
Abstract
Inflammation is a defense mechanism that the immune system uses in response to harmful stimuli such as pathogens, damaged cells, toxic compounds, or irradiation. These stimuli may induce inflammatory response and potentially tissue damage in respiratory, cardiovascular, digestive, nervous, endocrine, urinary, or reproductive systems. Inflammatory diseases include a broad array of disorders and conditions that are characterized by inflammation, ranging from autoimmune disease, atopic dermatitis, asthma, chronic obstructive pulmonary disease, inflammatory bowel disease, glomerulonephritis, hepatitis, reperfusion injury, transplant rejection, diabetes, cancer, Parkinson’s disease, multiple sclerosis, to depression. Epigenetic mechanisms play crucial roles in many biological processes by regulating transcriptional activation or repression. Histone post-translational modifications have emerged as prospective therapeutic targets. Methylation of histone 3 at lysine 9 is one of the most highly conserved epigenetic marks that correlate well with gene silencing. The methylation status of H3K9 modulates immune cell differentiation and immune responses and therefore influences the outcome of cancer, infection, and other inflammatory diseases. Here, we review the high impact and innovate discoveries in this field, highlight the critical role of the H3K9 methylation/de-methylation in human inflammatory diseases, discuss potential new therapeutic strategies based on a better understanding of the biology of H3K9 methylation modifications.
Abbreviations: 2OG: 2-oxoglutarate; AOL: Amine oxidase-like; ChIP: Chromatin immunoprecipitation; CNS2: conserved noncoding sequence 2; COPD: Chronic obstructive pulmonary disease; CoREST: Corepressor of RE1 silencing transcription factor; CPP: Cell-permeant protein; CTLA4: Cytotoxic T-lymphocyte antigen 4; DCs: Dendritic cells; DN: Diabetic nephropathy; ESRD: End-stage renal disease; ETDB: SET domain bifurcated histone lysine methyltransferase; FAD: Flavin adenine dinucleotide; FEV1%: The percentage of forced expiratory volume in one second; FEV1/FVC: Forced expiratory volume in one second (FEV1)/Forced vital capacity (FVC) value; FOXP3: forkhead/winged helix transcription factor 3; GLP: G9a like protein; GZMB: Granzyme B; H3K27me3: Trimethylation of lysine 27 on histone H3; H3K4me3: Trimethylation of lysine 4 on histone H3; H3K9: Histone 3 Lysine 9; HG: Hyperglycemia; HDAC3: Histone deacetylase 3; HP1: Heterochromatin protein 1; I/R: Ischemia/Reperfusion; IBD: Inflammatory bowel disease; IFN-: Interferon-; IFNG: Interferon Gamma; IL-2: Interleukin-2; ILC2s: Group 2 innate lymphoid cells; JMJD: Jumonji domain-containing protein; iTreg: inducible Treg; KDM: Lysine-specific demethylase; LPS: Lipopolysaccharide; LSD: Lys-specific demethylase; MCP-1: Monocyte chemoattractant protein-1; MCSF: Macrophage colony-stimulating factor; MIP-1: Macrophage inflammatory protein-1; MS: Metabolic syndrome; NAFLD: Non-alcoholic fatty liver disease; NKRF: NF-kappaB repressing factor; NOR: Norisoboldine; NuRD: Nucleosome remodeling and deacetylase; OxLDL: Oxidized low-density lipoprotein; PBMCs: Peripheral blood monocytes; PHD: Plant homology domain; Poly I:C: Polyinosinic:polycytidylic acid; PPARγ: Peroxisome proliferator-activated receptor gamma; PTMs: Post-translational modifications; SUV3-9: Suppressor of variegation3-9; T1D: Type 1 diabetes; TLRs: Toll like receptors; TSDR: TGF-β sensitive region and Treg cell specific demethylated region; Treg: Regulatory T cell; VSMCs: Vascular smooth muscle cells.
Introduction
Inflammation triggers inflammatory diseases including pulmonary diseases, Alzheimer’s disease and diabetes, cancer, atopic dermatitis, arthritis, asthma, gout, psoriasis, anemia, Parkinson’s disease, multiple sclerosis, and depression (Bruno et al. Citation2020; Ahmad et al. Citation2021; Carranza-Naval et al. Citation2021; Choi et al. Citation2021; Kinoshita and Goto Citation2021; Romano et al. Citation2021). These diseases have become increasingly threatening to human health (Wu et al. Citation2020; Placha and Jampilek Citation2021). During the development of inflammatory diseases, epigenetic mechanisms are responsible for expression or repression of different genes associated with cell development or immune responses (Alashkar Alhamwe et al. Citation2020; Fiuza et al. Citation2021). Histone modification, such as histone acetylation, methylation, or phosphorylation, plays a crucial role in early events which influence cognitive and other developmental outcomes as well as well-being (Bacon and Brinton Citation2021). For example, prenatal exposures to allergens from the environment can affect the placental histone modification in immune regulatory genes, and these genes are associated with the development of sensitization to allergens in young children (Acevedo et al. Citation2019; Harb et al. Citation2019). Eukaryotic DNA is wrapped around histone octamers that are comprised of two copies each of the H2A, H2B, H3 and H4 histone proteins to form nucleosomes (Mozzetta et al. Citation2015). The N-terminal tail of each core histone and the C-terminal tail of the two H2A proteins are the major sites for post-translational modifications (PTMs). Histone methylation occurs on the side chain lysine and arginine, gives rise to mono-, di-, or tri-methylated status, results in gene silencing or activation (Kang et al. Citation2017). The methylation of histone 3 Lysine 9 (H3K9), the most highly conserved epigenetic mark related to a heritable heterochromatic state (Du et al. Citation2015), regulates chromatin structure and gene expression by interactions among histones, DNA and chromatin-binding proteins (El Gazzar et al. Citation2007; Kang et al. Citation2017). H3K9me2 is reported as a repressive marker of euchromatin and hallmark feature of heterochromatin (Miao et al. Citation2008b). The dynamic regulation of H3K9 relies on the coaction of histone methyltransferase and demethylase. Currently, findings in H3K9 methylation modification have highly impacted in cancer and inflammation research (Lutgens Citation2019; Yoo et al. Citation2020). This review aims to assess the role of H3K9 methyltransferases and demethylases in inflammation and related diseases (Figure ).
Figure 1. H3K9 methyltransferases and demethylases in inflammatory diseases.
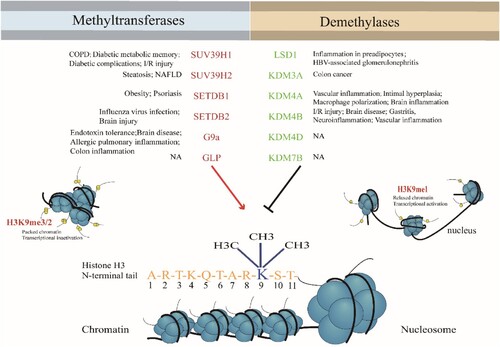
Properties of H3K9 methyltransferases and demethylases
Methylation of H3K9 is a hallmark of gene silencing, H3K9me2 and H3K9me3 contribute differently to heterochromatin formation. Comparing to transcriptionally silent H3K9me3 domains, H3K9me2 domains are relatively transcriptionally active (Jih et al. Citation2017). The H3K9 methylation modification is ubiquitous and is not only localized to the promoter but also to the inner region (body) of the silent transcription unit, thereby regulating gene expression in different states of the cell17, (El Gazzar et al. Citation2008, Citation2009; Chen et al. Citation2009). H3K9 methylases, such as the suppressor of variegation3-9 homolog (SUV39) family members, including SUV39H1, SUV39H2, SET domain bifurcated histone lysine methyltransferase (SETDB) 1, SETDB2, Euchromatic histone-lysine N-methyltransferase 2 (EHMT2, known as G9a) and G9a like protein 1(GLP), have a conserved SET domain. The acronym SET is derived from the Drosophila Su(var)3-9, Enhancer-of-zeste and Trithorax, which was identified as a part of conserved region in Drosophila Trithorax protein. The function of SET-domain is to transfer a methyl group from S-adenosyl-L-methionine (AdoMet) to the amino group of a lysine residue on the histone, leaving a methylated lysine residue and the cofactor byproduct S-adenosyl-L-homocysteine (AdoHcy).
