
ABSTRACT
The emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) disease (COVID-19) at the end of 2019 has caused a large global outbreak and now become a major public health issue. However, there is currently a lack of data underlying how the human host interacts with SARS-CoV-2 virus. In the current study, We performed Venn-analysis, Gene ontology (GO), KEGG pathway analysis and Protein-protein interaction analysis of whole transcriptome studies with the aim of clarifying the genes and pathways potentially altered during human respiratory tract cell infection with SARS-CoV-2. We found 36 overlapping upregulated genes among different types of cells after viral infection. Further functional enrichment analysis revealed these Differential Expressed Genes (DEGs) are most likely involved in biological processes related to inflammatory response and response to cytokine, cell component related to extracellular space and I-kappa B/NF-kappa B complex, molecular function related to protein binding and cytokine activity. KEGG pathways analysis highlighted altered conical and casual pathways related to TNF, NF-kappa B, Cytokine-cytokine receptor interaction and IL-17 signaling pathways during SARS CoV-2 infection with CXCL1, CXCL2, CXCL3, CXCL8, CXCL10, IL32, CX3CL1, CCL20, IRF1, NFKB2 and NFKB1A up-regulated which may explain the inflammatory cytokine storms associated with severe cases of COVID-19.
Introduction
The outbreak of Coronavirus Disease 2019 (COVID-19) caused by the novel coronavirus named severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), was first reported at the end of 2019 (Tan et al. Citation2020; Zhu et al. Citation2020). The clinical characteristics of COVID-19 include respiratory symptoms, fever, cough, dyspnea, pneumonia (Chen et al. Citation2020; Huang et al. Citation2020; Li et al. Citation2020; Wang et al. Citation2020), acute respiratory distress syndrome (ARDS), and even multiple organ dysfunction in severe cases (Singhal Citation2020). Clinical characteristics varied in different COVID-19 patients, and a number of patients showed asymptomatic infection (Chan et al. Citation2020; Guan et al. Citation2020; Xu et al. Citation2020) Recent studies have revealed that asymptomatic cases could also transmit the virus to contacts around them, which makes it more difficult to control the spread of the virus (Rothe et al. Citation2020).
Several anti-viral drugs have been used to target the SARS-CoV-2 infection, including lopinavir/ritonavir, penciclovir, chloroquine, hydroxychloroquine, remdesivir, and ribavirin. However, most of them have limited efficacy to reduce the severity of COVID-19 patients. In addition, some also have serious side effects (Cao et al. Citation2020; Wang et al. Citation2020). The shortcomings of current tested drugs have resulted in scientists and physicians continuously seeking alternative, effective treatments for COVID-19 infection. Transcriptional profiling has provided remarkable opportunities for understanding the relationship between cellular function and metabolic pathways, as well as to define the possible implications of genetic variability and environmental conditions in many tissues and organisms after viral infection (Nowakowski et al. Citation2016; Li et al. Citation2019), which could contribute to the screening of drug targets against viruses. In bioinformatics, RNA-Seq analyses are challenging due to its complexity and voluminosity. However, many computational methods and techniques have been developed for the use of the analysis of transcriptomics data. In addition, network-based approach is also extensively used to analyze and identify the potential candidate genes after viral infection (Gratton et al. Citation2020).
The clinical picture of COVID-19 patients are characterized by an over-exuberant immune response with lung lymphomononuclear cells infiltration and proliferation that may account for tissue damage more than the direct effect of viral replication (Azkur et al. Citation2020; Blanco-Melo et al. Citation2020; Yang et al. Citation2020). Evidence also showed that SARS-CoV-2 infects human respiratory tract cells, prompting inflammation and tissue damage/loss as well as in other different cell types (Wong et al. Citation2020). Despite recent advances, little is known about the pathways involved in viral pathogenesis (Ziegler et al. Citation2020). In the present study, we performed an analysis of several transcriptome studies, aiming to clarify which genes and cellular networks were up- or down-regulated during SARS-CoV-2 infection in human lung cells. Moreover, we assessed a comprehensive GO enrichment and KEGG pathway analysis to predict how the modulation of these genes could affect the outcome of the disease.
