
ABSTRACT
Accumulating evidence suggests that cardiomyocyte autophagy is relevant to the onset of cardiac hypertrophy (CH). Several miRNAs are involved in the occurrence of heart failure, and relevant therapeutic treatments are currently being developed. MicroRNA-377 (miR-377) is known to correlate with the progression of various cancers, however, its function in CH has not been determined. Therefore, this study aimed to evaluate miR-377 expression in H2C9 hypertrophic cardiomyocytes in vitro and in a murine model of CH. Gene expression changes were verified via qRT-PCR. Western blotting was used for evaluation of alterations in the expression of signaling pathway-related proteins. Our results indicated that miR-377 expression was markedly upregulated in mice with hypertrophic cardiomyopathy. Autophagy markers were downregulated in these mice and in hypertrophic cardiomyocytes following miR-377 transfection. In addition, we demonstrated that miR-377 acts by targeting peroxisome proliferator-activated receptor γ (PPARγ). PPARγ overexpression promoted autophagy and suppressed cyclosporine A-induced CH. In contrast, PPARγ knockdown further suppressed CH and autophagy. In conclusion, our findings indicated that miR-377 accelerates CH by inhibiting autophagy via targeting PPARγ. This newly identified miR-377/PPARγ axis improves our understanding of the molecular mechanisms underlying CH, and provides a potential new therapeutic target for its treatment.
Introduction
Cardiac hypertrophy (CH) refers to the cellular changes that occur because of increased mechanical stress, such as extrinsic and intrinsic stress (Banerjee et al. Citation2007). The heart consists of cardiomyocytes and non-myocytes, including vascular cells (Banerjee et al. Citation2007; Tirziu and Simons Citation2009), and accumulating evidence suggests an important role played by non-myocytes in CH pathogenesis (Tirziu and Simons Citation2009; Kamo et al. Citation2015). Various studies have focused on the pathogenesis of CH from diverse perspectives, including alterations in molecular signaling pathways and genetic mutations in structural proteins (Berlo et al. Citation2013; Zhou et al. Citation2013).
Autophagy, a highly conserved process involving the transport and degradation of dysfunctional cellular organelles and proteins, is induced by cellular stress and nutrient deficiency (Klionsky and Emr Citation2000; Gottlieb and Mentzer Citation2010; Efeyan et al. Citation2015). Basal autophagy is conducive to cell survival, but autophagy can also accelerate cell death (Gottlieb and Mentzer Citation2010). Autophagic dysfunction is involved in various diseases, including cancer (Levine and Kroemer Citation2008; Green and Beth Citation2014), and multiple lines of evidence have linked it to CH (Li et al. Citation2015).
MicroRNAs (miRNAs) can bind to specific sequences in target mRNAs and modulate their transcription, which subsequently blocks translation or induces degradation (Campard et al. Citation2006; Bushati and Cohen Citation2007). In addition to their critical roles in various biological functions, diverse miRNA expression patterns are observed in various cancers, and miRNAs can affect cancer development (Zamore and Haley Citation2005; Calin and Croce Citation2006). Although specific functions have not yet been elucidated for all identified miRNAs, research shows that several of them play critical roles in regulating cell signaling pathways and tumorigenesis (Guo et al. Citation2008; Esteller Citation2011; Han et al. Citation2011), and miRNAs are the focus of many ongoing cancer-related studies (Ahmed et al. Citation2013; Schultz et al. Citation2014; Zhu et al. Citation2014). A previously studied miRNA of potential interest is miR-377, located at 14q32.31. This miRNA was first shown to modulate epithelial–mesenchymal transition genes (Formosa et al. Citation2014). Subsequently, it was also shown to suppress the activity of specificity protein 1 in glioblastoma, thereby leading to decreased cell invasion (Zhang et al. Citation2014). However, the potential function of miR-377 in CH has not been studied.