SUV39 SET family has been characterized in the most detail. The pre-SET domain of SUV39-family proteins contains nine invariant cysteine residues that are grouped into two segments of five and four cysteines separated by various numbers of amino acids. The nine cysteines of the pre-SET domain coordinate three zinc ions to form a cluster, Zn3Cy9. The function of the pre-SET domain is maintaining the structural stability. The post-SET domain was discovered in the C-terminal forming a zinc-binding site, it forms part of the active sites by coupling to a fourth conserved cysteine in the knot-like structure close to the SET domain (Mozzetta et al. Citation2015). SUV39H1 and SUV39H2 have a chromatin organization modifier domain (chromodomain) and catalyze H3K9 di- and tri-methylation in constitutive heterochromatin, including the pericentromeric region. G9a and GLP have ankyrin repeating domains. A heterodimer of G9a and GLP (G9a-GLP) can mono- and di-methylate H3K9 in euchromatin regions which leads to gene repression. In vitro analyses have shown that G9a and GLP can form homodimers which exhibit H3K9 mono-, di- and tri-methylation activity (Hyun et al. Citation2017). The SET domain of SETDB1 and SETDB2 is interrupted by the insertion of 347 amino acids. SETDB1 also has two consecutive Tudor domains that contribute to methyl-lysine binding (Mozzetta et al. Citation2015; Rao et al. Citation2017). (Table ) H3K9 di- or tri-methylation exerts repressive effects by influencing DNA methylation and heterochromatin formation, prohibiting acetylation, the ‘activating’ modification, and actively recruiting transcriptional repressors of the Heterochromatin protein 1 family (Nakayama et al. Citation2001).
Table 1. The domain compositions and substrate specificities of H3K9 methyltransferases.
H3K9 demethylation activates gene expression. Histone methylation had long been considered stable until Lysine-specific demethylase 1A (KDM1A) / Lysine-specific demethylase 1 (LSD1) was identified as the first histone demethylase (Shi et al. Citation2004). Lysine-specific demethylases (KDMs) or Lysine-specific demethylase (LSD) subfamily contain a flavin adenine dinucleotide (FAD)-dependent C-terminal amine oxidase-like (AOL) domain which utilizes a FAD-dependent oxidase mechanism to generate an unstable hemiaminal intermediate which then fragments into a de-methylated product and formaldehyde (Nowak et al. Citation2016). LSD1 is the first defined histone demethylase which interacts with androgen receptor and promotes transcription of target genes by de-methylation of H3K9me1/2 (Alam et al. Citation2015). The incorporation of LSD1 into protein complexes such as CoREST(corepressor of RE1 silencing transcription factor) or NuRD (nucleosome remodeling and deacetylase) is essential for the ability of LSD1 to demethylate nucleosomes. The CoREST complex contains both histone demethylase and deacetylase enzymes, LSD1 and HDAC1, which are functionally and structurally coupled by the RCOR1 scaffold protein (Maiques-Diaz and Somervaille Citation2016; Song et al. Citation2020); Another class of lysine demethylases (KDMs, KDM3,4, and 7) are Jumonji domain (JmjC)- containing proteins. The catalytic JmjC domain has double-stranded and antiparallel β-sheet structure. And the JmjC proteins are Fe(II) and 2-oxoglutarate (2-OG, a-ketoglutarate)-dependent 30; The KDM4 family contains four genes (KDM4A-KDM4D) and two pseudogenes (KDM4E and KDM4F), specifically targets H3K9me2/3. Other KDM4 family members, including KDM4A, KDM4B and KDM4C, are structurally similar and contain JmjN domain, JmjC domain, two plant homology domains (PHD) and two Tudor domains. In contrast to other KDM4 members, KDM4D and KDM4E are structurally divergent, lacking the C-terminal PHD and Tudor domains, which serve as epigenome ‘readers’ (Black et al. Citation2012; Wilson and Krieg Citation2019). The KDM3 subfamily contains three members, namely KDM3A, KDM3B and JMJD1C, which are responsible for removing the methyl groups from H3K9 with a preference for H3K9me2. In the KDM3 family, a JmjC catalytic center and a C6 zinc finger are the two conserved evolutionary domains required for catalytic activity (Yoo et al. Citation2020; Sui et al. Citation2021). The KDM7 subfamily of enzymes PHF8 (KDM7B) and KIAA1718 (KDM7A) are human JmjC 2OG-dependent lysine demethylases. KDM7B and KDM7A are involved in demethylating lysine residues in H3K9me2/1, H3K27me2/1 and H4K20me1 (Chaturvedi et al. Citation2019b). The KDM7 enzymes have a PHD domain at the N-terminal, and JmjC at the C-terminal, both domains are dispensable for removing methyl group from H3K91/2. The KDM7A and KDM7B differ in flexibility and the length of their linker region (Chaturvedi et al. Citation2019a, Citation2019b) (Table ). To be brief, H3K9 methyltransferases and demethylases act as ‘writers’ and ‘erasers’ to precisely balance H3K9 methylation status. Aberrant histone methylation of H3K9 has been implicated in several human diseases (Greer and Shi Citation2012; Iacono et al. Citation2018). Therefore, more detailed understanding of H3K9 methylation is necessary for elucidating complex biological processes, developing and improving disease treatment.
Table 2. The domain composition and substrate specificities of H3K9 demethylases.
H3K9 methylation status in immune response
Epigenetic regulation has attracted increasing interest in the field of immune response. The epigenetic factors involved in initiation, maintenance, and innate immune memory were investigated intensively. The enrichment of H3K9 methylation was found at the promoters of a subset of inducible genes, such as IL12b and CCL22, and this repressive mark is removed rapidly following Lipopolysaccharide (LPS) stimulation (Saccani and Natoli Citation2002). The H3K9 methyltransferase Setdb1-deficiency leads to decreased basal H3K9 methylation levels and augments TLR4-mediated NF-κB recruitment on the proximal promoter region of IL-6, thereby accelerating IL-6 promoter activity (Hachiya et al. Citation2016; Yang et al. Citation2019). The expression of type I interferon (IFN) and IFN-stimulated genes (ISGs) are quantitatively controlled in a cell lineage-specific manner by H3K9me2, a negative histone marker. Moreover, deposition of H3K9me2 at ISGs by G9a results in reduced type I IFN response. Furthermore, GLP catalyzes H3K9 methylation at ISGs to suppress transcription, and GLP knockdown enhances antiviral immunity (Fang et al. Citation2012). Weak producers of IFN, such as fibroblasts, cardiac myocytes or neuroblastoma cells, or dendritic cells (DCs), have significantly lower levels of H3K9me2 at IFN and ISG gene promoters, yet the induction of the ISG genes is still regulated by the dynamic methylation of H3K9 (Saccani and Natoli Citation2002; Mehta and Jeffrey Citation2015). In macrophages, however, the decrease of jumonji domain-containing protein (JMJD) activity and high level of H3K9 methylation lead to a decrease in inflammatory chemokine production (Tausendschön et al. Citation2011). Alteration of H3K9 methylation status in inflammatory factor promoters or enhancers changes context-specific gene expression accordingly (Villeneuve et al. Citation2008).
In the studies of trained innate immunity, epigenetic modifications were evidenced to lead cell reprogramming by sustained changes in gene expression and cell physiology without permanent genetic changes. Polyinosinic:polycytidylic acid (Poly I:C) or retroviral vector encoding GFP accelerated the loss of H3K9me3 at the Sox2 promoter in cell-permeant protein (CPP)-treated fibroblasts via activating TLR3-induced chromatin remodeling and nuclear reprogramming (Lee et al. Citation2012a). Upon certain vaccinations or infections, human innate immune cells underwent extensive metabolic and epigenetic reprogramming, resulting in enhanced immune responses upon heterologous reinfection (Mourits et al. Citation2018). Hence, by targeting specific epigenetic mechanisms, inhibition of trained immunity can reduce excessive immune activation in chronic inflammatory conditions, and novel therapeutic approaches could be developed.
In adaptive immunity, lymphocyte lineage development, activation and movement are controlled by dynamic repression and activation of gene expression (Zhang et al. Citation2016). Epigenetic regulations are crucially involved in establishing cell type-specific gene-expression patterns (Taniuchi et al. Citation2004; Green et al. Citation2006; Falvo et al. Citation2013; Kakaradov et al. Citation2017; Tumes et al. Citation2017; Liu et al. Citation2019). Histone methylation is considered to play critical roles in restricting cytokine production to the appropriated lineages heritably in T cell differentiation (Rodriguez et al. Citation2017). Upon differentiation into Th1 cells in primary immune response, increased H3K9me3 in the promoter of IL4 and GATA3 genes was catalyzed by the histone methyltransferase, SUV39H1. Epigenetic modifications were found to play redundant role in both repression and activation of IFNG transcription: (1) the regulatory region of IFNG showed a reduction of trimethylation of lysine 27 on histone H3 (H3K27me3) and an increase in expression trimethylation of lysine 4 on histone H3 (H3K4me3) (Wei et al. Citation2009), which are sign of active marks, however, the repressive mark, H3K9me2 also increased (Chang and Aune Citation2007); (2) In CD4+ T cells cultured in Th1 conditions, loss of G9a, a histone methyltransferase catalyzing H3K9me2 and mediates gene repression, led to decrease of H3K9me2 at IFNG gene loci; However, the functional assessment did not indicate that G9a repressed IFNγ production in CD4+T cells from the G9a conditional knockout mice cultured in Th1 conditions. Furthermore, loss of G9a did not cause inappropriate IFNG transcription in CD4+T cells under Th2 conditions (Chang and Aune Citation2007).
