
Abstract
Accumulation of amyloid-β (Aβ) is implicated in the pathogenesis and development of Alzheimer's disease (AD) and it is a prime suspect for causing neuronal loss and cognitive deficits during AD. To explore the mechanisms of Aβ-induced neurodegeneration, we used Aβ-treated primary cortical neuron culture as a cell model of AD and investigated the function of miR-137 in this process. qRT-PCR and western blot were used to examine levels of miR-137, AMPA receptors (AMPARs), p-ERK1/2, and ERK1/2. MTT assay and caspases 3 activity assay were employed to examine cell viability and apoptosis. Dual-luciferase assay was used to validate the interaction between miR-137 and ERK1/2. We found that Aβ decreased cell viability, AMPAR and miR-137 levels, but increased caspase 3 activity in a dose- and time-dependent manner. Overexpression of miR-137 suppressed the Aβ-induced neurotoxicity. MiR-137 directly bound to ERK1/2 mRNA and negatively regulated its expression. Further, overexpression of ERK1/2 blocked the effects of miR-137 mimics on Aβ-induced neurotoxicity. These results provide strong evidence that miR-137 protects neurons against Aβ-induced neurotoxicity via targeting ERK1/2.
Introduction
Alzheimer’s disease (AD) is a devastative neurodegenerative disorder characterized by progressive loss of memory and impaired cognitive functions (Mayeux and Stern Citation2012; Collaborators GBDD Citation2019). It is the most common form of dementia and one of the leading causes of morbidity and mortality in the elderly (Mayeux and Stern Citation2012; Collaborators GBDD Citation2019). The hallmarks of AD are progressive deposition of amyloid beta (Aβ) plagues and formation of intracellular neurofibrillary tangles (Reiss et al. Citation2018; Panza et al. Citation2019). The large accumulation of extracellular Aβ peptide causes neurotoxicity in the brain, leading to neuronal dysfunction and loss and eventually cognitive deficits (Reiss et al. Citation2018; Panza et al. Citation2019). Currently, there is no effective treatment for AD due to its unclear pathogenesis. Therefore, exploring the molecular mechanisms of AD is very critical for the development of noveltherapy.
Micro RNAs (miRNAs) are a class of endogenous non-coding RNAs with a length of 18–22 nucleotides (O'Brien et al. Citation2018; Gebert and MacRae Citation2019). They usually bind target mRNAs to inhibit their expressions. A huge body of studies have shown that miRNAs play important roles in many biological processes including development, differentiation, and growth (O'Brien et al. Citation2018; Gebert and MacRae Citation2019). Dysregulated expression of miRNAs has been observed in many diseases (Giza et al. Citation2014). In AD, many miRNAs have been identified as regulators of Aβ production or the development of the disease, such as miR-124, miR-200b (Wang et al. Citation2019). miR-137 is a miRNA of 23-nucleotide long and located within a long non-coding gene, MIR137HG (Mahmoudi and Cairns Citation2017). Many studies have shown that miR-137 play crucial roles in neural development and brain function (Mahmoudi and Cairns Citation2017). Further, aberrant expression of miR-137 is associated with mental illness such as schizophrenia (He et al. Citation2018; Kandratsenka et al. Citation2018). Nevertheless, the role of miR-137 in AD is not clear.
In this study, we sought to investigate the function of miR-137 in AD. We used a cell model of AD by treating primary cultured neurons with Aβ peptide. We showed that Aβ treatment induced neurotoxicity in a dose- and time-dependent manner. Further, we found miR-137 was greatly reduced following Aβ treatment. Overexpression of miR-137 significantly ameliorated the neurotoxicity caused by Aβ. Mechanistically, we found miR-137 directly targeted ERK1/2 and protected neurons against Aβ-induced neurotoxicity via ERK1/2. Our study reveals an essential role of miR-137/ERK1/2 signaling in AD, which sheds light on the molecular mechanisms of AD and provides avenues for the development of therapeutic strategies for AD.