Materials and methods
Study search and RNA-Seq data collection
We searched for RNA-Seq experiments deposited in the Gene Expression Omnibus (GEO) database and NCBI PubMed related to SARS-CoV-2 infection in different types of human respiratory tract cells: 1. Calu-3 was infected with SARS-CoV-2 (USA-WA1/2020/NR-52281) at an MOI of 2 for 24 h, sequencing libraries were sequenced on an Illumina NextSeq 500 platform. ID: doi/10.3390/biology9090260 (Shaath and Alajez Citation2020); 2. A549ACE2 was infected with SARS-CoV-2 (USA-WA1/2020/ NR-52281) at an MOI of 0.2 for 24 h, sequencing libraries were sequenced on an Illumina NextSeq 500 platform. ID: doi/10.1101/2020.07.12.199687 (GSE154613) (Daisy et al. Citation2020); 3. A lung organoid model driving by human pluripotent stem cells was infected with SARS-CoV-2 (USA-WA1/2020/ NR-52281) at an MOI of 0.01 for 24 h, sequencing libraries were sequenced on an Illumina NovaSeq 6000 platform. ID: doi/10.1038/s41586-020-2901-9 (GSE155241) (Tatsuya et al. Citation2020); 4. Human bronchial organoids generated by primary human bronchial epithelial cells was infected with SARS-CoV-2 (Hu/DP/Kng/19-020) of 5.0 × 104 PFU for 5 days, sequencing libraries were sequenced on an Illumina NextSeq 500 platform. ID: doi/10.1101/2020.05.25.115600v2 (GSE150819) (Han et al. Citation2020) which carried out in human cells (ex vivo) or human cell lines (in vitro). Genes with fold change >1.3 or <0.77 and p-values <0.05 were considered to be statistically significant.
Functional analysis of differential expressed genes
The Go terms enrichment, KEGG pathways and protein–protein interaction analysis of the DEGs were performed using OmicsBean (http://www.omicsbean.cn) and Cytoscape v. 3.1.0 software (http://www.cytoscape.org/) (Saito et al. Citation2012) as a complementary and more comprehensive approach for identifying the central hub genes. Gene ID and Fold change are required to upload to complete the relevant analysis, the results were visualized using the ClueGO v. 2.1.1 application in Cytoscape, which creates clusters of functionally related genes using the following GO databases: KEGG, Wiki Pathways, REACTOME, and GO.
Results
Overlapping and functional enrichment analysis of differential expression genes among different lung cells infected with SARS-CoV-2
To highlight the changes in cellular host genes in response to SARS-CoV-2 infection and further explore the mechanism of viral infection and pathogenesis, we searched the electronic databases (GEO databases or PubMed) to identify the related studies. We included 4 studies in our qualitative study, they are all about transcriptome profiling of human respiratory tract cells (Calu-3, doi:10.3390/biology9090260; ACE2-A549, GSE154613; Lung organoid model driving by human pluripotent stem cells, GSE155241; Primary human bronchial epithelial cells, GSE150819).
Using Venn diagram analysis for the groups’ data, the following was found: there were 36 overlapping up-regulated and 0 overlapping down-regulated mRNAs among the four infection groups (Figure (A,B)). We further performed a functional enrichment analysis using the 36 up-regulated genes. Results revealed 2,506 terms associated with biological processes (BP), 164 terms associated with cell components (CC), 226 terms associated with molecular functions (MF), and 102 terms associated with Kyoto Encyclopedia of Genes and Genomes pathways (KEGG). Of these, a number of terms were significantly enriched including 1,907 for BP, 78 for CC, 98 for MF and 39 for KEGG (Figure (A)). The 10 most enriched terms are presented in detail for BP, CC and MF (Figure (B)).
Figure 1. Comparative analysis of differentially expressed mRNAs in SARS-CoV-2 infected lung cells. A,B Venn analysis of all up/down expressed mRNAs among the four SARS-CoV-2 infection groups. a: Calu-3 infected with SARS-CoV-2 (doi:10.3390/biology9090260), b: ACE2-A549 infected with SARS-CoV-2 (GSE154613), c: Lung organoid model using human pluripotent stem cells infected with SARS-CoV-2 (GSE155241), d: Primary human bronchial epithelial cells infected with SARS-CoV-2 (GSE150819).