In Th2 differentiation, H3K9me2 enriched in the IFNG locus, while the Granzyme B (GZMB) locus was heavily marked with H3K4me3 and H3K9me3 during an early stage. The SUV39H1-H3K9me3-heterochromatin protein 1 (HP1a) pathway is involved in maintaining the silencing of the Th1 locus (TBX21 and IFNG), ensuring the stability of the Th2 lineage (Bosch-Presegué et al. Citation2011; Allan et al. Citation2012). These findings showed the repressive effects of SUV39H1 on IFNG transcription in Th2 cells and on Th2 cytokines in Th1 cells. However, the levels of H3K9me3 at these gene loci and Th1 cells or Th2 cells generation were not affected in the loss of SUV39H1 during primary responses (Allan et al. Citation2012). Histone methyltransferase G9a was found to have an activating effect on Th2 cytokine production through a mechanism without catalyzing histone methylation (Chang and Aune Citation2007). All these observations suggest that the regulation of the repressive marker H3K9 is way more comprehensive, we believe that with a better understanding of the chromatin machinery, new therapeutic opportunities can be developed to boost or subdue the innate immune response in disease.
Regulatory T cells (Treg) represent a unique subset of T helper cells and are crucial for maintenance of immune equilibrium. Tregs has been known to promote homeostasis, self-tolerance, and tumor evasion/escape. As a regulator of Tregs, the transcription factor forkhead/winged helix transcription factor 3 (Foxp3) is instrumental in dermatitis, cancers, type 1 diabetes (T1D), and IBD (Agrawal et al. Citation2011; Fukuyama et al. Citation2012; Lesiak et al. Citation2012; Mayne and Williams Citation2013; Bayry and Gautier Citation2016; Tanaka and Sakaguchi Citation2019). In NOR(Norisoboldine) promoted Treg differentiation, SUV39H1 catalyzes the H3K9me3 modification, and this modification is associated with transcription repression of Foxp3. NOR promoted anti-ulcerative action by regulating AhR/glycolysis axis and subsequent NAD+/SIRT1/SUV39H1/H3K9me3 signaling pathway. Gene sequence analysis showed that Foxp3 loci had three highly conserved structures: promoter, TGF-β sensitive region and Treg cell specific demethylated region (TSDR, or conserved noncoding sequence 2, CNS2) (Zhang et al. Citation2013). Wang et. al undertook chromatin immunoprecipitation (ChIP) assays of HDAC3–/– and wild type T cells cultured under the iTreg (inducible Treg) polarizing conditions, HDAC3-deficient T cells showed increased H3K9me3 at the Foxp3 promoter and CNS2 regions comparing with wild type T cells, which indicate that H3K9 methylation status at Foxp3 promoter and CNS2 regions influences Treg cell development (Wang et al. Citation2015). H3K9 methyltransferases Suv39h1 was reported to be recruited by Smads to the IL-2 promoter, thereby inducing suppressive histone methylation and inhibiting T cell receptor-mediated IL-2 transcription (Wakabayashi et al. Citation2011), and finally suppressing Treg cell development, stability, and function. In a murine T cell transfer model of colitis, scholars found that G9a-dependent H3K9me2 is a homeostatic epigenetic checkpoint that regulates Th17 and Treg responses by limiting chromatin accessibility and TGF-β1 responsiveness. These findings suggested G9a as a therapeutic target for treating intestinal inflammation (Antignano et al. Citation2014). In T1D patients’ white blood cells, investigators discovered a subset of genes in diabetic lymphocytes displaying increased H3K9me2 linked to immune and inflammatory pathways often associated with T1D and its complications. The T1D candidate gene cytotoxic T-lymphocyte antigen-4 (CTLA4) has been displayed higher H3K9me2 at the promoter region (Miao et al. Citation2008a; Nano et al. Citation2018). In general, H3K9 methylating modification is essential in the regulation of immune cell differentiation, development, maintenance, and function. H3K9 methyltransferases or demethylates would be potential therapeutic targets for the treatment of inflammatory diseases including infections, T1D, IBD, dermatitis, and cancers.
The role of H3K9 methyltransferases in inflammatory diseases
SUV39H1
Chronic obstructive pulmonary disease (COPD) is a common, preventable and treatable disease that is associated with an enhanced chronic inflammatory response in the airways and lungs to noxious particles or gases (Vestbo et al. Citation2013). It was found that the levels of SUV39H1 and H3K9me3 were decreased in the lung tissue, peripheral blood monocytes (PBMCs) and primary human small airway epithelial cells in COPD patients (Chen et al. Citation2017). By Pearson correlation analysis, it was concluded that the levels of IL-6 and IL-8 in COPD patients were negatively correlated with SUV39H1 expression. In addition, there was a significant correlation between the percentage of forced expiratory volume in one second (FEV1%) predicted, or FEV1/forced vital capacity (FVC) value (FEV1/FVC), and SUV39H1 in all subjects (Chen et al. Citation2017). Other studies also confirmed that the PBMCs from COPD patients produce more pro-inflammatory cytokines than those from healthy controls, especially IL-8 (Lee et al. Citation2012b). These data suggest that the reduction of SUV39H1-H3K9me3 plays an important role in COPD inflammation (Chen et al. Citation2017). However, in the ECLIPSE (COPD) cohort, only a subset of patients showed a decrease in lung function (Kim et al. Citation2015). Similarly, merely 16% of patients showed persistent inflammation (Agustí et al. Citation2012). The reduction in SUV39H1 is likely to occur in the late stages of the disease. Some patients may develop susceptibilities to other harmful gases, leading to the rapid development of serious diseases and inflammation. Whether SUV39H1 is related to these patients remains unclear. Since the mutation or polymorphism of the SUV39H1 gene has not been reported in the genetic study of COPD (Wu et al. Citation2014), the regulation of transcription or post-transcription and the mechanism of disease progression are still unknown. Nevertheless, the maintenance of SUV39H1 expression could be a potential therapeutic target for COPD by the chromatin repressive effects of SUV39H1/ H3K9me3 on the downstream IL-8 gene (Chen et al. Citation2017).
In addition, SUV39H1/H3K9me3 disorders have been shown to be a potential mechanism for persistent inflammatory phenotypes, such as diabetic metabolic memory and vascular complications in diabetes patients (Villeneuve et al. Citation2008; Yu et al. Citation2012; Yang et al. Citation2014; Li et al. Citation2016; Wang et al. Citation2018). Relevant evidence from clinical trials and animal experimental models suggests that cardiovascular complications are currently the leading cause of disability and death in populations with diabetes (Cooper and Johnston Citation2000; King Citation2008). Although patients with type 1 and type 2 diabetes can control their blood sugar levels through medication, diet, and exercise, reducing their risk of vascular complications, most patients still cannot escape these fatal complications. Studies have shown that patients with poor early glycemic control, even after achieving glycemic control, continue to develop inflammation and cardiovascular complications, which is the phenomenon of ‘metabolic memory’ caused by hyperglycemia (HG) (Control Citation2005; Group Citation2008; Ceriello Citation2009); metabolism memory remains the main challenge in the treatment of complications from diabetes (Ceriello et al. Citation2009). Studies have shown that there is a link between chromatin histone methylation and metabolic memory, and H3K9me3 may be a key factor in metabolic memory (Villeneuve and Natarajan Citation2010). It is also suggested that enhancing H3K9me3, decreasing IL-6 expression and raising SUV39H1 in vascular smooth muscle cells may be effective strategies for preventing metabolic memory and cardiomyopathy in patients with diabetes (Yu et al. Citation2012), which can reduce its hypersensitivity to inflammatory stimulation (Villeneuve et al. Citation2008). A ChIP assay showed a significant decrease in the H3K9me3 level at the promoter of IL-6 in the cultured high glucose (HG) myocardial cell model (Yu et al. Citation2012); in the aortic vascular smooth muscle cells of diabetic mice, the levels of H3K9me3 at the promoters of IL-6, macrophage colony-stimulating factor (MCSF) and macrophage inflammatory protein-1 (MCP-1) and the enhancer of MCP-1 were significantly decreased, and the protein level of the H3K9me3 methyltransferase SUV39H1 was also reduced (Villeneuve et al. Citation2008). It is suggested that the chromatin modification of the histone methyltransferase SUV39H1 caused by high glucose contributes to the transcriptional activation of the IL-6 promoter. This is consistent with the increased expression of IL-6 in a diabetic state (Devaraj et al. Citation2006). After removing the HG conditions from the cell medium, the effect of HG on the expression of IL-6 and the level of H3K9me3 is irreversible, and the continuous increase in the expression of inflammatory genes indicates that inflammation may be the primary mechanism of ‘metabolic memory’. Previous experimental data on SUV39H1 gene silencing and overexpression indicate its negative regulation in inflammatory genes, such as IL-6, MCSF and MCP-1 (Villeneuve et al. Citation2008).