Peroxisome proliferator-activated receptors (PPARs) are members of a nuclear receptor protein family and have three isotypes, namely α, β and γ, which have been identified to date. Once activated by their corresponding cognate ligands, PPARs modulate the transcriptional rate of many genes associated with a variety of physiological functions, such as cellular and tissue metabolism (Lee et al. Citation2003) and inflammation (Staels and Fruchart Citation2005). Many ongoing studies have paid close attention to the roles played by PPARs in heart tissue. The level of PPARγ expression in heart is low, but a recent study reported that insufficient PPARγ expression can alter heart function (Duan et al. Citation2005). High PPARα expression levels have been demonstrated to be connected with the oxidation of fatty acids. In addition, PPARα is associated with enhanced inflammatory marker expression and extracellular matrix remodeling, indicating that PPARα exerts salutary effects during CH (Barger and Kelly Citation2000).
Herein, by measuring miR-377 levels in hypertrophic cardiomyopathy, we investigated whether miR-377 could regulate CH through suppressing autophagy by targeting PPARγ. We also examined atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) levels in cardiomyocyte hypertrophy via immunoradiometric assays with high sensitivity and specificity.
We found that miR-377 was highly expressed in hypertrophic cardiomyocytes and its expression was negatively associated with autophagy in hypertrophic cardiomyocytes. This miR-377-modulated reduction in autophagy was mediated by PPARγ. PPARγ overexpression increased autophagy and rescued CH induced by cyclosporine A.
Materials and methods
Animals
In this study, SPF C57BL/6 mice (n = 40; Beijing Sifubei Biotechnology Co.) were used, and all animal experiments have been approved by the First People’s Hospital of Wenling. A total of 40 8-week-old mice were grouped randomly: 20 in a sham group and 20 in a transverse aortic constriction (TAC) group. A CH model was established by TAC (Gómez-Hurtado et al. Citation2017; Tan et al. Citation2018). Briefly, the mice were anesthetized with ketamine and xylazine in the supine position. A mid-neck incision was made, and an 8 mm section of the trachea was then carefully dissected. After obtaining the transverse section, the aorta was ligated. Following surgery, all mice were placed in a culture room to recover (37°C). Control mice underwent the same operations except for aortic ligation. All mice were closely observed for any signs of pain or breathing problems. Once abnormal pain occurred, the mice were subcutaneously injected with 0.15–3.0 mg/kg of bucinnazine hydrochloride every 6 h as needed.
Cardiomyocyte culture and treatment
H2C9 cardiomyocyte cells (ATCC) were cultured and then treated with 200 nM cyclosporine A (Sigma) for 24 h to induce the autophagy phenotype. The cells are from rat. The cells were cultivated in DMEM containing 10% FBS inactivated by heat (Invitrogen), 5% horse serum, 100 units/ml of penicillin and 100 μg/ml of streptomycin. Incubation was conducted at 37°C under a humidified atmosphere containing 5% CO2. The cells are good in vitro model for mimicking mice CH.
qRT-PCR
RNA was extracted using TRIzol (TaKaRa). Then, miR-377 expression was measured by qRT-PCR (Qiagen, UK), and U6 snRNA was used as a control. TransScript® Reverse Transcriptase (Transgen Biotech, Beijing, China) was used to reverse-transcribe RNA into cDNA. The miR-377 primers were purchased from Thermo Fisher Scientific (Waltham, MA, USA). The following specific primers were used for qRT-PCR: mouse PPARγ (F: 5′-GAG TGT GAC GAC AAG ATT TG-3′ and R: 5′-GGT GGG CCA GAA TGG CAT CT-3′), mouse GAPDH (F: 5′-TTG TTG CCA TCA ATG ACCC-3′ and R: 5′-CTT CCC GTT CTC AGC CTTG-3′), and U6 snRNA (F: 5′-CTC GCT TCG GCA GCACA-3′ and R: 5′-AAC GCT TCA CGA ATT TGC GT-3′).