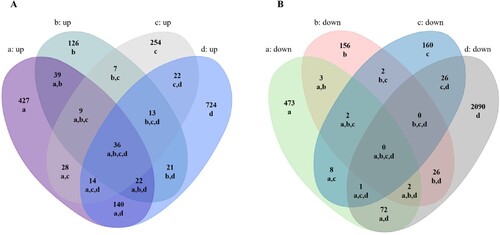
Figure 2. GO term enrichment analysis of SARS-CoV-2 induced up-regulated genes in all four groups. A, Number of enriched GO terms and KEGG pathways. B, Overview of significantly enriched biological processes, cell components, and molecular functions.
Biological process analysis of overlapping all up-regulated genes
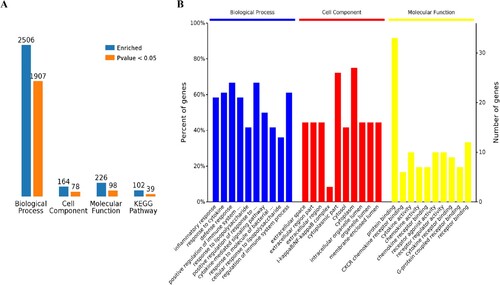
The 1,907 different biological process GO terms were further analyzed using OmicsBean software. According to the different levels of the GO terms (Figure (A,C)), most differential genes were related to positive regulation of immune system process, regulation of immune system process and positive regulation of response to stimulus (level 3 GO), and continued subdivision of the grades showed that the differential genes were involved in the defense response, response to cytokine and cytokine-mediated signaling pathways (level 3–7 GO). According to the number of enriched genes (Figure (B)), they mainly enriched in signal transduction (29 differential genes), defense response (24 differential genes), regulation of cell communication (24 differential genes), response to organic substance (23 differential genes), multicellular organism development (22 differential genes) and cellular response to chemical stimulus (20 differential genes). A sorting of the different GO terms based on the P-value and gene number (Figure (C–E)) revealed that these up-regulated genes are most likely involved in biological processes related to inflammatory response and response to cytokine, which indicates that SARS-CoV-2 infection could cause a strong host immune response.
Figure 3. Analysis of 1907 biological process significantly enriched by all-up-regulated genes. A, Levels of enriched biological processes. B, Percent of genes associated with enriched biological processes. C, Top 10 of significantly enriched biological processes. D, Expressed genes associated with enriched biological processes. E, Pie chart of expressed proteins of enriched biological processes.
Cell component analysis of all overlapping up-regulated genes
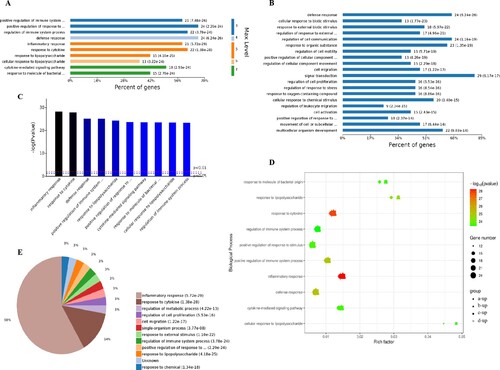
The analysis of the 78 different cell components-related GO terms showed that according to the classification of the different GO levels (Figure (A)), most up-regulated genes were related to continued subdivision identified extracellular region part, membrane-enclosed lumen (level 2 GO) and organelle lumen (level 3 GO), cytoplasm, cytoplasmic part and cytosol (level 4–6 GO). According to the number of enriched genes (Figure (B)), the most abundantly enriched up-regulated genes were associated with cytoplasm (27 genes), intracellular organelle (22 genes), intracellular organelle part (20 genes) and vesicle (9 genes). By distinguishing the difference in the P-value of the GO terms and gene number (Figure (C,D)), we found that these up-regulated genes were enriched in extracellular space and I-kappa B/NF-kappa B complex of cell components.
Figure 4. Analysis of 78 cell components significantly enriched by all up-regulated genes. A, Levels distribution of enriched cell components. B, Percent of genes associated with enriched cell components. C, Top 10 of significantly enriched cell components. D, Pie chart of expressed proteins of enriched cell components.