Recent studies have raised the importance of how chromatin levels are regulated for the pathogenesis of diabetes and its complications (Castelnau et al. Citation1998; Li et al. Citation2008; Intine and Sarras Citation2012; Reddy et al. Citation2013). Diabetic complications are associated with many inflammatory cytokines, such as TNF-α, MCP-1 and IL-6 (Kiyici et al. Citation2006; Dinh et al. Citation2009). Experiments have shown that high glucose level increases the expression of inflammatory cytokines in macrophages by reducing H3K9me3 level, which is partially mediated by SUV39H1. In high glucose cell media cultured macrophages, SUV39H1 was inhibited by chaetocin, which leads to increased levels of IL-6, IL-12p40, MIP-1a and MIP-1b. Furthermore, macrophages overexpressing SUV39H1 showed a decrease in aforementioned cytokines (Li et al. Citation2016). Approximately 40% of patients with diabetes develop diabetic nephropathy (DN), which is the leading cause of end-stage renal disease (ESRD) (Cooper Citation2012). Experiments have indicated that DN progression is related to SUV39H1 dysregulation. High glucose induces IL-6 and monocyte chemoattractant protein-1 (MCP-1) expression in renal tubular epithelial cell, HK-2, in a dose-dependent manner through upregulation of SUV39H1 and then downregulation. This is considered to be a negative feedback mechanism; the transcription and expression of inflammatory factors were inhibited with enriched H3K9me3 in the promoter region of the genes. However, the inhibitory effect of high level glucose on SUV39H1 was limited over time and the negative feedback loop broke (Wang et al. Citation2018). Clinical data suggested that patients with diabetes had an increased risk of ischemic events; diabetes is a risk factor for poor prognosis after coronary artery revascularization (Schnell et al. Citation2012). Researched indicated that SUV39H1 expression was reduced during myocardial ischemia-reperfusion (I/R) injury. Inflammation after I/R injury in patients with diabetes may be partially regulated by SUV39H1 expression and subsequent MAPK and NF-κB activation (Yang et al. Citation2014). SUV39H1 was found inhibiting the expression of inflammatory genes by significantly reducing the phosphorylation for p38, JNK, ERK1/2 and NF-κB pathways. In summary, SUV39H1 was proven to regulate inflammatory gene expression in vascular smooth muscle cells (VSMCs), macrophages, cardiomyocytes and renal tubular epithelial cells induced by high glucose via recruitment in the promoters of these genes. Still, more research needs to be carried out to address the link between inflammatory cytokine signaling and SUV39H1 as well as the mechanisms underlying the repressing effects of high glucose on SUV39H1 expression.
SUV39H2
Recent studies have suggested that H3K9 methyltransferase SUV39H2 plays a role in promoting the pathogenesis of steatosis and non-alcoholic fatty liver disease (NAFLD). Metabolic syndrome (MS) is a disease that is affected by genetic and environmental factors (Alberti et al. Citation2009). Steatosis is a typical manifestation of MS, characterized by the accumulation of lipid cells in the liver, the apparent infiltration of immune cells, the increase in the synthesis and release of pro-inflammatory mediators, and accelerating liver fibrosis in the later stages of the disease (Kang et al. Citation2017). SUV39H2 is found upregulated in a mouse model of steatosis, the level of inflammatory factors was downregulated, macrophage infiltration was reduced, and liver fibrosis was attenuated in SUV39H2 knockout mice. Further analysis revealed that SUV39H2 inhibited the expression of Sirtuin 1 (SIRT1) in the liver by inducing H3K9me3 around the SIRT1 promoter. SIRT1 is known to be a deacetylase of the Sir2 family and deacetylates NF-κB and SMAD3, thereby inhibiting its activity (Vaquero et al. Citation2004; Tachibana et al. Citation2005; El Gazzar et al. Citation2007). NAFLD is rapidly becoming the most common liver disease in the world due to high-fat, high-sugar diet and lack of physical activity, (Williams Citation2006; Fan and Farrell Citation2009). Clinical studies have shown that most cases of NAFLD are considered to be hepatic manifestations of MS (Marchesini et al. Citation2003; Bellentani and Marino Citation2009). Hepatocytes and macrophages are sources of the over-synthesis of pro-inflammatory mediators in the pathogenesis of NAFLD (Sato et al. Citation2014). In hepatocytes, SUV39H2 inhibits Sirt1 transcription by binding to its promoters. SUV39H2 deficiency normalizes Sirt1 expression, thereby inhibiting NF-κB-dependent transcription of pro-inflammatory mediators. In macrophages, the inhibition of Peroxisome proliferator-activated receptor gamma (PPARγ) transcription mediated by SUV39H2 promotes pro-inflammatory M1 phenotypic polarization.
SETDB1 and SETDB2
Studies suggest that SETDB1 is an anti-adipogenic factor (Okamura et al. Citation2010). SETDB1 acts on H3K9me3 in the promoter of PPARγ, a major regulator of lipogenesis (Morrison and Farmer Citation1999), resulting in chromatin inactivation and adipogenic gene silencing. Decrease of SETDB1 mRNA in was observed in obese mouse models (Huang Citation2002). Whole-genome expression profiling of a large sample size analysis of psoriasis skin biopsies revealed that the psoriasis tissues had differential SETDB1 expression compared to normal tissues (Swindell et al. Citation2013). Further experimental results indicate the potential role of SETDB1 as a mediator of transcriptional inhibition in the epidermis of psoriasis tissues (Swindell et al. Citation2013). SETDB2 was the only protein lysine methyltransferase induced during influenza infection and was induced by type I interferon signaling. The expression of the gene encoding the neutrophil attractant CXCL1 and other NF-κB downstream genes were repressed by SETDB2, which leaded to secondary bacterial infections after the flu (Schliehe et al. Citation2015).
G9a and GLP
Euchromatic histone-lysine N-methyltransferase 2 (EHMT2) or G9a and its related molecule GLP mediate H3K9 mono- and di-methylation on euchromatin to inhibit transcription and inhibit induced inflammatory gene expression (Saccani and Natoli Citation2002; Tachibana et al. Citation2005). The study found that after the initial activation phase of severe systemic inflammation or sepsis, H3K9me2 is involved in reprogramming the endotoxin-tolerant phenotype of leukocytes, leading to the silencing of acute pro-inflammatory genes, such as TNF-α and IL-1β (El Gazzar et al. Citation2008). In an LPS-stimulated human THP-1 cell endotoxin tolerance model, G9a mediated H3K9 di-methylation at the promoters of TNF-α and the IL-1β and facilitated a combination of heterogeneous chromatin protein 1 (HP1). The HP1 combination led to methyltransferase Dnmt3a/b binding and CpG methylation at TNF-α and IL-1β gene promoters (El Gazzar et al. Citation2008), therebysilencing acute proinflammatory gene expression. This silencing effect required induction of NF-κB factor RelB, which directed H3K9 dimethylation, disrupted the assembly of NF-κB p65, and induced the switch from the euchromatin to heterochromatin (El Gazzar et al. Citation2009). A complex of RelB, G9a and HP1 formed at the promoters of TNF-α and IL-1β induced the formation of facultative heterochromatin. Knockdown of G9a reversed this silencing (Chen et al. Citation2009). In another study, G9a regulated tolerance to viral infections by shaping the Jak-Stat pathway in Drosophila, thereby reducing the mortality rate of viral infections (Merkling et al. Citation2015). Moreover, G9a-dependent H3K9 di-methylation is also associated with the genetic silencing of innate immune-related factors and the innate immune memory of macrophages after the recovery from endotoxin tolerance (Yoshida et al. Citation2015). These studies demonstrate the role of G9a in the inhibition of inflammatory gene expression and reveal its involvement in tolerance mechanisms.
G9a also has effects on the progression of inflammatory diseases by regulating the development and differentiation of other immune cells. For example, G9a regulated the function of group 2 innate lymphoid cells (ILC2s) activated by an allergen in the lungs. The ILC2s produce type 2 interleukins IL-5 and IL-13, which act on different types of cells promoting type 2 pulmonary inflammation characterized by eosinophilia, mucus production, smooth muscle contraction and airway hyper-reaction. With G9a knockout, the number of eosinophilic granulocytes in the bronchial alveolar lavage washout decreased significantly and the expression of IL-5 and IL-13 in the lungs also reduced significantly (Antignano et al. Citation2016). The pathogenesis of inflammatory bowel disease (IBD) is associated with a dysregulated CD4+ T cell response, and studies have found that G9a-mediated di-methylation of H3K9 in naive T cells affects the balance of Th17 and Treg differentiation by regulating chromatin accessibility and the sensitivity of TGF-β1 cytokines. The G9a knockout in T cells leaded to severe colonic inflammation. A descriptive genome-wide analysis of H3K9me2 markers in resting human lymphocytes of patients with severe diabetes mellitus and healthy control subjects show significant differences, suggesting that the H3K9me2-dependent regulation of G9a-mediated T cell responses may be related to T cell function and the development of inflammatory diseases, such as diabetes mellitus (Miao et al. Citation2008a).