Materials and methods
Primary cortical neuron culture and Aβ treatment
Primary rat cortical neurons were prepared as previously described (Tan et al. Citation2015, Citation2020). Briefly, the plates were pre-coated with poly-L-lysine (Invitrogen, USA) overnight and rat embryonic (E18) cortical neurons were plated onto the precoated plates at a density of 60, 000 cells/cm2 in the Neurobasal medium supplemented with 2% (vol/vol) B-27, 2 mM GlutaMAX, 1% horse serum, and 50 U/ml Penn Strep. At days in vitro (DIV) 5, neurons were treated with 5 μM 5-FdU (5′-Fluoro-2′-deoxyuridine, Sigma, USA) and then fed twice a week with the culture medium until DIV 15–18.
Aβ1-42 oligomers (Invitrogen, USA) were prepared as previously described (Ryan et al. Citation2013) and ammonium hydroxide was used to pretreat Aβ1-42 to generate a seed-free solution of Aβ1-42. The pretreated Aβ1-42 was added into the culture medium at indicated concentrations when neurons were around 2 weeks old and incubated with the neurons for indicated periods before the cells were harvested for further analysis. Same amount of vehicle was added as the control group.
Cell transfection
Transfection was performed by using Lipofectamine 2000 (Invitrogen, USA) based on manufacturer’s protocol. Briefly, half of the old medium was saved and then 1 μg plasmid together with 1 μl Lipofectamin 2000 was mixed in fresh Neurobasal medium (100 μL) first for 20 mins and later added into the culture medium for 2 h. The medium was then discarded and the saved old medium was added back to the plates with the same amount of new culture medium.
RNA extraction and RT-qPCR
Total RNA was extracted from cultured neurons using Trizol reagent (Invitrogen, Missouri, USA) according to the manufacturer's instructions. Approximately 1 μg total RNA from each sample was subjected to reverse transcription before PCR amplification. PCR was conducted as follows: 95 °C for 10 min, amplification for 40 cycles (95 °C for 20 s, 60 °C for 20 s, 75 °C for 20 s). Relative expression level of miR-137 was normalized to 18S RNA and U6 snRNA as internal controls. The following primers were used: miR-137 forward: 5′-TGACAGCGGTAGCAGAGGCAGAG-3′; miR-137 reverse: 5′-CCGCTGCCCGCCTGCCGCTGGTA-3′; 18S RNA forward primer: 5′-CAGGATTGAC AGATTGATAGC-3′; 18S RNA reverse primer: 5′-GAGTCTCGTTCGT TATCGGAA-3′. U6 snRNA forward primer: 5′-GCTTCGGCAGCACATATACTAAAAT-3′; U6 snRNA reverse primer: 5′-CGCTTCACGAATTTGCGTGTCAT-3′. No significant difference was observed between 18S RNA normalization and U6 snRNA normalization. Data were plotted as the mean of the two calculations.
MTT assay
Primary cultured neurons were treated with Aβ at indicated concentrations for a period of time as indicated in figures. 15 μl of 3-(4,5-Dimethyltjiazol-2-yl)-2,5-diphenltetrazolium bromide (MTT, 5 mg/ml) was added into the medium and incubated with neurons for 3 h at 37 °C followed by incubation with 150 μl dimethyl sulfoxide (DMSO, Sigma-Aldrich, MO, USA) to stop the reaction. The absorbance in each condition was analyzed by a 590 nm light.
Caspase-3 activity assay
Caspase-3 activity was measured with the Caspase-3 assay kit (Abcam, USA) and performed according to the manufacturer’s protocol. Briefly, drug-treated neurons were harvested and incubated with the reaction buffer and Asp-Glu-Val-Asp (DEVD) – pNA substrate for 1 h at 37°C followed by the measurement of absorbance (405 nm).