Molecular function analysis of overlapping all upregulated genes
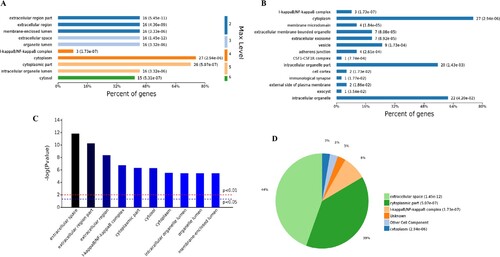
The analysis of the 98 different molecular functions according to the number of the different GO classification levels (Figure (A)), the genes showing the highest differential expression are associated with protein binding (level 3 GO), and further subdivisions showed that the up-regulated genes are associated with receptor binding, cytokine receptor binding, G-protein coupled receptor binding, chemokine receptor binding and CXCR chemokine receptor binding (level 4–7 of GOs). Further analysis of enriched genes (Figure (B)) revealed that the most abundant up-regulated genes were enriched in receptor binding (12 genes), identical protein binding (10 genes) and enzyme binding (6 genes). Sorting the different GO terms based on ascending P-values and gene number (Figure (C,D)) revealed that the genes showing the greatest differential expression are likely enriched in the molecular functions of the protein binding and cytokine activity.
Figure 5. Analysis of 98 molecular function enriched by all up-regulated genes. A, Levels distribution of enriched molecular functions. B, Percent of genes associated with enriched molecular functions. C, Top 10 of significantly enriched molecular functions. D, Pie chart of expressed proteins of enriched molecular functions.
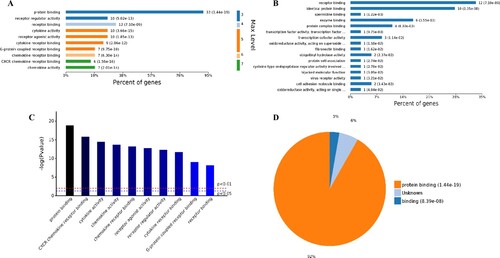
Based on the above results, it can be hypothesized that SARS-CoV-2 infection could activate the immune response in human respiratory tract cells. A statistical analysis of immune regulation GO terms and pathways (Supplemental Tables 1–4) showed that many up-regulated genes, such as, PTGS2, LIF, NFKBIA, ICAM1, CSF1, NFKB2, NFKBIZ, CX3CL1, CCL20, CXCL3, BIRC3 and so on, are involved in.
KEGG pathway and protein–protein interaction analysis of overlapping all up-regulated genes
Through a KEGG pathway cluster analysis, we further found that the up-regulated genes mainly regulated the TNF signaling pathway, cytokine-cytokine receptor interaction, viral protein interaction with cytokine and cytokine receptor and IL-17 signaling pathway (Figure (A)). Sorting the different KEGG pathways based on ascending P-values and gene number (Figure (B,C)) revealed that the up-regulated genes mainly enriched in TNF signaling pathway, NF-kappa B signaling pathway and IL-17 signaling pathway.
Figure 6. KEGG pathway cluster analysis and correlation analysis of all up-regulated genes. A, Percent of genes associated with enriched KEGG pathway. B, Distribution of enriched KEGG pathways. C, Pie chart of significant enriched KEGG pathway. D, Correlation analysis of up-regulated genes associated with key pathways. E, SARS-CoV-2 infection regulated the IL-17 signaling pathway. The key up-regulated factors are marked in red.
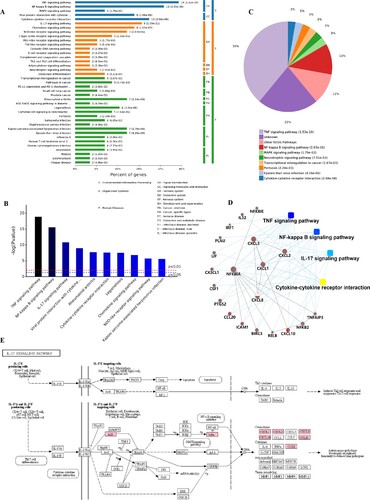
A protein–protein interaction analysis (Figure (D)) was also explored. The results showed that the key genes regulating these processes are CXCL1, CXCL2, CXCL3, CXCL8, CXCL10, IL32, CX3CL1, CCL20, IRF1, NFKB2 and NFKB1A, NFKB1E and their certain interaction relationship was also shown. Furthermore, an analysis of the most significant IL-17 signaling pathway in KEGG (Figure (E)) revealed that the TNFAIP3, NFKBIA (upstream) gene in the pathway and the CXCL1, CXCL2, CXCL8. CXCL10, CCL20 and PTGS2 (downstream) genes in the pathway play an important role in immunity of host cells to SARS-CoV-2 infection. In addition, the details of NF-kappa B and TNF signaling pathway activated by SARS-CoV-2 infection were also shown in Figure S1–2. This finding is consistent with the results of the GO analysis, which showed that SARS-CoV-2 infection mainly activated the ability of respiratory tract cells to respond to the invasion of extracellular pathogens.
