, which mean they can change the amino acid they encode in corresponding polypeptides.
Here in this study we assessed the structural impact of T613M, S641P and G648S mutations on biophysical properties of KCNH2 protein properties using different computational approaches (Tester et al. Citation2005; Kapplinger et al. Citation2009; Saprungruang et al. Citation2018). We first screened them with SIFT and PolyPhen-2, both of which involves genetic sequence conservation and protein structure properties to identify the deleterious potential of missense mutations. A study by Hicks et al. (Citation2011) has demonstrated that out of different test tools, SIFT and PolyPhen-2 tools showed better performance in identifying the damaging potential of missense mutations (Hicks et al. Citation2011). The accuracy of both tools was further tested by few other studies as well (Gnad et al. Citation2013; Shaik et al. Citation2018; Shaik et al. Citation2019). In case if the true positive mutation is predicted as benign by computational tools, then one should carefully interpret variant pathogenicity based on the clinical assessment. The variant prediction results, when combined with 3D structure information of protein molecule can help us to take a better insight into the variant induced effects. However, not all KCNH2 mutations are mapped in the 3D space of the KCNH2 protein, because it is technically infeasible. Therefore, we constructed the computational 3D model of KCNH2 protein and tried to explore the molecular connections between KCNH2 gene mutations and CA. The superposing of 3D structures of wild type and mutant residues allows us to examine them in different confirmations by estimating the RMSD differences in atomic coordinates. We observed that T613M, S641P and G648S variants bring minor structure related changes in the KCNH2 protein molecule at the site of amino acid residue they affected. The differences in the RMSDs could be attributed to chemical side chain, volume, molecular weight, orientation of amino acids. These RMSD differences disturb the hydrogen bonds, ionic bonds Van der Waals force, and electrostatic charges, existing between the amino acids which maintain the conformation of the polypeptide chains in KCNH2 molecule (Shaik et al. Citation2018).
The KCNH2 protein contains six transmembrane segments (S1-S6) where S1-S4 maintains voltage sensor domain (VSD) and S5-S6 in addition to intervening pore loop forms the Pore domain (Morais Cabral et al. Citation1998). The tetrameric functional channel containing pore domain responsible for ion conduction in the cell. The KCNH2 protein contains 3 functional domains. i.e. cytoplasmic NH2 terminal, COOH-terminal and transmembrane domains. The N-terminal contains a Per-Arnt-Sim (PAS) domain that defines a subfamily of VGK channel proteins. The C-terminal segment contains cyclic nucleotide binding domain (cNBD) (Warmke and Ganetzky Citation1994). The KCNH2 gene sequence in vertebrates showed a significant correlation between exon boundaries and protein domains. The exon 1 region encodes for 5’ UTR and 26 amino acid residues with a known role in ion channel deactivation kinetics. Exon-2&-3 encompasses the PAS domain. The cytoplasmic NH2 terminal region and transmembrane segments are encoded by exon 6. The fourth transmembrane segment of protein is encoded by exon-7 region. This segment is known as the principle voltage-sensing region within domain VSD along with the majority of pore domain. It is noticeable that the boundary region between exon −7 & −8 consists of glycine residue which is by and large seen to be highly conserved in all potassium channels and also determines the boundaries in between pore cavity upper and lower proportions. The c-linker and cNBD domains are encoded by exons 8 and 10, whereas the remaining 5 exons encode distal COOH terminal part of the protein (Jiang et al. Citation2002).
