
Abstract
Cell cycle inhibitors are considered as hallmark strategy for cancer treatment due to their relatively higher selectivity and efficacy on various cancer types in comparison to cytotoxic agents. Small molecules target dividing cells in G1/S, G2 or M phases of cells to arrest and eventually trigger cancer cells to enter apoptosis and/or inhibit tumor growth. Cell cycle arrest at G2/M phase is a widely used approach to inhibit proliferating cancer cells. We report a novel angiotensin II receptor type I (AT1R) antagonist that also targets CDC2 (CDK1) kinase thereby showing a putative anti-cancer activity. The molecule named 19D was tested in various cancer cell lines at different concentrations. Molecular mechanisms of action triggered by the treatment of 19D were investigated for anti-cancer activity at cellular and molecular level. 19D molecule showed a potent cell cycle arrest with anti-proliferation activity. Cells treated with 19D showed nuclear deteriorations and apoptotic induction on cancer cells. Moreover, the mechanism of cell cycle inhibition was revealed as G2/M arrest via suppressing the CDC2 kinase cell cycle check point inhibitor. In conclusion, 19D (an AT1R antagonist) is a novel small molecule/imidazolone derivative which has been demonstrated as a fairly potent anti-cancer molecule.
Abbreviations: AT1R: angiotensin II receptor type 1; BSA: bovine serum albumine; CDC2: cyclin-dependent kinase 1; CDK: cyclin-dependent kinase; DMEM: Dulbecco’s modified eagle medium; DMSO: dimethylsulfoxide; EDTA: ethylenediaminetetraacetic acid; FBS: fetal bovine serum; HBSS: Hank’s balanced salt solution; IC50: half maximal inhibitory concentration; PBS-T: phosphate buffered saline – TritonX; PBS: phosphate buffered saline; PDB: protein data bank; SDS: sodium dodecyl sulfate; TBS-T: Tris buffered saline – TritonX
Introduction
Cancer is one of the leading causes of death worldwide and there are numerous efforts to develop novel effective drugs for cancer treatment. Small molecule inhibitors are extensively pursued as potential anti-cancer molecules in many cancer types. To date, more than 80 small molecule inhibitors have been advanced to the clinical evaluation stage in the drug development pipeline (Zhang et al. Citation2009). Despite high interest in oncology research and investments, currently available chemotherapy drugs fall short of expectations due to the heterogeneous characteristics of cancers and patients. Therefore, it is important to find new anti-cancer drug candidates that may provide alternatives to existing drugs.
Checkpoint inhibitors are always attractive target molecules for the development of anti-cancer molecules. If the cells, are arrested in any step of the cell cycle, undergo cell death to terminate the propagation of damaged DNA-containing cells, thus apoptosis is induced. This control mechanism provides initiation of hallmark strategies against cancer cells (Hanahan and Weinberg Citation2011). The use of cell cycle arrest agents against abnormally proliferating cancer cells has been shown to be more effective than conventional cytotoxic drugs. (Debatin Citation2000). Therefore, small molecules that can induce cell cycle arrest have great potential in anti-cancer therapy (Spanò, Frasson, et al. Citation2016; Spanò, Pennati, et al. Citation2016; Barreca et al. Citation2020; Li Petri et al. Citation2020; Spanò et al. Citation2020; Sahin et al. Citation2021). One of the most promising and widely used examples of a small molecule inhibitor – Paclitaxel – arrests cells in G2/M phase by inducing microtubule instability and activating DNA damage responsive checkpoints (Jordan et al. Citation1996; Weaver Citation2014). It is now one of the leading anti-cancer molecules against ovarian cancer, breast cancer and lung cancer.