Discussion
As of 13 Oct 2020, a total of 37,423,660 confirmed cases of COVID-19 with 1,074,817 deaths had been reported (WHO Citation2020), the case numbers are still increasing rapidly due to the second wave of epidemics worldwide. The underlying biological processes and mechanisms impacted by this viral infection of the host are still not clear. Analyzing the gene expression profiles of host cells infected with SARS-CoV-2 will be necessary to decipher the subcellular functions perturbed by this virus and to inform drug development strategies.
RNA-Seq has contributed to increase the knowledge relative to the biological and cellular processes involved during viral infection, especially in the context of an emergent pathogen (Li et al. Citation2019; Gratton et al. Citation2020), such as SARS-CoV-2. Venn analysis of several RNA-Seq assays increases the statistical power of samples and we applied this method for re-examining past evidence to better understand the pathogenesis of infections. In our current study, we explored recent SARS-CoV-2 infected cell-derived transcriptome datasets to dissect the cellular and molecular changes in response to SARS-CoV-2 infection. We finally included four available studies and analyzed the gene expression patterns using Venn analysis (Figure ).
The RNA-Seq results of respiratory tract cells infected by SARS-CoV-2 showed that the up-regulated genes identified from the comparison of two groups such as CXCL1, CXCL2, CXCL3, CXCL8, CXCL10, IL32, CX3CL1, CCL20, IRF1, NFKB2 and NFKB1A, NFKB1E are involved in the regulation of TNF signaling pathway, NF-kappa B signaling pathway and IL-17 signaling pathway. Tumor necrosis factor (TNF) was first described as a glycoprotein induced in response to endotoxin and has the capacity to kill tumor cells (Carswell et al. Citation1975). Then, TNF was shown to be a T helper type 1 (Th1) cytokine produced by several cell types, and to be involved in pro-inflammatory responses like interleukin (IL)-1β and IL-6 (Beutler and Cerami Citation1986). In concert with chemokines, TNF signaling pathway is involved in the regulation of inflammatory processes of infectious diseases including HIV (Human immunodeficiency virus), JEV (Japanese encephalitis virus), WNV (West Nile virus) and USUV (Ustuuvirus) infection (Cheng et al. Citation2004; Bradley Citation2008; Pasquereau et al. Citation2017; Obdulio et al. Citation2019). The nuclear factor NF-kappa B pathway has also long been considered a prototypical pro-inflammatory signaling pathway, largely based on the role of NF-kappa B signaling pathway in the expression of pro-inflammatory genes including cytokines and chemokines (Lawrence et al. Citation2001). Therefore, NF-kappa B has been considered as a target for new anti-inflammatory drugs for a long time. In our study, we found SARS-CoV-2 infection could activate NF-kappa B signaling pathway and up-regulate some pro-inflammatory genes, we further speculate drugs targeting the NF-kappa B signaling pathway may be one of the candidate drugs for the treatment severe COVID-19 patients.
It is worth mentioning that, IRF1 in respiratory tract cells was up-regulated by SARS-CoV-2 infection. IRF1 is a member of the IFN regulatory factor (IRF) family that plays an important role in immunomodulation. IRF1 prominently participates in antiviral defense by regulating early expression of IFNs (Panda et al. Citation2019). In addition, as a transcription factor, IRF1 regulates constitutive expression of other antiviral genes by binding to their promoter directly. Studies revealed that IRF1 deficient mice are more susceptible to RNA virus infections (Kimura et al. Citation1994; Reis et al. Citation1994; Maloney et al. Citation2012; Mboko et al. Citation2014; Nair et al. Citation2017). Thus, we think IRF1 may play a critical role in regulating constitutive antiviral gene networks to confer resistance against SARS-CoV-2 infections in human respiratory epithelial cells.