The KB-R7943 is a known pharmacological agent that can inhibit Na+/Ca2+ across plasma membranes. It reversibly inhibits the Na(+)/Ca(2+) inward and outward movements in a dose-related fashion and does not affect sodium-dependent transport systems (Iwamoto et al. Citation1996). KB-R7943 is also known to suppress arrhythmias (induced by ouabain) by blocking the reverse-mode Na+ and Ca2+ exchanger (NCX). The anti-arrhythmic and cardioprotective effects of KB-R7943 are well documented in multicellular and intact heart preparations (Nakamura et al. Citation1998; Mukai et al. Citation2000). Further anti-arrhythmic effects were shown in Na+ overload induced by glycosides or sodium channel facilitators (Amran et al. Citation2004). When administered in-vivo, the KB-R7943 was able to prevent the development of atrial fibrillation (Miyata et al. Citation2002). Currently the therapeutic administration of KB-R7943 to CA patients is independent of their mutational status of KCNH2. We sought to check if KCNH2 mutations could account the variations in responding to anti-arrhythmic drug molecules. Therefore, we performed molecular docking to understand the differences in binding efficiencies of wild type and mutant KCNH2 forms with commonly used arrhythmic drug inhibitor i.e. KB-R7943. Our molecular analysis revealed that the binding energy range of KB-R7943 inhibitor with KCNH2 wild type and mutant forms [Thr613Met, Ser641Phe and Gly648Ser] is in between 7.4 Kcal to 7.7 Kcal/mol. The altered amino acid sequences in KCNH2 (Thr613Met, Ser641Phe and Gly648Ser) may lead to some changes in the hydrophobic, electrostatic and Van der Waals interactions between the ligand and protein molecules. Interestingly, both wild type and mutant forms showed same binding energies, even though the position of binding is different in different mutated forms. So, from our analysis we conclude that KB-R7943, a Na+/Ca2+ exchange inhibitor can effectively function even in CA patients with KCNH2 mutations. This concludes that the mutant KCNH2 (even with residue level structural deviation) shows same binding affinity towards KB-R7943 as that of its native protein form. Furthermore, we predict that these comparative studies create a direction to development of competitive peptide inhibitors for KCNH2 mutation positive patients.
In summary, this study has used different computational methods to screen the pathogenicity of cardiac arrhythmia linked KCNH2 missense (Thr613Met, Ser641Phe and Gly648Ser) mutations. The SIFT and PolyPhen-2 methods, which depends on sequence conservation across species, and structure-based information have correctly attributed pathogenicity to these variants. By superimposing the 3D structures of KCHN2 mutant proteins we found that these missense mutations induce some minor structural alterations at the amino acid residue level. These mutations are also seen to destabilize the KCNH2 protein molecule, hence its function. This study also points that the mutant KCNH2 (even with residue level structural deviation) shows same binding affinity towards KB-R7943 drug molecule as that of its native protein form. However, the real impact of Thr613Met, Ser641Phe and Gly648Ser missense mutations on the biophysical and chemical properties of KCNH2 protein can be better interpreted when the computational protein phenotype findings are confirmed by wet laboratory experiments. Nonetheless, the computational approaches we used in this study could help to quickly study the CA linked KCNH2 mutations before initiating time-consuming lab assays.
Supplemental Material
Download MS Word (14.4 KB)Acknowledgements
The authors, therefore, acknowledge with thanks DSR technical and financial support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Related Research Data
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010 Apr. A method and server for predicting damaging missense mutations. Nat Methods. 7:248–249. Epub 2010/04/01. doi: https://doi.org/10.1038/nmeth0410-248
- Afzal MR, Savona S, Mohamed O, Mohamed-Osman A, Kalbfleisch SJ. 2019 Oct. Hypertension and arrhythmias. Heart Fail Clin. 15:543–550. Epub 2019/09/02. doi: https://doi.org/10.1016/j.hfc.2019.06.011
- Amran MS, Hashimoto K, Homma N. 2004 Jul. Effects of sodium-calcium exchange inhibitors, KB-R7943 and SEA0400, on aconitine-induced arrhythmias in Guinea pigs in vivo, in vitro, and in computer simulation studies. J Pharmacol Exp Ther. 310:83–89. Epub 2004/03/19. doi: https://doi.org/10.1124/jpet.104.066951