In other inflammatory models, G9a also played a role in the induction of inflammatory factors. In TNF-α-induced vascular inflammation, G9a was involved in reducing ICAM1 and VCAM-1 expression, leukocyte adhesion, and migration in endothelial cells122. Similarly, in a skin inflammation induced by TNF-α, the decrease of the G9a/GLP protein and H3K9me2 in keratinocytes led to increased IL-23 expression (Li et al. Citation2018; Li et al. Citation2019) (Table ).
Table 3. Target Inflammatory genes and related inflammation models of H3K9 methylases.
The role of H3K9 demethylases in the inflammatory response
KDM4 (JMJD2) family
Many efforts have been focused on the effects of KDM4 family in various of inflammatory models. KDM4A, a member of KDM4 family, was revealed to play a role in regulating brain inflammation and tumorigenesis by global gene expression analysis in an LPS-induced NE-4C, a KDM4A-knockdown p53-null neuroectodermal stem cell (Das et al. Citation2014). Also, KDM4A was found to promote the migration and inflammation of vascular smooth muscle by regulating the level of H3K9me3 at promoters of MCP-1 and IL-6 in a diabetic balloon injury model (Qi et al. Citation2015). Similarly, in a vascular endothelial inflammatory model, KDM4A-mediated H3K9 methylation was required for SET1A to bind to NF-κB targeted promoters, and then promoted LPS-induced transactivation of pro-inflammatory cytokines (Zhang et al. Citation2019). In addition, KDM4A was identified to be a new epigenetic target for oxidized low-density lipoprotein (oxLDL)-induced M1 macrophage polarization. KDM4A promoted the expression of iNOS (inducible nitric oxide synthase, a typical marker for M1 macrophages) and inflammatory cytokines (e.g. TNF-α, MCP-1, and IL-1β). These would provide new strategies to the treatment of atherosclerotic diseases (Wang et al. Citation2017).
Another member of KDM4 family, KDM4B, regulates the expression of inflammation-associated transcription factors and cytokines. Studies show that KDM4B promoted demethylation of H3K9me3 in the promoters of Notch1, IL-1b and IL-2, led to upregulation of abovementioned genes and activation of Notch and NF-κB in LPS-stimulated NE-4C cells (Das et al. Citation2013). Scholars proved that with exposure to environmental pollutants, such as polychlorinated biphenyls, KDM4B enriched within the p65 promoter and NF-κB signaling translocated into nuclear and activated, resulted in increased release of IL-6, ICAM-1, VCAM-1 and other inflammatory factors from endothelial cells (Liu et al. Citation2015). Moreover, the modification of H3K9me2 by G9a and KDM4B upregulated vascular adhesion molecules and promoted inflammation-induced leukocyte extravasation (Choi et al. Citation2017; Zhang et al. Citation2018).
Other H3K9 demethylases
LSD1, the first identified demethylase, facilitates H3K9 and H3K4 demethylation and is associated with nuclear receptors. LSD1 was reported to inhibit inflammation via H3K4 demethylation (Reddy et al. Citation2008; Janzer et al. Citation2012). By coordinating with LSD1, CoREST1 promoted the expression of VEGF-A and the pro-inflammatory factors CCL2/MCP-1 and CXCL16 which contributed to angiogenesis and tumor-induced inflammatory responses in breast cancer (Mazumdar et al. Citation2015). In HBV-induced renal inflammation, LSD1 promoted TLR4 transcription and the TLR4-NF-ΚB/JNK signaling cascade by eliminating the mono-methylation or di-methylation of H3K9 in the TLR4 promoter (Yang et al. Citation2019).
KDM7B, or PHF8, is a dual demethylase for H3K9 and H3K27, functioned as an eraser of silencing marks on chromatin (Tsukada et al. Citation2010). By removing transcriptional inhibitor H3K9me2, KDM7B is capable of upregulating NF-κB-dependent pro-inflammatory genes in macrophages. Acute LPS-induced cytokine/chemokine productions and many other immune response-related proteins were proved to be KDM7B -dependent1 (Erdoğan et al. Citation2016) (Table ).
Table 4. Target genes and related inflammation models of H3K9 demethylases.
Conclusion
Dynamic modification of histone residue H3K9 methylation plays a crucial role in regulation of inflammatory gene expression. Knowledge of how transcriptionally permissive and repressive histone marks are regulated in physiological and pathological state remain limited. Mis-regulation of H3K9 methylation has been implicated in various inflammatory diseases. Here, we show that H3K9 methyl-modifying enzymes are key epigenetic regulatory features in the development of inflammation and the outcome of the corresponding diseases. How the cell ‘reads’ the H3K9 methylation marks correctly and how H3K9 methylation interacts with other epigenetic modifying partners in certain context remain unanswered questions. More detailed understanding of how H3K9 methyl-modification precisely controls the expression of inflammatory mediators, such as inflammatory cytokines and chemokines, is necessary for developing and improving disease treatments. Specific inhibitors targeting histone methyl-modifying enzymes have been developed in cancer treatment. However, translational studies with H3K9 methyl-modification as therapeutic target in inflammatory disease remain staying at a very preliminary stage. Newly developed genetic and chemical engineering techniques (epigenetic editing) will help to find more evidence, broadening our repertoire of histone modifiers and ultimately ignite discoveries of potential therapeutic targets.
Declaration of conflict of interest
None.
Data availability statement
The data supporting this review are from previously reported studies and datasets, which have been cited. The processed data are available.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Acevedo N, Frumento P, Harb H, Alashkar Alhamwe B, Johansson C, Eick L, Alm J, Renz H, Scheynius A, Potaczek DP. 2019. Histone acetylation of immune regulatory genes in human placenta in association with maternal intake of olive Oil and fish consumption. Int J Mol Sci. 20(5):1060.
- Agrawal R, Wisniewski JA, Woodfolk JA. 2011. The role of regulatory T cells in atopic dermatitis. Curr Probl Dermatol. 41:112–124.
- Agustí A, Edwards LD, Rennard SI, MacNee W, Tal-Singer R, Miller BE, Vestbo J, Lomas DA, Calverley PM, Wouters E. 2012. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS One. 7(5):e37483.
- Ahmad S, Manzoor S, Siddiqui S, Mariappan N, Zafar I, Ahmad A, Ahmad A. 2021. Epigenetic underpinnings of inflammation: connecting the dots between pulmonary diseases, lung cancer and COVID-19. Semin Cancer Biol. S1044-579X(21)00008-0. https://doi.org/10.1016/j.semcancer.2021.01.003.
- Alam H, Gu B, Lee MG. 2015. Histone methylation modifiers in cellular signaling pathways. Cell Mol Life Sci. 72(23):4577–4592.
- Alashkar Alhamwe B, Miethe S, Pogge von Strandmann E, Potaczek DP, Garn H. 2020. Epigenetic regulation of airway epithelium immune functions in asthma. Front Immunol. 11:1747.
- Alberti KGMM, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, Fruchart JC, James WP, Loria CM, Smith SC Jr. 2009. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 120(16):1640–1645.
- Allan RS, Zueva E, Cammas F, Schreiber HA, Masson V, Belz GT, Roche D, Maison C, Quivy JP, Almouzni G, et al. 2012. An epigenetic silencing pathway controlling T helper 2 cell lineage commitment. Nature. 487(7406):249–253.
- Antignano F, Braam M, Hughes MR, Chenery AL, Burrows K, Gold MJ, Oudhoff MJ, Rattray D, Halim TY, Cait A. 2016. G9a regulates group 2 innate lymphoid cell development by repressing the group 3 innate lymphoid cell program. J Exp Med. 213(7):1153–1162.
- Antignano F, Burrows K, Hughes MR, Han JM, Kron KJ, Penrod NM, Oudhoff MJ, Wang SK, Min PH, Gold MJ, et al. 2014. Methyltransferase G9A regulates T cell differentiation during murine intestinal inflammation. J Clin Invest. 124(5):1945–1955.
- Bacon ER, Brinton RD. 2021. Epigenetics of the developing and aging brain: mechanisms that regulate onset and outcomes of brain reorganization. Neurosci Biobehav Rev. 125:503–516.
- Bayry J, Gautier JF. 2016. Regulatory T cell immunotherapy for type 1 diabetes: a step closer to success? Cell Metab. 23(2):231–233.
- Bellentani S, Marino M. 2009. Epidemiology and natural history of non-alcoholic fatty liver disease (NAFLD). Ann Hepatol. 8(Suppl 1):S4–S8.
- Black JC, Van Rechem C, Whetstine JR. 2012. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 48(4):491–507.