Construction of a luciferase reporter system
To perform reporter assays, we cloned the regulatory region of genes of interest upstream of the luciferase gene in an expression vector, introduced the resulting vector DNA into cells, and allowed the cells to grow for a period of time. Then, we collected and lysed the cells to release all proteins (including luciferase), added luciferin and all the necessary cofactors, and measured enzymatic activity using a luminometer. The gene of interest binds to the luciferase reporter gene and, therefore, its expression is directly correlated with luciferase activity. In this study, we used the TargetScan online tool (http://www.targetscan.org) to identify potential miR-377 targets. The analysis identified PPARγ as a downstream target. The 3′-UTR sequence of PPARγ was cloned and inserted into the PsicheckTM-2 vector to generate PsicheckTM-2-PPARγ-WT. The miR-377 binding site in PPARγ was then mutated, and the mutant fragment was inserted into PsicheckTM-2 to generate PsicheckTM-2-PPARγ-MT. Forty-eight hours after the aforementioned constructs were transfected, the luciferase activities were determined. These experiments were repeated three times.
Plasmid constructs and transfection
PPARγ siRNA was designed and purchased from Invitrogen. In addition, a miR-377 mimic, miR-377-NC, and anti-377, or anti-NC, were synthesized by and purchased from Invitrogen. Plasmids containing these RNAs were transfected into H2C9 cardiomyocytes with Lipofectamine 2000. After incubation for 48 h, the cardiomyocytes were collected for subsequent experimentation.
Determination of cell surface area
After fixation in 4% paraformaldehyde, primary cardiomyocytes were treated with Triton X-100 and stained with DAPI. Then, these cardiomyocytes were observed via a fluorescence microscope and surface area was calculated using ImageJ. Calculations were performed using 100 cells from not fewer than nine fields selected randomly. The cardiomyocyte surface area was calculated by assuming a prolate ellipsoid cell shape via a Matlab function for image analysis.
Western blotting assay
Total protein was extracted, and its concentration was measured. Proteins were separated and then transferred onto a membrane. After blocking with skimmed milk, the membrane was incubated separately with rabbit anti-PPARγ (1:1000) and rabbit anti-GAPDH (1:1000) antibodies. The antibody-bound proteins were then visualized and observed using a Gel Doc XR system (Bio-Rad, California, USA).
Statistical analysis
The data obtained are shown as means ± SD, and GraphPad, one-way ANOVA and Tukey’s test were applied to analyze differences among groups, and an independent Student’s t-test for intergroup difference. P < 0.05 was considered statistically significant.
Results
High upregulation of miR-377 expression in hypertrophic cardiomyopathy in TAC mice
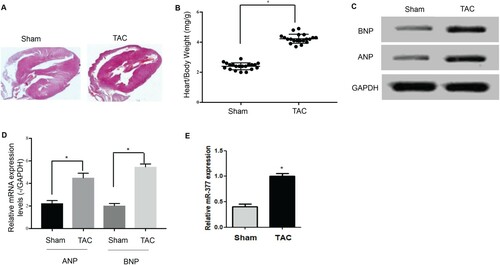
Hypertrophic cardiomyopathy induced significant increases in hypertrophic responses, including increased heart/body weight ratio (Figure (A)) as well as ANP and BNP levels (Figure (B,C)). miR-377 expression was assessed via qPCR following TAC surgery, and the results showed that cardiac miR-377 expression in mice in the TAC group was dramatically higher than that in the sham group (Figure (D)), which indicated that miR-317 might play a crucial role in CH.
Figure 1. Increased miR-377 expression in hypertrophic cardiomyopathy. (A) Heart/body weight ratio in the TAC and sham groups. (B) Protein expression of ANP and BNP in the TAC and sham groups. (C) mRNA expression of ANP and BNP in the TAC and sham groups. (D) Levels of miR-377 expression in the hearts of mice in the TAC and sham groups as detected by qRT-PCR.
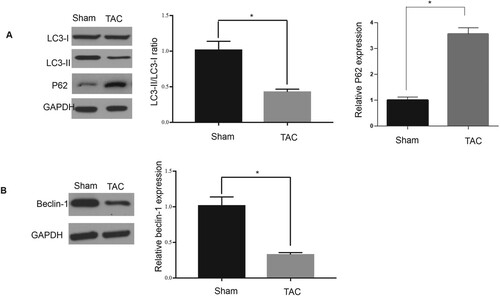
Decreased expression of autophagy-related factors in hypertrophic cardiomyopathy
The results presented in Figure demonstrated that autophagy could be dramatically suppressed due to the induction of hypertrophic cardiomyopathy, which was demonstrated by the fact that the protein levels of LC3-II and beclin-1, as well as the ratio of LC3-II to LC3-I in the sham mouse group, were obviously higher than those in the TAC mouse group (Figure (A)). This was in contrast to the inhibition of autophagy, miR-377 expression, and hypertrophic markers observed in the TAC group (Figure (B)). These findings revealed that the progression of CH could suppress autophagy, which might be related to increased miR-377 expression.