- Antoons G, Willems R, Sipido KR. 2012 Apr. Alternative strategies in arrhythmia therapy: evaluation of Na/Ca exchange as an anti-arrhythmic target. Pharmacol Ther. 134:26–42. Epub 2011/12/27. doi: https://doi.org/10.1016/j.pharmthera.2011.12.001
- De Zio R, Gerbino A, Forleo C, Pepe M, Milano S, Favale S, Procino G, Svelto M, Carmosino M. 2019 Sept. Functional study of a KCNH2 mutant: novel insights on the pathogenesis of the LQT2 syndrome. J Cell Mol Med. 23:6331–6342. Epub 2019/07/31. doi: https://doi.org/10.1111/jcmm.14521
- Fan LL, Chen YQ, Huang H, Yuan ZZ, Jin JY, Hu M, Xiang R. 2019 Mar. Exome sequencing identifies a novel nonsense mutation of Ring Finger protein 207 in a Chinese family with long QT syndrome and syncope. J Hum Genet. 64:233–238. Epub 2018/12/14. doi: https://doi.org/10.1038/s10038-018-0549-1
- Fu DG. 2015 Nov. Cardiac arrhythmias: diagnosis, symptoms, and treatments. Cell Biochem Biophys. 73:291–296. Epub 2015/03/05. doi: https://doi.org/10.1007/s12013-015-0626-4
- Gianulis EC, Trudeau MC. 2011 Jun. Rescue of aberrant gating by a genetically encoded PAS (Per-Arnt-Sim) domain in several long QT syndrome mutant human ether-a-go-go-related gene potassium channels. J Biol Chem. 24(286):22160–22169. Epub 2011/05/04. doi: https://doi.org/10.1074/jbc.M110.205948
- Gnad F, Baucom A, Mukhyala K, Manning G, Zhang Z. 2013. Assessment of computational methods for predicting the effects of missense mutations in human cancers. BMC Genomics. 14(Suppl 3):S7. Epub 2013/07/17. doi: https://doi.org/10.1186/1471-2164-14-S3-S7
- Guillen Sacoto MJ, Chapman KA, Heath D, Seprish MB, Zand DJ. 2015 Jun. An uncommon clinical presentation of relapsing dilated cardiomyopathy with identification of sequence variations in MYNPC3, KCNH2 and mitochondrial tRNA cysteine. Mol Genet Metab Rep. 3:47–54. Epub 2016/03/05. doi: https://doi.org/10.1016/j.ymgmr.2015.03.007
- Hicks S, Wheeler DA, Plon SE, Kimmel M. 2011 Jun. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum Mutat. 32:661–668. Epub 2011/04/12. doi: https://doi.org/10.1002/humu.21490
- Imberti JF, Underwood K, Mazzanti A, Priori SG. 2016 Aug. Clinical challenges in catecholaminergic polymorphic ventricular tachycardia. Heart Lung Circ. 25:777–783. Epub 2016/03/08. doi: https://doi.org/10.1016/j.hlc.2016.01.012
- Ittisoponpisan S, Islam SA, Khanna T, Alhuzimi E, David A, Sternberg MJE. 2019 May. Can predicted protein 3D structures provide reliable insights into whether missense variants are disease associated? J Mol Biol. 17(431):2197–2212. Epub 2019/04/18. doi: https://doi.org/10.1016/j.jmb.2019.04.009
- Iwamoto T, Watano T, Shigekawa M. 1996 Sept. A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J Biol Chem. 13(271):22391–22397. Epub 1996/09/13. doi: https://doi.org/10.1074/jbc.271.37.22391
- Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. 2002 May. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 30(417):515–522. Epub 2002/05/31. doi: https://doi.org/10.1038/417515a
- Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris-Kerr C, Pollevick GD, Wilde AA, Ackerman MJ. 2009 Sept. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 6:1297–1303. Epub 2009/09/01. doi: https://doi.org/10.1016/j.hrthm.2009.05.021
- Kufareva I, Abagyan R. 2012. Methods of protein structure comparison. Methods Mol Biol. 857:231–257. Epub 2012/02/11. doi: https://doi.org/10.1007/978-1-61779-588-6_10
- Laskowski RA, Jablonska J, Pravda L, Varekova RS, Thornton JM. 2018 Jan. PDBsum: structural summaries of PDB entries. Protein Sci. 27:129–134. Epub 2017/09/07. doi: https://doi.org/10.1002/pro.3289
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 26:283–291. doi: https://doi.org/10.1107/S0021889892009944
- Li M, Petukh M, Alexov E, Panchenko AR. 2014 Apr. Predicting the impact of missense mutations on protein-protein binding affinity. J Chem Theory Comput. 8(10):1770–1780. Epub 2014/05/08. doi: https://doi.org/10.1021/ct401022c
- Li YG, Benditt DG, Klingenheben T, Hu K, Feng D. 2016. Cardiac arrhythmias: update on mechanisms and clinical managements. Cardiol Res Pract. 2016:8023723. Epub 2016/08/25. doi: https://doi.org/10.1155/2016/8023723
- Lieve KV, Wilde AA. 2015 Oct. Inherited ion channel diseases: a brief review. Europace. 17(Suppl 2):ii1–ii6. Epub 2016/02/05. doi: https://doi.org/10.1093/europace/euv105
- Miyata T, Kobayashi Y, Araki H, Ohto T, Shin K. 2002 May–Jun. The influence of controlled occlusal overload on peri-implant tissue. part 4: a histologic study in monkeys. Int J Oral Maxillofac Implants. 17:384–390. Epub 2002/06/21.