- Bosch-Presegué L, Raurell-Vila H, Marazuela-Duque A, Kane-Goldsmith N, Valle A, Oliver J, Serrano L, Vaquero A. 2011. Stabilization of Suv39H1 by SirT1 is part of oxidative stress response and ensures genome protection. Mol. Cell. 42(2):210–223.
- Bruno A, Dolcetti E, Rizzo FR, Fresegna D, Musella A, Gentile A, De Vito F, Caioli S, Guadalupi L, Bullitta S, et al. 2020. Corrigendum: inflammation-associated synaptic alterations as shared threads in depression and multiple sclerosis. Front Cell Neurosci. 14:647259.
- Carranza-Naval MJ, Vargas-Soria M, Hierro-Bujalance C, Baena-Nieto G, Garcia-Alloza M, Infante-Garcia C, Del Marco A. 2021. Alzheimer's disease and diabetes: role of diet, microbiota and inflammation in preclinical models. Biomolecules. 11(2):262.
- Castelnau PA, Garrett RS, Palinski W, Witztum JL, Campbell IL, Powell HC. 1998. Abnormal iron deposition associated with lipid peroxidation in transgenic mice expressing interleukin-6 in the brain. J Neuropathol Exp Neurol. 57(3):268–282.
- Ceriello A. 2009. Hypothesis: the “metabolic memory”, the new challenge of diabetes. Diabetes Res Clin Pract. 86(Suppl 1):S2–S6.
- Ceriello A, Ihnat MA, Thorpe JE. 2009. Clinical review 2: The “metabolic memory": is more than just tight glucose control necessary to prevent diabetic complications? J Clin Endocrinol Metab. 94(2):410–415.
- Chang S, Aune TM. 2007. Dynamic changes in histone-methylation ‘marks’ across the locus encoding interferon-gamma during the differentiation of T helper type 2 cells. Nat Immunol. 8(7):723–731.
- Chaturvedi SS, Ramanan R, Waheed SO, Ainsley J, Evison M, Ames JM, Schofield CJ, Karabencheva-Christova TG, Christov CZ. 2019a. Conformational dynamics underlies different functions of human KDM7 histone demethylases. Chemistry. 25(21):5422–5426.
- Chaturvedi SS, Ramanan R, Waheed SO, Karabencheva-Christova TG, Christov CZ. 2019b. Structure-function relationships in KDM7 histone demethylases. Adv Protein Chem Struct Biol. 117:113–125.
- Chen T-T, Wu S-M, Ho S-C, Chuang H-C, Liu C-Y, Chan Y-F, Kuo L-W, Feng P-H, Liu W-T, Chen K-Y. 2017. SUV39H1 reduction is implicated in abnormal inflammation in COPD. Sci Rep. 7:46667.
- Chen X, El Gazzar M, Yoza BK, McCall CE. 2009. The NF-κB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J Biol Chem. 284(41):27857–27865.
- Choi JY, Yoon SS, Kim SE, Ahn Jo S. 2017. KDM4B histone demethylase and G9a regulate expression of vascular adhesion proteins in cerebral microvessels. Sci Rep. 7:45005.
- Choi N, Yang G, Jang JH, Kang HC, Cho YY, Lee HS, Lee JY. 2021. Loganin alleviates gout inflammation by suppressing NLRP3 inflammasome activation and mitochondrial damage. Molecules. 26(4):1071.
- Control D. 2005., interventions CTEoD, group CSR. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 353(25):2643–2653.
- Cooper ME. 2012. Diabetes: treating diabetic nephropathy-still an unresolved issue. Nat Rev Endocrinol. 8(9):515–516.
- Cooper ME, Johnston CI. 2000. Optimizing treatment of hypertension in patients with diabetes. JAMA. 283(24):3177–3179.
- Das A, Chai JC, Jung KH, Das ND, Kang SC, Lee YS, Seo H, Chai YG. 2014. JMJD2A attenuation affects cell cycle and tumourigenic inflammatory gene regulation in lipopolysaccharide stimulated neuroectodermal stem cells. Exp Cell Res. 328(2):361–378.
- Das ND, Choi MR, Jung KH, Park JH, Lee HT, Das A, Kim SH, Chai YG. 2013. Functional analysis of histone demethylase Jmjd2b on lipopolysaccharide-treated murine neural stem cells (NSCs). Neurotox Res. 23(2):154–165.
- Devaraj S, Glaser N, Griffen S, Wang-Polagruto J, Miguelino E, Jialal I. 2006. Increased monocytic activity and biomarkers of inflammation in patients with type 1 diabetes. Diabetes. 55(3):774–779.
- Dinh W, Füth R, Nickl W, Krahn T, Ellinghaus P, Scheffold T, Bansemir L, Bufe A, Barroso MC, Lankisch M. 2009. Elevated plasma levels of TNF-alpha and interleukin-6 in patients with diastolic dysfunction and glucose metabolism disorders. Cardiovasc Diabetol. 8(1):58.
- Du J, Johnson LM, Jacobsen SE, Patel DJ. 2015. DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol. 16(9):519–532.
- El Gazzar M, Yoza BK, Chen X, Garcia BA, Young NL, McCall CE. 2009. Chromatin-specific remodeling by HMGB1 and linker histone H1 silences proinflammatory genes during endotoxin tolerance. Mol Cell Biol. 29(7):1959–1971.
- El Gazzar M, Yoza BK, Chen X, Hu J, Hawkins GA, McCall CE. 2008. G9a and HP1 couple histone and DNA methylation to TNFα transcription silencing during endotoxin tolerance. J Biol Chem. 283(47):32198–32208.
- El Gazzar M, Yoza BK, Hu JY-Q, Cousart SL, McCall CE. 2007. Epigenetic silencing of tumor necrosis factor α during endotoxin tolerance. J Biol Chem. 282(37):26857–26864.
- Erdoğan Ö, Xie L, Wang L, Wu B, Kong Q, Wan Y, Chen X. 2016. Proteomic dissection of LPS-inducible, PHF8-dependent secretome reveals novel roles of PHF8 in TLR4-induced acute inflammation and T cell proliferation. Sci Rep. 6:24833.
- Falvo JV, Jasenosky LD, Kruidenier L, Goldfeld AE. 2013. Epigenetic control of cytokine gene expression: regulation of the TNF/LT locus and T helper cell differentiation. Adv Immunol. 118:37–128.
- Fan J-G, Farrell GC. 2009. Epidemiology of non-alcoholic fatty liver disease in China. J Hepatology. 50(1):204–210.
- Fang TC, Schaefer U, Mecklenbrauker I, Stienen A, Dewell S, Chen MS, Rioja I, Parravicini V, Prinjha RK, Chandwani R, et al. 2012. Histone H3 lysine 9 di-methylation as an epigenetic signature of the interferon response. J Exp Med. 209(4):661–669.
- Fiuza BSD, Fonseca HF, Meirelles PM, Marques CR, da Silva TM, Figueiredo CA. 2021. Understanding asthma and allergies by the lens of biodiversity and epigenetic changes. . Front Immunol. 12:623737.
- Fukuyama T, Kosaka T, Miyashita L, Nishino R, Wada K, Hayashi K, Ueda H, Harada T. 2012. Role of regulatory T cells in the induction of atopic dermatitis by immunosuppressive chemicals. Toxicol Lett. 213(3):392–401.
- Green MR, Yoon H, Boss JM. 2006. Epigenetic regulation during B cell differentiation controls CIITA promoter accessibility. J Immunol. 177(6):3865–3873.
- Greer EL, Shi Y. 2012. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 13(5):343–357.
- Group AC. 2008. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 358(24):2560–2572.
- Hachiya R, Shiihashi T, Shirakawa I, Iwasaki Y, Matsumura Y, Oishi Y, Nakayama Y, Miyamoto Y, Manabe I, Ochi K, et al. 2016. The H3K9 methyltransferase Setdb1 regulates TLR4-mediated inflammatory responses in macrophages. Sci Rep. 6:28845.
- Harb H, Alashkar Alhamwe B, Acevedo N, Frumento P, Johansson C, Eick L, Papadogiannakis N, Alm J, Renz H, Potaczek DP, et al. 2019. Epigenetic modifications in Placenta are associated with the child's sensitization to allergens. Biomed Res Int. 2019:1315257.
- Huang S. 2002. Histone methyltransferases, diet nutrients and tumour suppressors. Nat Rev Cancer. 2(6):469–476.
- Hyun K, Jeon J, Park K, Kim J. 2017. Writing, erasing and reading histone lysine methylations. Exp Mol Med. 49(4):e324.
- Iacono G, Dubos A, Meziane H, Benevento M, Habibi E, Mandoli A, Riet F, Selloum M, Feil R, Zhou H, et al. 2018. Increased H3K9 methylation and impaired expression of protocadherins are associated with the cognitive dysfunctions of the kleefstra syndrome. Nucleic Acids Res 46(10):4950–4965.