Angiotensin and its receptors are members of renin angiotensin system and one of the most popular classes of molecules against hypertension to treat cardiovascular diseases. Due to its central role in many diseases, various antagonists have been developed against angiotensin II type 1 receptor (AT1R) (Bauer and Reams Citation1995). Furthermore, there is increasing evidence showing the role of AT1R in cancer via affecting cell proliferation, angiogenesis, inflammation and tissue remodeling (Deshayes and Nahmias Citation2005). Various reports showed the activity of RAS pathways in different cancers including glioblastoma (Juillerat-Jeanneret et al. Citation2004), astrocytoma (Fogarty et al. Citation2002), breast cancer (De Paepe et al. Citation2002) and prostate cancer (De Paepe et al. Citation2002). Therefore, the activity of AT1R antagonists has been shown to be effective on various cancer cell types (Chen et al. Citation2013). In efforts to develop AT1R inhibitors, we have previously reported the putative role of an AT1R small molecule antagonist, named 19D, in blocking the cell proliferation potential of different cancer cells (Figure ) (Durdagi et al. Citation2018). In our previous study, the cell proliferation inhibition role of the 19D molecule was shown, but the mode of action was not characterized extensively. The increased population of G2/M phase cells and increased number of Go phase cells were shown on breast cancer (MCF-7) and human epithelial cell lines (HUVEC). Here, we hypothesize that 19D has great potential as a cell cycle inhibitor molecule by inhibiting the AT1R and we compared its anti-cancer activity by comparing currently used commercial drugs in chemotherapy. Further, we investigated the additional potential targets of 19D molecule. Various cancer cells were studied like human colon (HCT), human glioblastoma (U-87 MG) and human epithelial cell line (HUVEC). Anti-cancer effects of 19D molecule was investigated by observation of cellular morphology changes, cell cycle arrest, proliferation restriction and apoptotic induction by using in vitro studies.
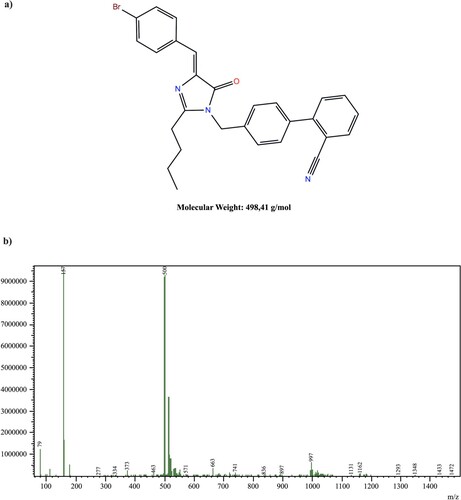
Figure 1. Chemical structure of 19D. (a) Chemical structure of 19D molecule. (b) LC-MS spectrum. Peak close to 499.4 m/z value indicates 19D molecule.
Materials and methods
Synthesis and characterization of 19D molecule
19D ((4′-((4-(4-Bromobenziliden)-2-butyl-5-oxo-4,5-dihidro-1H-imidazol-1-il)methyl)biphenyl-2-carbonitril)) molecule is an imidazolone derivative small molecule that has been discovered and reported in our previous study (Durdagi et al. Citation2018). Briefly, imidazolone derivatives refluxed with bromomethyl-2-biphenylcarbonitrile in the presence of potassium carbonate in acetonitrile under nitrogen atmosphere and 19D was purified as previously described (Durdagi et al. Citation2018).
Cell culture conditions and reagents
Human colon (HCT), human glioblastoma (U-87 MG) and human epithelial cell lines (HUVEC) were used to test the efficacy of molecule 19D. All cell lines were grown in Dulbecco’s modified Eagle’s medium (DMEM) high glucose with 10% heat-inactivated FBS and 1% penicillin–streptomycin in 37°C humidified environment with 5% CO2. 19D molecule was solubilized in dimethyl sulfoxide (DMSO) as 50 mM stock solution, Paclitaxel and Staurosporine as 1 mM stock solution and stored at −20°C until use. Final DMSO in each experiment did not exceed 0.2%.
Cell viability assay
All cells were seeded at 20 × 103 cells/well density in 24-well plates. Following 24 h of preincubation, cells were treated with 25, 50 and 100 µM concentrations of 19D molecules, 100 nM Paclitaxel and 0.2% DMSO as a vehicle control. All conditions were studied in triplicate wells. Cells were analyzed with MTT colorimetric assay for 3 days following the treatment of molecules. About 50 µl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (5 mg/ml) was added to each well and incubated for 4 h at 37°C humidified environment with 5% CO2. Formazan precipitates were solubilized with SDS buffer. Following 15 min incubation at 37°C, absorbance was measured at 570 nm using PG Instruments T80 UV/VIS spectrophotometer.