Figure 2. Decreased autophagy in hypertrophic cardiomyopathy. (A–E): Ratio of LC3-II to I, as well as levels of LC3-II and beclin-1, in heart samples; *P < 0.05.
Promotion of CH and inhibition of autophagy by miR-377
Consistent with our in vivo results, we also found highly upregulated miR-377 expression in H2C9 hypertrophic cardiomyocyte cells. The Ang-II-induced pro-hypertrophic effect was almost entirely reversed by miR-377 knockdown (P < 0.05) (Figure (A)), whereas miR-377 overexpression could enhance the progression of CH via the transfection of a miR-377 mimic, as evidenced by changes which occurred in terms of the cell surface area (Figure (B)) as well as BNP and ANP levels (Figure (C,D)).
Figure 3. Promotion of CH and inhibition of autophagy by miR-377. (A) Levels of miR-377 expression in cultured H2C9 cardiomyocytes after transfection with miR-NC, anti-377, and anti-NC as determined by qRT-PCR. The presence of CH was determined with cell surface area measurement (B) and by analyzing ANP (C) and BNP (D) expression via qPCR and western blotting assays (E, upper). In addition, changes in the ratio of LC3-II to I (F), and levels of LC3-II and beclin-1, P62 and beclin-1 were also detected (E,G,H).
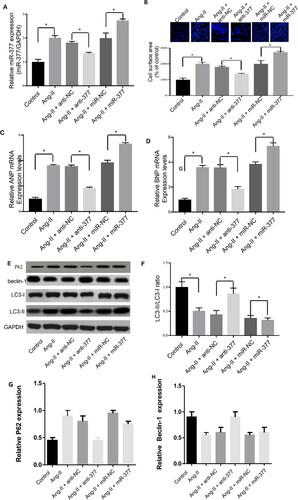
Autophagy was obviously suppressed in cardiomyocytes treated with Ang-II. Similarly, autophagy was suppressed by transfection of a miR-377 mimic, which was evidenced by the fact that LC3-II and beclin-1 were downregulated (Figure (E)), and the ratio of LC3-II to LC3-I was decreased (Figure (F)). Cyclosporine A treatment significantly increased autophagy, as evidenced by the strong upregulation of beclin-1 and LC3-II (Figure (G)). In contrast, downregulating miR-377 with antagomir dramatically increased the ratio of LC3-II to I via upregulating beclin-1and LC3-II (Figure (E,F)). These observations mentioned above suggested that miR-377 could suppress autophagy in cardiomyocytes, thereby affecting CH.
Inhibition of PPARγ expression in hypertrophied cardiomyocytes by miR-377
We evaluated the effects elicited by miR-377 on PPARγ expression by detecting miR-377 and PPARγ expression levels in murine hypertrophic cardiomyocytes following Ang-II induction. We found that miR-377 upregulation (observed in the TAC mouse group) was associated with lower PPARγ expression (Figure (G)). We took advantage of the TargetScan online tool to identify PPARγ as a possible target of miR-377 (Figure (A)).
Figure 4. miR-377 directly targets PPARγ. (A) Levels of miR-377 and PPARγ in control and cardiomyocytes treated with Ang-II were detected. (B) The graphic demonstrates how the luciferase reporter plasmids were constructed. (C) Luciferase activity of a reporter clone containing the 3′-UTR of PPARγ transfected into cardiomyocytes together with the miR-377 and miR-NC plasmid vectors; *P < 0.05.