Dual-luciferase report assay
cDNAs that contain the wild-type sequences or mutated binding sites of miR-137 in ERK1/2 mRNA 3’ UTR were cloned into downstream of the luciferase report gene of psiCHECK2. Neuron transfection was performed as described above. The co-transfected cells were lysed with Reporter Lysis Buffer, and the luciferase activity was detected using a Luciferase Reporter Gene Assay Kit (Promega, USA).
Western blot
Total proteins were extracted from cultured neurons with RIPA lysis buffer (Beyotime Institute of Biotechnology, China) containing protease inhibitor. Protein concentration was determined by Pierce BCA protein Assay (Thermo Fisher Scientific, USA). Equal amounts of protein from each sample were loaded and separated by SDS-PAGE electrophoresis followed by transferring to PVDF membranes (Millipore, USA). The membranes were blocked with 3% BSA for 1 h at room temperature and then incubated with primary antibodies at 4°C overnight. The membranes with incubated with secondary antibodies after washes with TBST and subsequently analyzed with the ECL kit (Thermo Fisher Scientific, USA). The following primary antibodies were used: Anti-GluA1 (1: 1000; Abcam. USA); Anti-p44/42 MAPK (ERK1/2) (1: 5000; Cell Signaling, USA); Anti-phospho-p44/42 MAPK (ERK1/2) (1:2500; Cell Signaling, USA); Anti-β-actin (1:2000; Santa Cruz, USA).
Statistical analysis
All experiments were performed with at least three biological replicates and all data were presented as mean, with error bars indicating ± SEM (Standard error of the mean). All statistical analyses were analyzed in GraphPad Prism 7. Statistical significance was determined by unpaired Student t test or one-way ANOVA post-hoc Bonferroni as indicated in figure legends.
Results
Aβ caused neurotoxicity and decreased mir-137 level in a dose- and time-dependent manner
To investigate the effects of Aβ on neurotoxicity, we treated DIV14 rat primary cortical neurons with Aβ (1, 5, 10 μM) or vehicle for 24 h or 48 h and then measured the effects on cell viability and the levels of AMPA receptors (AMPARs) in neurons as AMPARs are the principle glutamate receptors that mediate fast excitatory transmission and are key to neuronal functions (Volk et al. Citation2015). With MTT assay, we showed that Aβ treatment greatly decreased the viability of cultured neurons compared to control vehicle treatment (Figure (A)). Moreover, we observed that the reduction in cell viability caused by Aβ treatment was dose and time-dependent (Figure (A)). Regarding AMPAR expression, we found Aβ significantly decreased total protein levels of AMPAR GluA1 subunit in a dose- and time-dependent manner (Figure (B,C)). Further, using Caspase 3 activity assay, we observed that Aβ treatment greatly enhanced Caspase 3 activity (Figure (D)), which is closely correlated with cell apoptosis (McIlwain et al. Citation2013). These results indicate that Aβ induces neurotoxicity in a concentration- and time-dependent way.
Figure 1. Aβ caused neurotoxicity and decreased miR-137 level in a dose- and time-dependent manner. (A) MTT assay to assess cell viability following Aβ treatment or control vehicle treatment (Con). (B) GluA1 level in neurons following Aβ treatment or control vehicle treatment (Con). (C) Quantification of (B). (D) Relative Caspase 3 activity in neurons following Aβ treatment or control vehicle treatment (Con). (E) Relative miR-137 level in neurons following Aβ treatment or control vehicle treatment (Con). (n = 4). One-way ANOVA post-hoc Bonferroni. **P < 0.01. (Comparisons were done between groups at same time point but with different concentrations). #P < 0.05; ###P < 0.001 (Comparisons were done between groups at different time points but with same concentrations).