Cell cycle analysis
Cell cycle analysis was performed to assess the frequencies of cell cycle phases in 19D treated and untreated cells by using the advantage of DNA content measurement via flow cytometry. Cells were cultured in 24-well plates as three replicates from each group, selected groups were treated with 19D molecule with different concentrations at 5, 10, 25, 50 and 100 µM. About 100 nM Paclitaxel was used as positive control to show G2/M arrest and 0.2% DMSO was used as negative control. Efficacy was tested for both 24 and 48 h of incubation times. After treatment, cells were harvested with HBSS and 5 mM EDTA, transferred into HBSS 2 mM EDTA 2% FBS media, washed with ice-cold PBS with 2% FBS and fixed with 70% (v/v) ethanol for 30 min on ice. Cells were washed twice with PBS with 2% FBS and pellets suspended in PBS containing 200 µg/ml RNaseA. After 30 min room temperature incubation, cells were stained with propidium iodide at 25 µg/ml concentration. Following 15 min incubation in dark, cells were analyzed in Flow Cytometry ACEA Biosciences (Novocyte).
Histochemistry and immunohistochemistry analysis
Cells were seeded into 6-well plates over the glass cover slips. After 24 h preincubation, all molecular concentrations and control groups were tested as previously stated. Following 24 h of treatment, cover slips were fixed in freshly prepared 4% (v/v) paraformaldehyde in PBS for 5 min at room temperature following ice-cold methanol for 5 min at −20°C. Fixative solutions were removed with repeated PBS washes. Cells stained with hematoxylin for 45 s and eosin for 10 s. After mounting, cells were visualized with Leica DM500 Microscope.
Cells were washed three times with PBS for immunofluorescence analysis, then permeabilized with 0.2% TritonX-100 in PBS at room temperature for 5 min. Blocking was done with 3% bovine serum albumin (BSA) in PBS at room temperature for 30 min. Primary antibodies used 1:500 for anti-lamin B1 (Thermo Fischer, PA519468) and 1:100 for beta tubulin (BT7R) (Thermo Fischer, MA516308) were diluted in 0.1% BSA in PBS and incubated overnight. Cells were washed three times with PBST and secondary antibodies were used in 1:500 goat anti-rabbit alexa flour 488 (Thermo Fischer, A11017) and 1:1000 goat anti-mouse IgG Alexa Flour 555 (Thermo Fischer, A21425) dilution in 0.1% BSA in PBS and incubated for 1 h in dark room. Cell nuclei were stained with 1:7000 DAPI (10 mg/ml) in PBS for 5 min in dark. Finally, slides were mounted and visualized under Leica Fluorescence Microscope.
Caspase 3/7 apoptotic marker analysis
Apoptotic activity of cells was tested by using in situ hybridization of the apoptotic markers, caspase 3 and caspase 7. Cells were seeded at 100 × 103 cells/well density to 6-well plates over the glass cover slips. About 100 µM concentration of 19D and 0.2% DMSO for 24 h and 1 µM Staurosporine for 4 h were introduced to cells. Caspase 3/7 antibody-containing PBS solution with 2% FBS was added in 1:125 dilution to live cells. Cells were incubated for 3 h at 37°C humidified environment with 5% CO2, then images were obtained.
Western Blot
Total protein extract (30 µg) was electrophoresed on 8–15% SDS polyacrylamide gel. Proteins were transferred into 0.45 µm pore-sized nitrocellulose membrane. Membranes were blocked with 5% nonfat dried milk powder in TBST for 1 h at room temperature. Primary antibodies CDC2 (CST, 9116), beta actin (CST, 3700) and caspase 3 (CST, 9662) were diluted 1:1000 concentration and incubated overnight. After 1 h of secondary antibody incubation at room temperature, proteins were detected by using Supersignal Pico Plus chemiluminescence substrate (Thermo Fischer) and visualized with ChemiDoc (Biorad) imaging system.
Molecular modeling
As human cyclin-dependent kinase 2 CDC2 (CDK1) was determined as an additional target protein for 19D binding, 6GU2 PDB coded structure was used. Target protein and the ligand were prepared before the molecular docking. Prime module of the Maestro molecular modeling package was used for fixing the missing loops and side chains of the target (Jacobson et al. Citation2004). Protonation states of the residues were calculated by PROPKA at the physiological pH. The target was relaxed with structural optimization using OPLS3 force field (0.3 Å of root mean square deviation convergence threshold for heavy atoms was considered). The protonation states of the ligand were calculated by EPIK at the neutral pH and OPLS3 force field is used in the geometry optimization (Shelley et al. Citation2007; Harder et al. Citation2016). A grid-based molecular docking program (Glide/XP) was used for the docking (Friesner et al. Citation2006). The coordinates of co-crystallized ligand were used at the grid map generation and water molecules within the 5 Å at the binding pocket are kept during the docking. Default parameters were used.