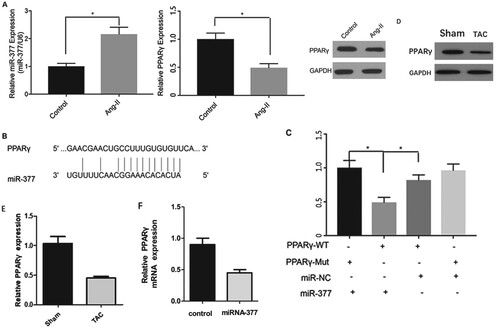
In order to investigate if PPARγ was a target of miR-377, we transfected H2C9 cardiomyocyte cells with the corresponding plasmids. As shown in Figure (B), this analysis identified a region complementary to miR-377 in the 3′-UTR of PPARγ. Moreover, these findings indicated that miR-377 suppressed luciferase activity of the wild-type construct PsicheckTM-2-PPARγ-WT. However, the suppressive effect of miR-377 on PsicheckTM-2-PPARγ-MT was less than that of the wild-type construct and, therefore, our speculation that PPARγ was a target of miR-377 was verified (Figure (C)). Considering these observations mentioned above, we can draw the conclusion that miR-377 can inhibit PPARγ expression in hypertrophied cardiomyocytes in vitro.
Inhibition of CH by PPARγ via promotion of autophagy in H2C9 cardiomyocytes
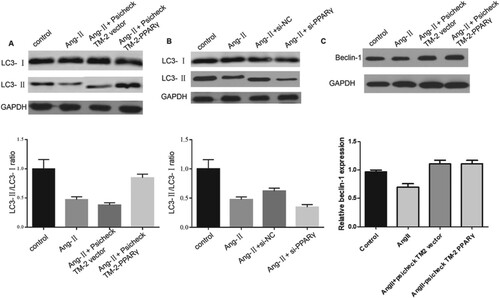
In a subsequent study, we investigated if PPARγ could suppress the progression of CH by enhancing autophagy. PPARγ overexpression significantly contributed to myocardial autophagy, which could be evidenced by the fact that the ratio of LC3-II to I, as well as protein levels of LC3-II and beclin-1, increased regardless of whether Ang-II was present or not (Figure (A–C)). These findings indicated that PPARγ exhibits pro-autophagic effects on H2C9 cardiomyocytes.
Figure 5. PPARγ inhibits CH by enhancing autophagy. (A, B, C) Levels of LC3-I/II and beclin-1 in cardiomyocytes were measured via western blotting assays in the presence of Ang-II, pCDNA-PPARγ, si-PPARγ, and corresponding negative controls.
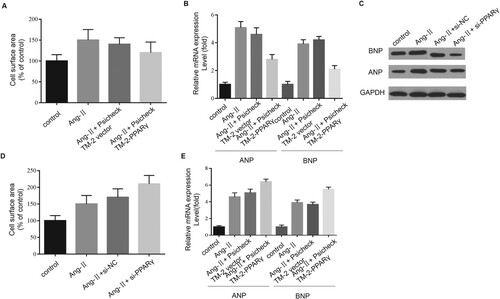
To thoroughly analyze the effects exerted by PPARγ on CH, we detected the hypertrophic responses of H2C9 cardiomyocyte cells and observed that PPARγ overexpression could obviously reverse Ang-II-induced CH, which could be proven by the fact that cell surface area decreased (Figure (A)), as well as ANP and BNP being downgraded (Figure (B,C)). In contrast, PPARγ knockdown further contributed to CH, which could be evidenced by the fact that hypertrophic markers and cell surface area decreased (Figure (D,E)). In summary, PPARγ attenuated CH by promoting autophagy.
Figure 6. Effects of PPARγ on CH. CH was evaluated via cell surface area measurement (A, D), and mRNA and protein expression levels of ANP and BNP (B, C and E) were evaluated via qPCR and western blotting assays. The data obtained in the study are presented as means ± SD, *P < 0.05.
Discussion
Various miRNAs have been implicated in the process of CH. For example, Cheng et al. reported that miR-21 is present in cultured hypertrophic cardiomyocytes (Cheng et al. Citation2007). Here, we showed that miR-377 was upregulated in hypertrophic cardiomyopathy and that miR-377 overexpression could contribute to the progression of CH.