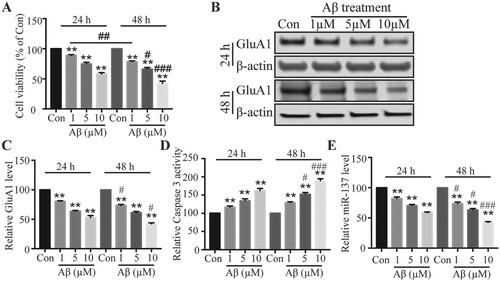
To study the role of miR-137 in AD, we examined miR-137 level following Aβ treatment. With qRT-PCR, we observed that Aβ remarkably diminished the level of miR-137 (Figure (D)). The level was lower with higher concentration or longer treatment period (Figure (D)), indicating that Aβ decreases miR-137 level in a dose- and time-dependent manner as well. Since the neurotoxicity is very robust after 2 days of 5 μM Aβ treatment, we used 5 μM Aβ to treat neurons for 2 days for further investigations.
Overexpression of mir-137 reduced the neurotoxicity causedby Aβ
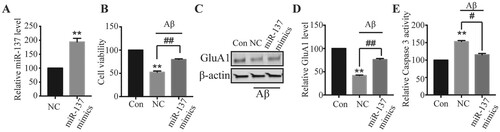
The lower level of miR-137 in neurons following Aβ treatment implied that miR-137 might have some protective roles. To test that hypothesis, we used miR-137 mimics to increase miR-137 level and assessed its effects on the Aβ-induced neurotoxicity. As expected, neurons transfected with miR-137 mimics had much higher level of miR-137 compared to cells transfected with mimic-NC (Figure (A)). In control transfected cells, consistently, we saw a remarkable decrease in cell viability and total AMPAR level but an increase of Caspase 3 activity following Aβ treatment (Figure (B–E)). However, in neurons transfected with miR-137 mimics, the decreases of cell viability and GluA1 and the increase of Caspase 3 activity induced by Aβ were much smaller (Figure (B–E)). Therefore, these results indicate that overexpression of miR-137 ameliorates the neurotoxicity caused by Aβ.
Figure 2. Overexpression of miR-137 reduced the neurotoxicity caused by Aβ. (A) Relative miR-137 level in neurons transfected with mimic-NC (NC) or miR-137 mimics. Student’s t-test. (B) Viability of neurons transfected mimic-NC or miR-137 mimics neurons following Aβ treatment (5 μM for 48 h) compared with control vehicle treated neurons (Con). (C, D) GluA1 protein level in transfected neurons following Aβ treatment (5 μM for 48 h) compared with control vehicle treated neurons (Con). One-way ANOVA post-hoc Bonferroni. (E) Relative Caspase 3 activity in transfected neurons following Aβ treatment (5 μM for 48 h) compared with control vehicle treated neurons (Con). One-way ANOVA post-hoc Bonferroni. (n = 4) ** P < 0.01.
miR-137 directly targeted ERK1/2
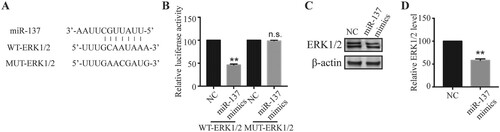
We next investigated the molecular mechanisms underlying the protective role of miR-137. miRNAs usually exert their functions via binding downstream mRNAs (O'Brien et al. Citation2018). Through the bioinformatic analysis, we identified some complementary binding sites between miR-137 and extracellular signal-regulated kinase 1/2 (ERK1/2) (Figure (A)). To confirm this interaction, we performed dual luciferase assay. miR-137 mimics significantly reduced the relative luciferase activity of WT-ERK1/2 but not the MUT-ERK1/2 wherein the predicted binding sites with miR-137 were mutated (Figure (A,B)), suggesting that miR-137 directly binds to ERK1/2 mRNA. Further, in neurons transfected with miR-137, we observed a lower level of ERK1/2 proteins compared to mimics NC-transfected neurons (Figure (C,D)), indicating that miR-137 negatively regulates ERK1/2 expression.