Results
19D molecule inhibited cancer cell proliferation
In our previous study (Durdagi et al. Citation2018) imidazolone derivative 19D molecule was shown to be specifically inhibiting the cell cycle progression. The molecule is relatively a small molecule with a molecular weight of 498.41 g/mol and has several side chains that increase its potential to target several molecules in the cell (Figure (a)). The purity and the yield of the synthesis were assessed by liquid chromatography–mass spectrometry (LC–MS). Although there are subfractions of the 19D molecule shown in 167 Kda peak, the average purity of the molecule was above 90%(Figure (b)).
The minimum inhibitory concentration of 19D molecule at the AT1R (Ki) was shown to be 24.8 µM with a competitive binding assay. Furthermore, it was also shown that 100 µM concentration of 19D molecule was inhibiting the binding of around 60% of AngII to human AT1 receptor (Durdagi et al. Citation2018). Therefore, we tested 19D concentrations in the range of 25–100 µM as an anti-cancer molecule by MTT cell proliferation and viability assay. First, we observed HUVEC and HCT cells require less time to duplicate and proliferate when compared to U-87 MG (Figure ). This finding also supported flow cytometric cell cycle analysis results which showed that the majority of all cell population in HUVEC and HCT cells were arrested at G2/M phase in 24 h 19D-treated cells, whereas almost 65% arrested cells were observed for U-87 MG cells (Figure ). However, the number of viable cells dramatically decreased in 48 h of treatment. In all cells, even the minimum inhibitory concentration (25 µM) was shown to significantly inhibit the cell proliferation, while higher doses of 19D showed a more prominent effect. Our results indicated that 19D treatment inhibited the cell proliferation in different cancer cells for 96 h without recovery of cells as well as mediate irreversible apoptoticinduction.
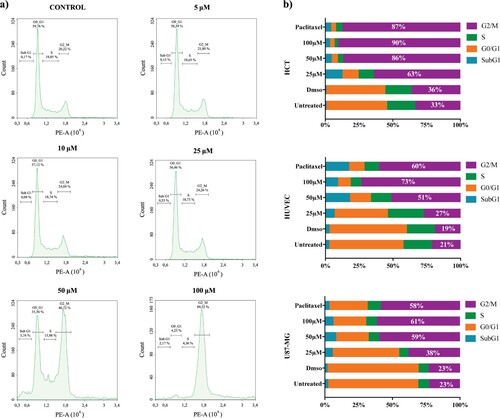
Figure 2. Cell cycle analysis of HCT, HUVEC and U-87 MG cells after 48 h of treatment with 19D and Paclitaxel. (a) Cell cycle histograms of U-87 MG cells after treatment with DMSO (control), 5, 10, 25, 50 and 100 µM 19D. (b) Cell cycle stage distributions of HCT, HUVEC and U-87 MG after treatment with 25, 50 and 100 µM concentration of 19D molecule compared with Paclitaxel and DMSO (control) All studies were triplicated.

Figure 3. Viability and proliferation of 19D-treated and control cells. Cells were treated by DMSO (control), 25, 50 and 100 µM 19D molecule and MTT assay was performed to determine cell viability. Cells were monitored for 4 days and cell viability was measured at 24, 48, 72 and 96 h.
19D induced G2/M arrest in various cancer cell lines
To further investigate the mechanism of action, we first analyzed the cell cycle inhibition on different cell lines; HUVEC, HCT, U-87 MG cells, which require various population doubling time. All cell lines were treated with 19D at 5, 10, 25, 50 and 100 µM concentrations and appropriate controls for both 24 and 48 h. The results showed specific accumulation of cell population at G2/M phase (doubled nuclear genome, 4n DNA content) of the cell cycle. (Figure (a)) DMSO-treated control, 5 and 10 µM 19D-treated cells showed an average of 15–20% G2/M cell population, whereas 25, 50 and 100 µM 19D-treated cells showed gradually increased G2/M phase cell population up to 60–80%. On the other hand, the mean level of G1/G0 phase cell population decreased correspondingly down to 10–20%, whereas the S phase cell population remained stable with a range of 5–10%. Furthermore, at higher concentrations of 19D-treated cells, apoptosis indicating SubG1 cell population was apparent.