- Intine RV, Sarras MP. 2012. Metabolic memory and chronic diabetes complications: potential role for epigenetic mechanisms. Curr Diabetes Rep. 12(5):551–559.
- Janzer A, Lim S, Fronhoffs F, Niazy N, Buettner R, Kirfel J. 2012. Lysine-specific demethylase 1 (LSD1) and histone deacetylase 1 (HDAC1) synergistically repress proinflammatory cytokines and classical complement pathway components. Biochem Biophys Res Commun. 421(4):665–670.
- Jih G, Iglesias N, Currie MA, Bhanu NV, Paulo JA, Gygi SP, Garcia BA, Moazed D. 2017. Unique roles for histone H3K9me states in RNAi and heritable silencing of transcription. Nature. 547(7664):463.
- Kakaradov B, Arsenio J, Widjaja CE, He Z, Aigner S, Metz PJ, Yu B, Wehrens EJ, Lopez J, Kim SH, et al. 2017. Early transcriptional and epigenetic regulation of CD8(+) T cell differentiation revealed by single-cell RNA sequencing. Nat Immunol. 18(4):422–432.
- Kang M, Mehrazarin S, Park NH, Wang CY. 2017. Epigenetic gene regulation by histone demethylases: emerging role in oncogenesis and inflammation. Oral Diseases. 23(6):709–720.
- Kim J, Yoon HI, Oh Y-M, Lim SY, Lee J-H, Kim T-H, Lee SY, Lee JH, Lee S-D, Lee C-H. 2015. Lung function decline rates according to GOLD group in patients with chronic obstructive pulmonary disease. Int J Chronic Obstructive Pulm Disease. 10:1819.
- King GL. 2008. The role of inflammatory cytokines in diabetes and its complications. J Periodontology. 79:1527–1534.
- Kinoshita T, Goto T. 2021. Links between inflammation and postoperative cancer recurrence. J Clin Med. 10(2):228.
- Kiyici S, Erturk E, Budak F, Ersoy C, Tuncel E, Duran C, Oral B, Sigirci D, Imamoglu S. 2006. Serum monocyte chemoattractant protein-1 and monocyte adhesion molecules in type 1 diabetic patients with nephropathy. Arch Med Res. 37(8):998–1003.
- Lee J, Sayed N, Hunter A, Au KF, Wong WH, Mocarski ES, Pera RR, Yakubov E, Cooke JP. 2012a. Activation of innate immunity is required for efficient nuclear reprogramming. Cell. 151(3):547–558.
- Lee K-Y, Ho S-C, Chan Y-F, Wang C-H, Huang C-D, Liu W-T, Lin S-M, Lo Y-L, Chang Y-L, Kuo L-W. 2012b. Reduced nuclear factor-κB repressing factor: a link toward systemic inflammation in COPD. Eur Respir J. 40(4):863–873.
- Lesiak A, Smolewski P, Sobolewska-Sztychny D, Sysa-Jedrzejowska A, Narbutt J. 2012. The role of T-regulatory cells and toll-like receptors 2 and 4 in atopic dermatitis. Scand J Immunol. 76(4):405–410.
- Li H, Petersen S, Garcia Mariscal A, Brakebusch C. 2019. Negative regulation of p53-induced senescence by N-WASP Is crucial for DMBA/TPA-induced skin tumor formation. Cancer Res 79(9):2167–2181.
- Li H, Yao Q, Mariscal AG, Wu X, Halse J, Pedersen E, Helin K, Waisman A, Vinkel C, Thomsen SF, et al. 2018. Epigenetic control of IL-23 expression in keratinocytes is important for chronic skin inflammation. Nat Commun. 9(1):1420.
- Li M-F, Zhang R, Li T-T, Chen M-Y, Li L-X, Lu J-X, Jia W-P. 2016. High glucose increases the expression of inflammatory cytokine genes in macrophages through H3K9 methyltransferase mechanism. J Interferon & Cytokine Research. 36(1):48–61.
- Li Y, Reddy MA, Miao F, Shanmugam N, Yee J-K, Hawkins D, Ren B, Natarajan R. 2008. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-κB-dependent inflammatory genes relevance to diabetes and inflammation. J Biol Chem. 283(39):26771–26781.
- Liu D, Perkins JT, Petriello MC, Hennig B. 2015. Exposure to coplanar PCBs induces endothelial cell inflammation through epigenetic regulation of NF-κB subunit p65. Toxicol Appl Pharmacol. 289(3):457–465.
- Liu H, Li P, Wei Z, Zhang C, Xia M, Du Q, Chen Y, Liu N, Li H, Yang XP. 2019. Regulation of T cell differentiation and function by epigenetic modification enzymes. Semin Immunopathol. 41(3):315–326.
- Lutgens E. 2019. Epigenetic quenching of VSMC inflammation in CVD: H3K9me2 in control. Arterioscler Thromb Vasc Biol. 39(11):2199–2200.
- Maiques-Diaz A, Somervaille TC. 2016. LSD1: biologic roles and therapeutic targeting. Epigenomics. 8(8):1103–1116.
- Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, Natale S, Vanni E, Villanova N, Melchionda N, et al. 2003. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 37(4):917–923.
- Mayne CG, Williams CB. 2013. Induced and natural regulatory T cells in the development of inflammatory bowel disease. Inflamm Bowel Dis. 19(8):1772–1788.
- Mazumdar S, Arendt LM, Phillips S, Sedic M, Kuperwasser C, Gill G. 2015. CoREST1 promotes tumor formation and tumor stroma interactions in a mouse model of breast cancer. PLoS One. 10(3):e0121281.
- Mehta S, Jeffrey KL. 2015. Beyond receptors and signaling: epigenetic factors in the regulation of innate immunity. Immunol Cell Biol. 93(3):233–244.
- Merkling SH, Bronkhorst AW, Kramer JM, Overheul GJ, Schenck A, Van Rij RP. 2015. The epigenetic regulator G9a mediates tolerance to RNA virus infection in drosophila. PLoS Pathogens. 11(4):e1004692.
- Miao F, Smith DD, Zhang L, Min A, Feng W, Natarajan R. 2008a. Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: an epigenetic study in diabetes. Diabetes. 57(12):3189–3198.
- Miao F, Wu X, Zhang L, Riggs AD, Natarajan R. 2008b. Histone methylation patterns are cell-type specific in human monocytes and lymphocytes and well maintained at core genes. J Immunol. 180(4):2264–2269.
- Morrison RF, Farmer SR. 1999. Role of PPARγ in regulating a cascade expression of cyclin-dependent kinase inhibitors, p18 (INK4c) and p21 (Waf1/Cip1), during adipogenesis. J Biol Chem. 274(24):17088–17097.
- Mourits VP, Wijkmans JC, Joosten LA, Netea MG. 2018. Trained immunity as a novel therapeutic strategy. Curr Opin Pharmacol. 41:52–58.
- Mozzetta C, Boyarchuk E, Pontis J, Ait-Si-Ali S. 2015. Sound of silence: the properties and functions of repressive Lys methyltransferases. Nat Rev Mol Cell Biol. 16(8):499–513.
- Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. 2001. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 292(5514):110–113.
- Nano J, Fernandez EP, Troup J, Ghanbari M, Franco OH, Muka T. 2018. Epigenetics of diabetes in humans. In: Tollefsbol TO, editor. Epigenetics in human disease Vol 6. 2nd ed.. Cambridge, MA: Academic Press; p. 457–488.
- Nowak RP, Tumber A, Johansson C, et al. 2016. Advances and challenges in understanding histone demethylase biology. Curr Opin Chem Biol. 33:151–159.
- Okamura M, Inagaki T, Tanaka T, Sakai J. 2010. Role of histone methylation and demethylation in adipogenesis and obesity. Organogenesis. 6(1):24–32.
- Placha D, Jampilek J. 2021. Chronic inflammatory diseases, anti-inflammatory agents and their delivery nanosystems. Pharmaceutics. 13(1):64.
- Qi H, Jing Z, Xiaolin W, Changwu X, Xiaorong H, Jian Y, Jing C, Hong J. 2015. Histone demethylase JMJD2A inhibition attenuates neointimal hyperplasia in the carotid arteries of balloon-injured diabetic rats via transcriptional silencing: inflammatory gene expression in vascular smooth muscle cells. Cell Physiol Biochem. 37(2):719–734.
- Rao VK, Pal A, Taneja R. 2017. A drive in SUVs: from development to disease. Epigenetics. 12(3):177–186.
- Reddy MA, Tak Park J, Natarajan R. 2013. Epigenetic modifications in the pathogenesis of diabetic nephropathy. Semin Nephrol. 33(4):341–353.
- Reddy MA, Villeneuve LM, Wang M, Lanting L, Natarajan R. 2008. Role of the lysine-specific demethylase 1 in the proinflammatory phenotype of vascular smooth muscle cells of diabetic mice. Circ Res. 103(6):615–623.