Inflammation is the first line of defense against tissue damage caused viral infection, involving activating both the innate and adaptive immune responses (D’Elia et al. Citation2013). However, exuberant immune responses after viral infection, which also called cytokine storm, has been found to be associated with excessive levels of pro-inflammatory cytokines and also some tissue damage (Thompson et al. Citation2017). Cytokine storm has been proved to exist after several influenza viruses infection (Wang et al. Citation2010; Zhou et al. Citation2013), as well as coronavirus (Zhang et al. Citation2004; Jiang et al. Citation2005; Ayman et al. Citation2019), and contributes to acute lung injury or ARDS. Preliminary studies have revealed that SARS-CoV-2 infection could also trigger cytokine storm in severe cases, accompanied by an increase of various cytokines (Liu et al. Citation2020). In the current study, we found that several cytokine-related genes were upregulated after SARS-CoV-2 infection which could provide explanation about the cytokine storms associated with severe COVID-19 cases.
Conclusions
Our results highlighted activation of TNF, NF-kappa B, cytokine-cytokine interaction, IL-17 signaling pathway and other inflammatory response as the main hallmark associated with SARS-CoV-2 infection in different types of human respiratory epithelial cells in vitro. We further identified several differentially expressed genes during the course of infection such as IRF1, CXCL1-3, IL32, CXCL10, CX3CL1, CCL20, NFKB2 and others. Upregulation of these genes which may serve as useful biomarkers of COVID-19 infection. Interferon and inflammatory response to SARS-CoV-2 infection might provide explanation to cytokine storms associated with severe COVID-19 cases. However, precise mechanisms of the host response to SARS-CoV-2 require further detailed investigation.
Supplemental Material
Download Zip (3.2 MB)Acknowledgements
This work was supported by Shenzhen Natural Science Foundation (JCYJ20190809152415652), China Postdoctoral Science Foundation (2019M660836), the National Science and Technology Major Project (2018ZX10711001, 2017ZX10103011, 2018ZX09711003, and 2020YFC0841700), Shenzhen Science and Technology Research and Development Project (202002073000001). Yingxia Liu, Yu Wang, Minghui Yang and Luping Lei contributed to the study design. Minghui Yang, Luping Lei, Yang Yang and Jun Wang contributed to the manuscript and figures. Qiumei Cao, Xiao Jiang, Jinzhi Lai, Ling Qing and Kun Huang contributed to the proof reading. All the authors have read and approved the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
We searched for RNA-Seq experiments deposited in the Gene Expression Omnibus (GEO) database and NCBI PubMed related to SARS-CoV-2 infection in different types of human respiratory tract cells (Calu-3, doi:10.3390/biology9090260; ACE2-A549, GSE154613; a lung organoid model driving by human pluripotent stem cells, GSE155241; Primary human bronchial epithelial cells, GSE150819) which carried out in human cells (ex vivo) or human cell lines (in vitro).
Additional information
Funding
Related Research Data
References
- Ayman M, Wael A, Maged GH. 2019. Middle East respiratory syndrome coronavirus (MERS-CoV): infection, immunological response, and vaccine development. J Immunol Res. doi:10.1155/2019/6491738.
- Azkur AK, Akdis M, Azkur D, Sokolowska M, Veen W, Brüggen M, O’Mahony L, Gao Y, Nadeau K, Akdis C. 2020. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy. 75(7):1564–1581.
- Beutler B, Cerami A. 1986. Cachectin and tumour necrosis factor as two sides of the same biological coin. Nature. 320:584–588.
- Blanco-Melo D, Nilsson-Payant BE, Liu W-C, Uhl S, Hoagland D, Møller R, Jordan TX, Oishi K, Panis M, Sachs D, et al. 2020. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 181(5):1036–1045.e9.
- Bradley JR. 2008. TNF-mediated inflammatory disease. J Pathol. 214:149–160.
- Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G, Ruan L, Song B, Cai Y, Wei M, et al. 2020. A trial of lopinavir-ritonavir in adults hospitalized with severe covid-19. N Engl J Med. 382(19):1787–1799.
- Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. 1975. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci USA. 72:3666–3670.
- Chan J, Yuan S, Kok K, Wang To K, Chu H, Yang J, Xing F, Liu J, Yip CC, Poon RW et al., 2020. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 395(10223):514–523.
- Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, Han Y, Qiu Y, Wang J, Liu Y et al., 2020. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 395(10223):507–513.