- Morais Cabral JH, Lee A, Cohen SL, Chait BT, Li M, Mackinnon R. 1998 Nov. Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell. 25(95):649–655. Epub 1998/12/09. doi: https://doi.org/10.1016/S0092-8674(00)81635-9
- Mukai M, Terada H, Sugiyama S, Satoh H, Hayashi H. 2000 Jan. Effects of a selective inhibitor of Na+/Ca2+ exchange, KB-R7943, on reoxygenation-induced injuries in Guinea pig papillary muscles. J Cardiovasc Pharmacol. 35:121–128. Epub 2000/01/12. doi: https://doi.org/10.1097/00005344-200001000-00016
- Nakamura A, Harada K, Sugimoto H, Nakajima F, Nishimura N. 1998 Feb. Effects of KB-R7943, a novel Na+/Ca2+ exchange inhibitor, on myocardial ischemia/reperfusion injury. Nihon Yakurigaku Zasshi. 111:105–115. Epub 1998/04/29. doi: https://doi.org/10.1254/fpj.111.105
- Ng PC, Henikoff S. 2003 Jul. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 1(31):3812–3814. Epub 2003/06/26. doi: https://doi.org/10.1093/nar/gkg509
- Pires DE, Ascher DB, Blundell TL. 2014 Jul. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 42:W314–W319. Epub 2014/05/16. doi: https://doi.org/10.1093/nar/gku411
- Rigsby RE, Parker AB. 2016 Sept. Using the PyMOL application to reinforce visual understanding of protein structure. Biochem Mol Biol Educ. 10(44):433–437. Epub 2016/06/01. doi: https://doi.org/10.1002/bmb.20966
- Roy A, Kucukural A, Zhang Y. 2010 Apr. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5:725–738. Epub 2010/04/03. doi: https://doi.org/10.1038/nprot.2010.5
- Saprungruang A, Khongphatthanayothin A, Mauleekoonphairoj J, Wandee P, Kanjanauthai S, Bhuiyan ZA, Wilde AAM, Poovorawan Y. 2018 Sept–Oct. Genotype and clinical characteristics of congenital long QT syndrome in Thailand. Indian Pacing Electrophysiol J. 18:165–171. Epub 2018/07/24. doi: https://doi.org/10.1016/j.ipej.2018.07.007
- Shaik NA, Awan ZA, Verma PK, Elango R, Banaganapalli B. 2018 Nov. Protein phenotype diagnosis of autosomal dominant calmodulin mutations causing irregular heart rhythms. J Cell Biochem. 119:8233–8248. Epub 2018/06/23. doi: https://doi.org/10.1002/jcb.26834
- Shaik NA, Bokhari HA, Masoodi AT, Shetty PJ, Ajabnoor GMA, Elango R, Banaganapalli B. 2019 Sept. Molecular modelling and dynamics of CA2 missense mutations causative to carbonic anhydrase 2 deficiency syndrome. J Biomol Struct Dyn. 23:1–20. Epub 2019/09/24. doi: https://doi.org/10.1080/07391102.2019.1671899
- Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. 2012 Jul. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40:W452–W457. Epub 2012/06/13. doi: https://doi.org/10.1093/nar/gks539
- Singh M, Morin DP, Link MS. 2019 May–Jun. Sudden cardiac death in long QT syndrome (LQTS), Brugada syndrome, and catecholaminergic polymorphic ventricular tachycardia (CPVT). Prog Cardiovasc Dis. 62:227–234. Epub 2019/05/13. doi: https://doi.org/10.1016/j.pcad.2019.05.006
- Tester DJ, Ackerman MJ. 2014 Jan–Mar. Genetics of long QT syndrome. Methodist Debakey Cardiovasc J. 10:29–33. Epub 2014/06/17. doi: https://doi.org/10.14797/mdcj-10-1-29
- Tester DJ, Will ML, Haglund CM, Ackerman MJ. 2005 May. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2:507–517. Epub 2005/04/21. doi: https://doi.org/10.1016/j.hrthm.2005.01.020
- Warmke JW, Ganetzky B. 1994 Apr. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci USA. 12(91):3438–3442. Epub 1994/04/12. doi: https://doi.org/10.1073/pnas.91.8.3438
- Witvliet DK, Strokach A, Giraldo-Forero AF, Teyra J, Colak R, Kim PM. 2016 May. ELASPIC web-server: proteome-wide structure-based prediction of mutation effects on protein stability and binding affinity. Bioinformatics. 15(32):1589–1591. Epub 2016/01/24. doi: https://doi.org/10.1093/bioinformatics/btw031
- Zemla A. 2003 Jul. LGA: a method for finding 3D similarities in protein structures. Nucleic Acids Res. 1(31):3370–3374. Epub 2003/06/26. doi: https://doi.org/10.1093/nar/gkg571