Figure 3. miR-137 directly targeted ERK1/2. (A) Complementary binding sites between miR-137 and ERK1/2 mRNA. (B) Relative luciferase activity of WT-ERK1/2 and MUT-ERK1/2 in transfected neurons. Student’s t-test (n = 4). (C, D) ERK1/2 protein level in neurons transfected with mimic-NC or miR-137 mimics. Student’s t-test. (n = 4) n.s., not significant; ** P < 0.01.
miR-137 protected neurons from Aβ-induced neurotoxicity via targeting ERK1/2
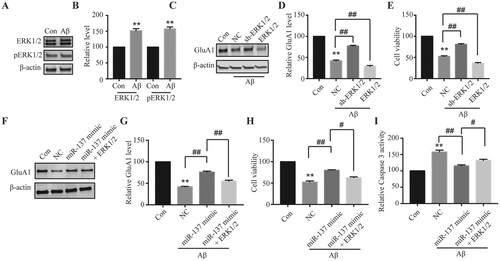
We finally evaluated the function of miR-137/ERK1/2 signaling in Aβ-induced neurotoxicity. First, we found ERK1/2 protein level was significantly up-regulated after Aβ treatment (Figure (A,B)), suggesting that it might contribute to this process. We then manipulated ERK1/2 level through overexpression or shRNA. As shown in Figure (C–E), knockdown of ERK1/2 partially reversed the effects of Aβ on AMPAR expression and Caspase 3 activity while overexpression of ERK1/2 further enhanced Aβ-induced neurotoxicity. Moreover, overexpression of ERK1/2 blocked the protective effects of miR-137 (Figure (F–H)). Taken together, these data demonstrate that miR-137 protects neurons against Aβ via targeting ERK1/2.
Figure 4. miR-137 protected neurons from Aβ-induced neurotoxicity via targeting ERK1/2. (A, B) ERK1/2 and pERK1/2 protein levels in neurons with Aβ treatment (5 μM for 48 h) compared with control vehicle treated neurons (Con). Student’s t-test. (C, D) GluA1 level in neurons transfected with NC, sh-ERK1/2, or ERK1/2 following Aβ treatment (5 μM for 48 h) compared with control vehicle treated neurons (Con). One-way ANOVA post-hoc Bonferroni. (E) Relative viabilities of neurons transfected with NC, sh-ERK1/2, or ERK1/2 following Aβ treatment (5 μM for 48 h) compared with control vehicle treated neurons (Con). One-way ANOVA post-hoc Bonferroni. (F, G) GluA1 level in neurons transfected with NC, sh-ERK1/2, or ERK1/2 following Aβ treatment (5 μM for 48 h) compared with control vehicle treated neurons (Con). One-way ANOVA post-hoc Bonferroni. (H) Relative viabilities of neurons transfected with NC, sh-ERK1/2, or ERK1/2 following Aβ treatment (5 μM for 48 h) compared with control vehicle treated neurons (Con). One-way ANOVA post-hoc Bonferroni. (I) Relative Caspase 3 activity in neurons transfected with NC, sh-ERK1/2, or ERK1/2 following Aβ treatment (5 μM for 48 h) compared with control vehicle treated neurons (Con). One-way ANOVA post-hoc Bonferroni. (n = 4). **P < 0.01; #P < 0.05; ##P < 0.01.
Discussion
As the most common cause of dementia, AD affects millions of aging people around the world and the incidence of AD has been increasing drastically in the past decades (Mayeux and Stern Citation2012; Collaborators GBDD Citation2019). Despite great advances in research, there is still no available satisfying treatments to AD due to its complex and elusive pathogenesis. Here, we fully elucidated the function of miR-137 in AD. We showed that Aβ significantly down-regulated miR-137 level in a concentration- and time-dependent manner, accompanied by decreased AMPARs and increased cell apoptosis. Rescue of miR-137 level with miR-137 mimics greatly reduced the neurotoxicity caused by Aβ. Moreover, we found that miR-137 bound ERK1/2 mRNA and protected neurons against Aβ through targeting ERK1/2. Our work clearly demonstrates that miR-137 plays a critical role in AD and could serve as a target for future therapy development.