- Cheng Y, Nicholas JC, Alison MK. 2004. The role of tumor necrosis factor in modulating responses of murine embryo fibroblasts by flavivirus, West Nile. Virology. 329(2):361–370.
- Daisy AH, Daniel JC, Rasmus M, Yuling H, Liuliu Y, Megan LW , Alexander L, Kasopefoluwa YO, Christian S, Benhur L, et al. 2020. Modulating the transcriptional landscape of SARS-CoV-2 as an effective method for developing antiviral compounds. Biorxiv. 7:13.
- D’Elia RV, Harrison K, Oyston PC, Lukaszewski RA, Clark GC. 2013. Targeting the ‘cytokine storm’ for therapeutic benefit. Clin Vaccine Immunol. 20:319–327.
- Gratton R, Tricarico PM, Agrelli A, Colaço da Silva HV, Coêlho Bernardo L, Crovella S, Campos Coelho AV, Rodrigues de Moura R, Cavalcanti Brandão LA. 2020. In vitro Zika virus infection of human neural progenitor cells: meta-analysis of RNA-Seq assays. Microorganisms. 8(2):270.
- Guan W, Ni Z, Hu Y, Liang W, Ou C, He J, Liu L, Shan H, Lei C, Hui D et al., 2020. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 382(18):1708–1720.
- Han Y, Duan X, Yang L, Benjamin EN, Wang P, Duan F, Tang X, Yaron TM, Zhang T, Uhl S, et al. 2020. Identification of SARS-CoV-2 inhibitors using lung and colonic organoids. Nature. 11:28.
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, et al. 2020. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 395(10223):497–506.
- Jiang Y, Xu J, Zhou C, Wu Z, Zhong S, Liu J, Luo W, Chen T, Qin Q, Deng P . 2005. Characterization of cytokine/chemokine profiles of severe acute respiratory syndrome. Am J Respir Crit Care Med. 171:850–857.
- Kimura T, Nakayama K, Penninger J, Kitagawa M, Harada H, Matsuyama T, Tanaka N, Kamijo R, Vilcek J, Mak TW . 1994. Involvement of the IRF-1 transcription factor in antiviral responses to interferons. Science. 264:1921–1924.
- Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA. 2001. Possible new role for NF-κB in the resolution of inflammation. Nat Med. 7:1291–1297.
- Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, Ren R, Leung K, Lau E, Wong J, et al. 2020. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med. 382(13):1199–1207.
- Li J, Zhang K, Fan W, Zhang S, Li Y, Gu J, Zhou J, Liu W. 2019. Transcriptome profiling reveals differential effect of interleukin-17A upon influenza virus infection in human cells. Front Microbiol. 10:2344.
- Liu Y, Zhang C, Huang F, Yang Y, Wang F, Yuan J, Zhang Z, Qin Y, Li X, Zhao D, et al. 2020. Elevated plasma level of selective cytokines in COVID-19 patients reflect viral load and lung injury. Natl Sci Rev. doi:10.1093/nsr/nwaa037.
- Maloney NS, Thackray LB, Goel G, Maloney NS, Thackray LB, Goel G. 2012. Essential cell-autonomous role for interferon (IFN) regulatory factor 1 in IFNgamma-mediated inhibition of norovirus replication in macrophages. J Virol. 86:12655–12664.
- Mboko WP, Mounce BC, Emmer J, Eric D, Shailendra BP, Vera LT. 2014. Interferon regulatory factor 1 restricts gammaherpesvirus replication in primary immune cells. J Virol. 88:6993–7004.
- Nair S, Poddar S, Shimak RM, Diamond MS. 2017. Interferon regulatory factor1 (IRF-1) protects against chikungunya virus induced immunopathology by restricting infection in muscle cells. J Virol. 91:e01419–17.
- Nowakowski T, Pollen A, Di Lullo E, Sandoval-Espinosa C, Bershteyn M, Kriegstein A. 2016. Expression analysis highlights AXL as a candidate Zika virus entry receptor in neural stem cells. Cell Stem Cell. 18(5):591–596.
- Obdulio GN, Marta L, Meret ER, Artur S. 2019. Monocyte-derived dendritic cells as model to evaluate species tropism of mosquito-borne flaviviruses. Front Cell Infect Microbiol. 28(9).
- Panda D, Gjinaj E, Bachu M, Squire E, Novatt H, Ozato K, Rabin RL. 2019. IRF1 maintains optimal constitutive expression of antiviral genes and regulates the early antiviral response. Front Immunol. 10:1019.