We observed significant miR-377 upregulation in vitro in Ang-II-treated H2C9 cardiomyocytes as well as in vivo in mice with CH. In mice with CH, ANP and BNP were also highly upregulated. The results of other previous studies are also notable. Zhang et al. (Citation2014) showed that CH could be induced by miR-22 overexpression, and Callis et al. (Citation2009) reported that miR-208a overexpression was essential for inducing CH in mice. These findings accord with our study. Thomas et al. found that arrhythmias could be induced by miR-280a overexpression (Callis et al. Citation2009). In the current study, we did not investigate the relevance between arrhythmias and miR-377, but we intend to perform similar experiments with miR-377 in the future. Another relevant previous study showed that miR-212/132 overexpression induced pathological CH and directly regulated FoxO3 (Ucar et al. Citation2012). Here, we analyzed ANP and BNP instead of FoxO3. In our study, we confirmed the important role played by miR-377 in CH, and the findings obtained indicated that miR-377 could be a new target for the treatment of heart disease.
Autophagy refers to a series of events that occur in response to nutritional insufficiency. This process plays a pivotal role in the constitutive turnover of mitochondria in highly oxidative tissues, including cardiomyocytes (Kim et al. Citation2007). However, dysfunctional autophagy promotes the pathogenesis of many diseases, including neurodegenerative disorders, various cancers, and bacterial infections (Levine and Kroemer Citation2008). The findings obtained in our study indicated that increased miR-377 expression diminished cardiomyocyte autophagy and promoted the progression of CH, which could be reflected by increased ANP and BNP expression levels.
Our data add to recent evidence implicating autophagic dysfunction in the development of pathological CH. Berberine has been reported to promote CH development by enhancing autophagy via suppressing ERK1/2 MAPK, mTOR, and the p38 signaling pathway (Li et al. Citation2015). However, we did not analyze the ERK1/2 MAPK and p38 MAPK pathways in our study. Cao et al. reported that histone deacetylase decreased CH by inhibiting autophagy. This observation is contrary to our findings. Those authors also found that histone deacetylase was essential for autophagic responses (Cao et al. Citation2011). In addition, sestrin-1 was identified as a downstream target of p53 that promoted CH by activating autophagy (Xue et al. Citation2017).
Our finding of links involving miR-377, CH and PPARγ also augments the increasing body of evidence demonstrating the pivotal part played by PPARγ in the pathophysiology and development of the heart, including cardiac dysfunction and cardiomyocyte length regulation (Son et al. Citation2007; Hinrichs et al. Citation2011). Ni-Huiping et al. showed that PPARγ overexpression induced cardiac dysregulation, including dilated cardiomyopathy, distorted architecture of the mitochondria, and disrupted cristae (Son et al. Citation2007). It was reported that PPARγ downregulation in adipocytes effectively attenuated CH induced by rosiglitazone in vivo (Duan et al. Citation2017). The findings obtained in the current study indicated that PPARγ expression in hypertrophic cardiomyocytes dramatically decreased, which suggested that miR-377 acts directly on PPARγ. PPARγ overexpression significantly promoted myocardial autophagy, which could be evidenced by the fact that protein levels of LC3-II and beclin-1 as well as the ratio of LC3-II to I increased regardless of whether Ang-II was present or not (P < 0.05). In contrast, PPARγ knockdown significantly inhibited autophagy, as evidenced by concomitant decreases in LC3-II, beclin-1, and in the ratio of LC3-II to I. Notably, Weiss et al. reported that PPARγ activation by pioglitazone did not alleviate left ventricular hypertrophy in rats (Weiss et al. Citation2010). However, consistent with our study, PPARγ-induced suppression of CH progression via autophagy activation has been shown previously (Yuan et al. Citation2017). The results of our current study further advocated that PPARγ could be investigated as a target in the treatment of CH.
In summary, we found that miR-377 promoted CH by targeting PPARγ. This novel miR-377/PPARγ axis may offer a potential therapeutic target for CH.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Due to the nature of this research, participants of this study did not agree for their data to be shared publicly, so supporting data is not available.
References
- Ahmed FE, Ahmed NC, Vos PW, Bonnerup C, Atkins JN, Casey M, Nuovo GJ, Naziri W, Wiley JE, Mota H, et al. 2013. Diagnostic microRNA markers to screen for sporadic human colon cancer in stool: I. Proof of principle. Cancer Genomics Proteomics. 10(3):93–113.
- Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. 2007. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol. 293(3):H1883–H1891.
- Barger PM, Kelly DP. 2000. PPAR signaling in the control of cardiac energy metabolism. Trends Cardiovasc Med. 10(6):238–245.
- Berlo JH, Van, Marjorie M, Molkentin JD. 2013. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 123(1):37–45.
- Bushati N, Cohen SM. 2007. microRNA functions. Annu Rev Cell Dev Biol. 23:175–205.
- Calin GA, Croce CM. 2006. MicroRNA-cancer connection: the beginning of a new tale. Cancer Res. 66(15):7390–7394.
- Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, Chen JF, Deng Z, Gunn B, Shumate J, et al. 2009. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest. 119(9):2772–2786.
- Campard D, Vasse M, Rose-John S, Poyer F, Lamacz M, Vannier JP. 2006. Multilevel regulation of IL-6R by IL-6-sIL-6R fusion protein according to the primitiveness of peripheral blood-derived CD133+ cells. Stem Cells. 24(5):1302–1314.
- Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, Rothermel BA, Gillette TG, Hill JA. 2011. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A. 108(10):4123–4128.
- Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. 2007. MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy? Am J Pathol. 170(6):1831–1840.
- Duan LJ, Ding M, Hou LJ, Cui YT, Li CJ, Yu DM. 2017. Long noncoding RNA TUG1 alleviates extracellular matrix accumulation via mediating microRNA-377 targeting of PPARgamma in diabetic nephropathy. Biochem Biophys Res Commun. 484(3):598–604.
- Duan SZ, Ivashchenko CY, Russell MW, Milstone DS, Mortensen RM. 2005. Cardiomyocyte-specific knockout and agonist of peroxisome proliferator-activated receptor-gamma both induce cardiac hypertrophy in mice. Circ Res. 97(4):372–379.
- Efeyan A, Comb WC, Sabatini DM. 2015. Nutrient-sensing mechanisms and pathways. Nature. 517(7534):302–310.
- Esteller M. 2011. Non-coding RNAs in human disease. Nat Rev Genet. 12(12):861–874.
- Formosa A, Markert EK, Lena AM, Italiano D, Finazzi-Agro E, Levine AJ, Bernardini S, Garabadgiu AV, Melino G, Candi E. 2014. MicroRNAs, miR-154, miR-299-5p, miR-376a, miR-376c, miR-377, miR-381, miR-487b, miR-485-3p, miR-495 and miR-654-3p, mapped to the 14q32.31 locus, regulate proliferation, apoptosis, migration and invasion in metastatic prostate cancer cells. Oncogene. 33(44):5173–5182.
- Gómez-Hurtado N, Domínguez-Rodríguez A, Mateo P, Fernandez-Velasco M, Val-Blasco A, Aizpún R, Sabourin J, Gómez AM, Benitah JP, Delgado C. 2017. Beneficial effects of leptin treatment in a setting of cardiac dysfunction induced by transverse aortic constriction in mouse. J Physiol. 595(13):4227–4243.
- Gottlieb RA, Mentzer RM. 2010. Autophagy during cardiac stress: joys and frustrations of autophagy. Annu Rev Physiol. 72:45–59.
- Green DR, Beth L. 2014. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 157(1):65–75.
- Guo Y, Chen Z, Zhang L, Zhou F, Shi S, Feng X, Li B, Meng X, Ma X, Luo M, et al. 2008. Distinctive microRNA profiles relating to patient survival in esophageal squamous cell carcinoma. Cancer Res. 68(1):26–33.
- Han Y, Chen J, Zhao X, Liang C, Wang Y, Sun L, Jiang Z, Zhang Z, Yang R, Chen J, et al. 2011. MicroRNA expression signatures of bladder cancer revealed by deep sequencing. PLoS One. 6(3):e18286.
- Hinrichs S, Heger J, Schreckenberg R, Wenzel S, Euler G, Arens C, Bader M, Rosenkranz S, Caglayan E, Schluter KD. 2011. Controlling cardiomyocyte length: the role of renin and PPAR-γ. Cardiovasc Res. 89(2):344–352.