It is well known that miRNAs play essential roles in diverse processes including physiological processes and pathological processes (Giza et al. Citation2014; Gebert and MacRae Citation2019). In the brain, miR-137 has drawn a lot of attention as many studies show that miR-137 has critical functions in neuronal development, as well as neurological diseases (Mahmoudi and Cairns Citation2017). It is highly expressed in the dentate gyrus, a sub-region of hippocampus, and has been implicated in schizophrenia, autism spectrum disorders, Parkinson’s disease (Sarachana and Hu Citation2013; Devanna and Vernes Citation2014; Kong et al. Citation2015; Wright et al. Citation2015; Mahmoudi and Cairns Citation2017; He et al. Citation2018). Further, in the ketamine-induced hippocampal neurodegeneration and memory loss, miR-137 was observed reduced, and overexpression of miR-137 decreased the effects of ketamine on neurodegeneration (Huang et al. Citation2014). Regarding AD, previous studies have reported a reduced level of miR-137 in APP/PS1 transgenic AD mice wherein spatial learning and memory was greatly compromised (Jiang et al. Citation2018), although the underlying mechanisms are unclear. Here, we also found that Aβ treatment diminished miR-137 level to induce neurotoxicity. Moreover, we showed that rescue of miR-137 level protected neurons against Aβ. Our study, together with previous studies, suggests that miR-137 is beneficial to neurons in a general way and that maintaining its level could be potentially used to treat neurodegenerative diseases.
MiRNAs negatively regulate gene expression of their targets. In the current study, we found that miR-137 directly bound ERK1/2 mRNA in neurons and inhibited its expression. ERK1/2 is a versatile protein kinase that has many important cellular functions (Subramaniam and Unsicker Citation2010; Roskoski R Citation2012). Emerging evidence suggests that ERK1/2 is largely involved in promoting neuronal death (Subramaniam and Unsicker Citation2010). For example, it has been shown that ERK1/2 activation leads to activation of Caspase 3 and promotes cell apoptosis (Chong et al. Citation2006). Here we saw similar results. Aβ treatment significantly increased ERK1/2 level and Caspase 3 activity. Knockdown of ERK1/2 partially blocked the Aβ-induced neurotoxicity and inhibited cell apoptosis. Moreover, we demonstrate that miR-137 protects neurons against Aβ through ERK1/2 in that overexpression of ERK1/2 blocked the effects of miR-137 mimics. Notably, ERK1/2 only partially reversed the effects of miR-137 mimics, suggesting that miR-137 might have other targets. Indeed, it has been shown that miR-137 targets varieties of mRNAs such as CDC42, MAPK3, and AKT2 (Zhu et al. Citation2013; Wright et al. Citation2015; Mahmoudi and Cairns Citation2017; Duan et al. Citation 2019). Therefore, it is very likely that miR-137 exert its protective functions through multiple downstream targets including ERK1/2. Further studies are necessary to examine other targets. Interestingly, GluA1 has been reported as a target of miR-137 as well and miR-137 represses AMPAR-mediated synaptic transmission by inhibiting GluA1 expression (Loohuis NF et al. Citation2015). However, in Aβ-treated neurons, overexpression of miR-137 mimics did not result in further reduction of GluA1, but instead, partially recovered the level of GluA1. These results suggest that ERK1/2 is a primary target of miR-137 while GluA1 is not during Aβ-induced neurotoxicity process. Also, our data support the notion that miRNA-mediated gene regulation is context-dependent (Erhard et al. Citation2014).
In summary, with a combination of primary neuron culture, genetic tools, and biochemistry methods, we show that miR-137/ERK1/2 signaling pathway plays a crucial role in Aβ-induced neurotoxicity. Rescue of miR-137 level is beneficial to neurons and could serve as a way to treat AD.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Chong YH, Shin YJ, Lee EO, Kayed R, Glabe CG, Tenner AJ . 2006. ERK1/2 activation mediates Abeta oligomer-induced neurotoxicity via caspase-3 activation and tau cleavage in rat organotypic hippocampal slice cultures. J Biol Chem. 281:20315–20325. doi:10.1074/jbc.M601016200. 2006/05/23.