- Pasquereau S, Kumar A, Herbein G. 2017. Targeting TNF and TNF receptor pathway in HIV-1 infection: from immune activation to viral reservoirs. Viruses. 9(4):64.
- Reis LF, Ruffner H, Stark G, Aguet M, Weissmann C. 1994. Mice devoid of interferon regulatory factor 1 (IRF-1) show normal expression of type I interferon genes. EMBO J. 13:4798–4806.
- Rothe C, Schunk M, Sothmann P, Bretzel G, Froeschl G, Wallrauch C, Zimmer T, Thiel V, Janke C, Guggemos W, et al. 2020. Transmission of 2019-nCoV infection from an asymptomatic contact in Germany. N Engl J Med. 382(10):970–971.
- Saito R, Smoot ME, Ono K, Ruscheinski J, Wang P-L, Lotia S, Pico AR, Bader GD, Ideker T. 2012. A travel guide to cytoscape plugins. Nat Methods. 9:1069–1076.
- Shaath H, Alajez NM. 2020. Computational and transcriptome analyses revealed preferential induction of chemotaxis and lipid synthesis by SARS-CoV-2. Biology (Basel). 9(9):260.
- Singhal T. 2020. A review of coronavirus disease-2019 (COVID-19). Indian J Pediatr. 87(4):281–286.
- Tan W, Zhao X, Ma X, Wang W, Niu P, Xu W, Gao GF, Wen G. 2020. A novel coronavirus genome identified in a cluster of pneumonia cases – Wuhan, China 2019− 2020. China CDC Weekly. 2(4):61–62.
- Tatsuya S, Yumi I, Yusuke S, Akatsuki S, Daisuke O, Daisuke M, Shohei M, Takeshi K, Takuya Y, Toru O, et al. 2020. Generation of human bronchial organoids for SARS-CoV-2 research. Biorxiv. 6:01.
- Thompson BT, Rachel CC, Kathleen DL. 2017. Acute respiratory distress syndrome. N Engl J Med. 377(6):562–572.
- Wang S, Le T, Kurihara N, Chida J, Cisse Y, Yano M, Kido H. 2010. Influenza virus-cytokine-protease cycle in the pathogenesis of vascular hyperpermeability in severe influenza. J Infect Dis. 202:991–1001.
- Wang Y, Zhang D, Du G, Du R, Zhao J, Jin Y, Fu S, Gao L, Cheng Z, Lu Q, et al. 2020. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 395(10236):1569–1578.
- WHO. 2020. Coronavirus disease 2019 (COVID-19) situation report.
- Wong CK, Hayes KH, Lai W, Lau Y, Zhang R, Wong A, Lo G, Chan K, Hung I, Tse H, et al. 2020. Human-induced pluripotent stem cell-derived cardiomyocytes platform to study SARS-CoV-2 related myocardial injury. Circ J. 84(11):2027–2031.
- Xu X, Wu X, Jiang X, Xu K, Ying L, Ma C, Li S, Wang H, Zhang S, Gao H, et al. 2020. Clinical findings in a group of patients infected with the 2019 novel coronavirus (SARS-Cov-2) outside of Wuhan, China: retrospective case series. Br Med J. 368. doi: 10.1136/bmj.m606.
- Yang Y, Shen C, Li J, Yuan J, Wei J, Huang C, Wang F, Li G, Li Y, Xing L, et al. 2020. Plasma IP-10 and mcp-3 levels are highly associated with disease severity and predict the progression of COVID-19. J Allergy Clin Immunol. 146(1):119–127.e4.
- Zhang Y, Li J, Zhan Y, Wu L, Yu X, Zhang W, Ye L, Xu S, Sun R, Wang Y, et al. 2004. Analysis of serum cytokines in patients with severe acute respiratory syndrome. Infect Immun. 72:4410–4415.
- Zhou J, Wang D, Gao R, Zhao B, Song J, Qi X, Zhang Y, Shi Y, Yang L, Zhu W, et al. 2013. Biological features of novel avian influenza A (H7N9) virus. Nature. 499:500–503.
- Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, et al. 2020. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 382(8):727–733.
- Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, Cao Y, Yousif AS, Bals J, Hauser BM, et al. 2020. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. 181(5):1016–1035.e19.