- Kamo T, Akazawa H, Komuro I. 2015. Cardiac nonmyocytes in the hub of cardiac hypertrophy. Circ Res. 117(1):89–98.
- Kim I, Rodriguez-Enriquez S, Lemasters JJ. 2007. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 462(2):245–253.
- Klionsky DJ, Emr SD. 2000. Autophagy as a regulated pathway of cellular degradation. Science. 290(5497):1717–1721.
- Lee CH, Olson P, Evans RM. 2003. Minireview: lipid metabolism, metabolic diseases, and peroxisome proliferator-activated receptors. Endocrinology. 144(6):2201–2207.
- Levine B, Kroemer G. 2008. Autophagy in the pathogenesis of disease. Cell. 132(1):27–42.
- Li Z, Wang J, Yang X. 2015. Functions of autophagy in pathological cardiac hypertrophy. Int J Biol Sci. 11(6):672–678.
- Schultz NA, Dehlendorff C, Jensen BV, Bjerregaard JK, Nielsen KR, Bojesen SE, Calatayud D, Nielsen SE, Yilmaz M, Hollander NH, et al. 2014. MicroRNA biomarkers in whole blood for detection of pancreatic cancer. JAMA. 311(4):392–404.
- Son NH, Park TS, Yamashita H, Yokoyama M, Huggins LA, Okajima K, Homma S, Szabolcs MJ, Huang LS, Goldberg IJ. 2007. Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J Clin Invest. 117(10):2791–2801.
- Staels B, Fruchart JC. 2005. Therapeutic roles of peroxisome proliferator-activated receptor agonists. Diabetes. 54(8):2460–2470.
- Tan WS, Mullins TP, Flint M, Walton SL, Bielefeldt-Ohmann H, Carter DA, Gandhi MR, McDonald HR, Li J, Moritz KM, et al. 2018. Modeling heart failure risk in diabetes and kidney disease: limitations and potential applications of transverse aortic constriction in high-fat-fed mice. Am J Physiol Regul Integr Comp Physiol. 314(6):R858–R869.
- Tirziu D, Simons M. 2009. Endothelium as master regulator of organ development and growth. Vascul Pharmacol. 50(1–2):1–7.
- Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S, Dangwal S, Kumarswamy R, Bang C, Holzmann A, et al. 2012. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. 3:1078.
- Weiss CS, Hagenmuller M, Pichler M, Munz S, Ochs M, Buss SJ, Bekeredjian R, Katus HA, Hardt SE. 2010. Activation of PPARgamma by pioglitazone does not attenuate left ventricular hypertrophy following aortic banding in rats. Naunyn Schmiedebergs Arch Pharmacol. 381(4):285–295.
- Xue R, Zeng J, Chen Y, Chen C, Tan W, Zhao J, Dong B, Sun Y, Dong Y, Liu C. 2017. Sestrin 1 ameliorates cardiac hypertrophy via autophagy activation. J Cell Mol Med. 21(6):1193–1205.
- Yuan S, Jin J, Chen L, Hou Y, Wang H. 2017. Naoxintong/PPARgamma signaling inhibits cardiac hypertrophy via activation of autophagy. Evid Based Complement Alternat Med. 2017:3801976.
- Zamore PD, Haley B. 2005. Ribo-gnome: the big world of small RNAs. Science. 309(5740):1519–1524.
- Zhang R, Luo H, Wang S, Chen W, Chen Z, Wang HW, Chen Y, Yang J, Zhang X, Wu W, et al. 2014. MicroRNA-377 inhibited proliferation and invasion of human glioblastoma cells by directly targeting specificity protein 1. Neuro Oncol. 16(11):1510–1522.
- Zhou LY, Liu JP, Wang K, Gao J, Ding SL, Jiao JQ, Li PF. 2013. Mitochondrial function in cardiac hypertrophy. Int J Cardiol. 167(4):1118–1125.
- Zhu W, He J, Chen D, Zhang B, Xu L, Ma H, Liu X, Zhang Y, Le H. 2014. Expression of miR-29c, miR-93, and miR-429 as potential biomarkers for detection of early stage non-small lung cancer. PLoS One. 9(2):e87780.