- Collaborators GBDD. 2019. Global, regional, and national burden of Alzheimer's disease and other dementias, 1990-2016: a systematic analysis for the Global burden of disease study 2016. Lancet Neurol. 18:88–106. doi: 10.1016/S1474-4422(18)30403-4. 2018/12/01.
- Devanna P, Vernes SC. 2014. A direct molecular link between the autism candidate gene RORa and the schizophrenia candidate MIR137. Sci Rep. 4:3994. doi: 10.1038/srep03994. 2014/02/07.
- Duan J, Lu G, Li Y, Zhou S, Zhou D, Tao H. 2019. miR-137 functions as a tumor suppressor gene in pituitary adenoma by targeting AKT2. Int J Clin Exp Pathol. 12:1557–1564. 2020/01/15.
- Erhard F, Haas J, Lieber D, Malterer G, Jaskiewicz L, Zavolan M, Dölken L, Zimmer R . 2014. Widespread context dependency of microRNA-mediated regulation. Genome Res. 24:906–919. doi:10.1101/gr.166702.113. 2014/03/29.
- Gebert LFR, MacRae IJ. 2019. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol. 20:21–37. doi: 10.1038/s41580-018-0045-7. 2018/08/16..
- Giza DE, Vasilescu C, Calin GA. 2014. Key principles of miRNA involvement in human diseases. Discoveries (Craiova). 2:e34. doi: 10.15190/d.2014.26. 2015/09/01..
- He E, Lozano MAG, Stringer S, Watanabe K, Sakamoto K, Oudsten FD, Koopmans F, Giamberardino SN, Hammerschlag A, Cornelisse LN et al. 2018. MIR137 schizophrenia-associated locus controls synaptic function by regulating synaptogenesis, synapse maturation and synaptic transmission. Hum Mol Genet. 27:1879–1891. doi: 10.1093/hmg/ddy089. 2018/04/11.
- Huang C, Zhang X, Zheng J, Chen C, CHen Y, Yi J. 2014. Upregulation of miR-137 protects anesthesia-induced hippocampal neurodegeneration. Int J Clin Exp Pathol. 7:5000–5007. 2014/09/10.
- Jiang Y, Xu B, Chen J, Sui Y, Ren L, Li J, Zhang H, Guo L, Sun X. 2018. Micro-RNA-137 inhibits Tau hyperphosphorylation in Alzheimer's disease and targets the CACNA1C gene in transgenic mice and human neuroblastoma SH-SY5Y cells. Med Sci Monit. 24:5635–5644. doi:10.12659/MSM.908765. 2018/08/14.
- Kandratsenka H, Nestsiarovich A, Goloenko I, Danilenko N, Makarevich A, Obyedkov V, Davydenko O, Waszkiewicz N. 2018. Association of MIR137 With symptom severity and cognitive functioning in Belarusian schizophrenia patients. Front Psychiatry. 9:295. doi: 10.3389/fpsyt.2018.00295. 2018/07/22.
- Kong Y, Liang X, Liu L, Zhang D, Wan C, Gan Z, Yuan L. 2015. High throughput sequencing identifies MicroRNAs mediating alpha-synuclein toxicity by targeting neuroactive-ligand receptor interaction pathway in early stage of drosophila Parkinson's disease model. PLoS One. 10:e0137432. doi: 10.1371/journal.pone.0137432. 2015/09/12.
- Loohuis NF O, Ba W, Stoerchel PH, Kos A, Jager A, Schratt G, Martens GJM, van Bokhoven H, Kasri NN, Aschrafi A. 2015. MicroRNA-137 controls AMPA-receptor-mediated transmission and mGluR-dependent LTD. Cell Rep. 11:1876–1884. doi:10.1016/j.celrep.2015.05.040. 2015/06/23.