- Rodriguez RM, Suarez-Alvarez B, Lavin JL, Mosen-Ansorena D, Baragano Raneros A, Marquez-Kisinousky L, Aransay AM, Lopez-Larrea C. 2017. Epigenetic networks regulate the transcriptional program in memory and terminally differentiated CD8+ T cells. J Immunol. 198(2):937–949.
- Romano S, Savva GM, Bedarf JR, Charles IG, Hildebrand F, Narbad A. 2021. Meta-analysis of the Parkinson's disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Parkinsons Dis. 7(1):27.
- Ross R. 1999. Atherosclerosis—an inflammatory disease. N Engl J Med. 340(2):115–126.
- Saccani S, Natoli G. 2002. Dynamic changes in histone H3 Lys 9 methylation occurring at tightly regulated inducible inflammatory genes. Genes Dev 16(17):2219–2224.
- Sato H, Taketomi Y, Ushida A, Isogai Y, Kojima T, Hirabayashi T, Miki Y, Yamamoto K, Nishito Y, Kobayashi T. 2014. The adipocyte-inducible secreted phospholipases PLA2G5 and PLA2G2E play distinct roles in obesity. Cell Metabolism. 20(1):119–132.
- Schliehe C, Flynn EK, Vilagos B, Richson U, Swaminathan S, Bosnjak B, Bauer L, Kandasamy RK, Griesshammer IM, Kosack L. 2015. The methyltransferase Setdb2 mediates virus-induced susceptibility to bacterial superinfection. Nat Immunol. 16(1):67.
- Schnell O, Erbach M, Hummel M. 2012. Primary and secondary prevention of cardiovascular disease in diabetes with aspirin. Diab Vasc Dis Res. 9(4):245–255.
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. 2004. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 119(7):941–953.
- Song Y, Dagil L, Fairall L, Robertson N, Wu M, Ragan TJ, Savva CG SA, Morone N, Kunze MBA, Jamieson AG, et al. 2020. Mechanism of crosstalk between the LSD1 demethylase and HDAC1 deacetylase in the CoREST complex. Cell Rep. 30(8):2699–2711.
- Sui Y, Gu R, Janknecht R. 2021. Crucial functions of the JMJD1/KDM3 epigenetic regulators in cancer. Mol Cancer Res. 19(1):3–13.
- Swindell WR, Johnston A, Xing X, Voorhees JJ, Elder JT, Gudjonsson JE. 2013. Modulation of epidermal transcription circuits in psoriasis: new links between inflammation and hyperproliferation. PLoS One. 8(11):e79253.
- Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, Sakihama T, Kodama T, Hamakubo T, Shinkai Y. 2005. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 19(7):815–826.
- Tanaka A, Sakaguchi S. 2019. Targeting Treg cells in cancer immunotherapy. Eur J Immunol. 49(8):1140–1146.
- Taniuchi I, Ellmeier W, Littman DR. 2004. The CD4/CD8 lineage choice: new insights into epigenetic regulation during T cell development. Adv Immunol. 83:55–89.
- Tausendschön M, Dehne N, Brüne B. 2011. Hypoxia causes epigenetic gene regulation in macrophages by attenuating Jumonji histone demethylase activity. Cytokine. 53(2):256–262.
- Tsukada Y-i, Ishitani T, Nakayama KI. 2010. KDM7 is a dual demethylase for histone H3 Lys 9 and Lys 27 and functions in brain development. Genes & Development. 24(5):432–437.
- Tumes DJ, Papadopoulos M, Endo Y, Onodera A, Hirahara K, Nakayama T. 2017. Epigenetic regulation of T-helper cell differentiation, memory, and plasticity in allergic asthma. Immunol Rev. 278(1):8–19.
- Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. 2004. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell. 16(1):93–105.
- Vestbo J, Hurd SS, Agustí AG, Jones PW, Vogelmeier C, Anzueto A, Barnes PJ, Fabbri LM, Martinez FJ, Nishimura M. 2013. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 187(4):347–365.
- Villeneuve LM, Natarajan R. 2010. The role of epigenetics in the pathology of diabetic complications. Am J Physiol Renal Physiol. 299(1):F14–F25.
- Villeneuve LM, Reddy MA, Lanting LL, Wang M, Meng L, Natarajan R. 2008. Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. PNAS. 105(26):9047–9052.
- Wakabayashi Y, Tamiya T, Takada I, Fukaya T, Sugiyama Y, Inoue N, Kimura A, Morita R, Kashiwagi I, Takimoto T, et al. 2011. Histone 3 lysine 9 (H3K9) methyltransferase recruitment to the interleukin-2 (IL-2) promoter is a mechanism of suppression of IL-2 transcription by the transforming growth factor-beta-Smad pathway. J Biol Chem. 286(41):35456–35465.
- Wang J, Yan W, Peng X, Jiang Y, He L, Peng Y, Chen X, Ye M, Zhuo H. 2018. Functional role of SUV39H1 in human renal tubular epithelial cells under high-glucose ambiance. Inflammation. 41(1):1–10.
- Wang L, Liu Y, Han R, Beier UH, Bhatti TR, Akimova T, Greene MI, Hiebert SW, Hancock WW. 2015. FOXP3+ regulatory T cell development and function require histone/protein deacetylase 3. J Clin Invest. 125(3):1111–1123.
- Wang X, Wang S, Yao G, Yu D, Chen K, Tong Q, Ye L, Wu C, Sun Y, Li H, et al. 2017. Identification of the histone lysine demethylase KDM4A/JMJD2A as a novel epigenetic target in M1 macrophage polarization induced by oxidized LDL. Oncotarget. 8(70):114442–114456.
- Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, et al. 2009. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 30(1):155–167.
- Williams R. 2006. Global challenges in liver disease. Hepatology. 44(3):521–526.
- Wilson C, Krieg AJ. 2019. KDM4B: a nail for every hammer? Genes. 10(2):134.
- Wu WH, Zegarra-Ruiz DF, Diehl GE. 2020. Intestinal microbes in autoimmune and inflammatory disease. Front Immunol. 11:597966.
- Wu X, Yuan B, López E, Bai C, Wang X. 2014. Gene polymorphisms and chronic obstructive pulmonary disease. J Cell Mol Med. 18(1):15–26.
- Yang B, Yang J, Bai J, Pu P, Liu J, Wang F, Ruan B. 2014. Suv39h1 protects from myocardial ischemia-reperfusion injury in diabetic rats. Cell Physiol Biochem. 33(4):1176–1185.
- Yang Y-T, Wang X, Zhang Y-Y, Yuan W-J. 2019. The histone demethylase LSD1 promotes renal inflammation by mediating TLR4 signaling in hepatitis B virus-associated glomerulonephritis. Cell Death Disease. 10(4):278.
- Yoo J, Jeon YH, Cho HY, Lee SW, Kim GW, Lee DH, Kwon SH. 2020. Advances in histone demethylase KDM3A as a cancer therapeutic target. Cancers (Basel). 12(5):1098.
- Yoshida K, Maekawa T, Zhu Y, Renard-Guillet C, Chatton B, Inoue K, Uchiyama T, Ishibashi K-i, Yamada T, Ohno N. 2015. The transcription factor ATF7 mediates lipopolysaccharide-induced epigenetic changes in macrophages involved in innate immunological memory. Nat Immunol. 16(10):1034.
- Yu X-Y, Geng Y-J, Liang J-L, Zhang S, Lei H-P, Zhong S-L, Lin Q-X, Shan Z-X, Lin S-G, Li Y. 2012. High levels of glucose induce “metabolic memory” in cardiomyocyte via epigenetic histone H3 lysine 9 methylation. Mol Biol Rep. 39(9):8891–8898.
- Zhang X, Cook PC, Zindy E, et al. 2016. Integrin alpha4beta1 controls G9a activity that regulates epigenetic changes and nuclear properties required for lymphocyte migration. Nucleic Acids Res. 44(7):3031–3044.
- Zhang X, Liu S, Weng X, Zeng S, Yu L, Guo J, Xu Y. 2018. Brg1 deficiency in vascular endothelial cells blocks neutrophil recruitment and ameliorates cardiac ischemia-reperfusion injury in mice. Int J Cardiol. 269:250–258.
- Zhang Y, Maksimovic J, Naselli G, Qian J, Chopin M, Blewitt ME, Oshlack A, Harrison LC. 2013. Genome-wide DNA methylation analysis identifies hypomethylated genes regulated by FOXP3 in human regulatory T cells. Blood. 122(16):2823–2836.
- Zhang Y, Yuan Y, Li Z, Chen H, Fang M, Xiao P, Xu Y. 2019. An interaction between BRG1 and histone modifying enzymes mediates lipopolysaccharide-induced proinflammatory cytokines in vascular endothelial cells. J Cell Biochem. 120(8):13216–13225.