- Mahmoudi E, Cairns MJ. 2017. MiR-137: an important player in neural development and neoplastic transformation. Mol Psychiatry. 22:44–55. doi: 10.1038/mp.2016.150. 2016/09/14.
- Mayeux R, Stern Y. 2012. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. doi: 10.1101/cshperspect.a006239. 2 2012/08/22..
- McIlwain DR, Berger T, Mak TW. 2013. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 5:a008656. doi: 10.1101/cshperspect.a008656. 2013/04/03.
- O'Brien J, Hayder H, Zayed Y, Peng C. 2018. Overview of MicroRNA biogenesis. Mech Actions Circ. Front Endocrinol (Lausanne). 9:402. doi: 10.3389/fendo.2018.00402. 2018/08/21.
- Panza F, Lozupone M, Logroscino G, Imbimbo BP. 2019. A critical appraisal of amyloid-beta-targeting therapies for Alzheimer disease. Nat Rev Neurol. 15:73–88. doi: 10.1038/s41582-018-0116-6. 2019/01/06.
- Reiss AB, Arain HA, Stecker MM, et al. 2018. Amyloid toxicity in Alzheimer's disease. Rev Neurosci. 29:613–627. doi: 10.1515/revneuro-2017-0063. 2018/02/16.
- Roskoski R J. 2012. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 66:105–143. doi:10.1016/j.phrs.2012.04.005. 2012/05/10.
- Ryan TM, Caine J, Mertens HD, Kirby N, Nigro J, Breheney K, Waddington LJ, Streltsov VA, Curtain C, Masters CL, et al. 2013. Ammonium hydroxide treatment of Abeta produces an aggregate free solution suitable for biophysical and cell culture characterization. PeerJ. 1:e73. doi: 10.7717/peerj.73. 2013/05/17.
- Sarachana T, Hu VW. 2013. Genome-wide identification of transcriptional targets of RORA reveals direct regulation of multiple genes associated with autism spectrum disorder. Mol Autism. 4:14. doi: 10.1186/2040-2392-4-14. 2013/05/24.
- Subramaniam S, Unsicker K. 2010. ERK and cell death: ERK1/2 in neuronal death. FEBS J. 277:22–29. doi:10.1111/j.1742-4658.2009.07367.x. 2009/10/22.
- Tan HL, Chiu SL, Zhu Q, Huganir RL. 2020. GRIP1 regulates synaptic plasticity and learning and memory. Proc Natl Acad Sci U S A. 117:25085–25091. doi: 10.1073/pnas.2014827117. 2020/09/20.
- Tan HL, Queenan BN, Huganir RL. 2015. GRIP1 is required for homeostatic regulation of AMPAR trafficking. Proc Natl Acad Sci U S A. 112:10026–10031. doi: 10.1073/pnas.1512786112. 2015/07/29.
- Volk L, Chiu SL, Sharma K, Huganir RL. 2015. Glutamate synapses in human cognitive disorders. Annu Rev Neurosci. 38:127–149. doi: 10.1146/annurev-neuro-071714-033821. 2015/04/22.
- Wang M, Qin L, Tang B. 2019. MicroRNAs in Alzheimer's disease. Front Genet. 10:153. doi: 10.3389/fgene.2019.00153. 2019/03/19.
- Wright C, Calhoun VD, Ehrlich S, Wang L, Turner JA, Perrone-Bizzozero NI. 2015. Meta gene set enrichment analyses link miR-137-regulated pathways with schizophrenia risk. Front Genet. 6:147. doi: 10.3389/fgene.2015.00147. 2015/05/06.
- Zhu X, Li Y, Shen H, Li H, Long L, Hui L, Xu W. 2013. miR-137 inhibits the proliferation of lung cancer cells by targeting Cdc42 and Cdk6. FEBS Lett. 587:73–81. doi:10.1016/j.febslet.2012.11.004. 2012/11/28.