We aimed to investigate the anti-cancer activity of 19D on different cell lines in order to show that cell cycle inhibition activity was cell proliferation capacity dependent. Strictly speaking, we hypothesized that the faster the cell is proliferating, the more G2/M-arrested cell population should be detected when treated with 19D. Different cell lines showed differential responses to 19D treatments. We compared the efficacy of 19D with regards to cell cycle inhibition by comparing it to Paclitaxel, a known G2/M blocking molecule. About 100 µM concentration of 19D led to a significant increase in the percentage of G2/M phase cells in HCT, HUVEC and U-87 MG (90%, 73% and 61% respectively) compared with the control (36%, 19% and 23% respectively) upon 24 h of treatment (Figure (b)). Besides, Paclitaxel, which is notably known as G2/M arrest-causing chemotherapeutic agent, caused either similar or a reduced level of accumulation of the cells in G2/M phase in HCT, HUVEC and U-87 MG (87%, 60% and 58% respectively). We also showed that 0.2% DMSO as a control of our molecules has no manipulative effect on cell cycle progress. After a 48 h treatment with 19D and Paclitaxel, agents triggered more potent DNA fragmentation in HUVEC and HCT and more G2/M arrested cells in U-87 MG, in which the population was increased from 58% to 69% (data not shown). Furthermore, the level of subG1 cell population, which is an indicator of apoptotic cell bodies, showed a significant increase in all cell types accordingly. These results indicated that treatment with 19D induced G2/M phase arrest and further DNA fragmentation (subG1 population) in various cell lines at a similar or even higher level than the commonly used anti-cancer drug Paclitaxel.
19D triggered cellular and nuclear aberrations
We further explored the effect of 19D molecule on morphological changes in HUVEC, HCT and U-87 MG cells. Apoptotic signs of cell death, cell cycle and cell proliferation assays triggered us to investigate the cell structure and shape upon 19D treatment. 19D-treated cells showed dramatic changes in both the cellular and nuclear structures. When compared to DMSO treatment, all 19D-treated cells showed apoptotic body formation with varying extents (Figure (a)). Results showed that 19D-treated cells were not capable of performing proliferation which caused a very low number of cells compared to the control. Furthermore, we detected treated cell populations express cell disintegrations and membrane blebbing morphologies, which can be associated with possible apoptosis mechanism (Figure (b)).
Figure 4. Microscopic evaluation of 19D-treated and control cells (a) Phase-contrast images of HUVEC, HCT and U-87 MG treated with DMSO (control) and 100 µM concentration of 19D. (b) 100× image of 100 µM 19D-treated cells showing membrane blebbing and apoptotic body formation. (c) Hematoxylin and Eosin staining of DMSO (control), 100 µM 19D and 100 nM Paclitaxel-treated cells at 24 h. (d) Immunofluorescence staining with beta-tubulin, Lamin B1 and DAPI of DMSO (control),100 µM 19D and 100 nM Paclitaxel-treated HUVEC cells at 24 h.
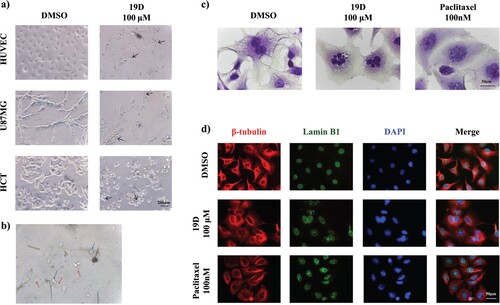
To further explore the nuclear aberrations, cells were stained with different dyes and antibodies. Hematoxylin and eosin staining revealed 19D-treated nucleus fragmentation into smaller nuclear bodies within a single cell. The effect of 19D compared with vehicle (DMSO) and Paclitaxel-treated cells. DMSO-treated cells show neither nuclear fragmentation nor multinucleated cells. In Paclitaxel-treated cells, multi-lobed nucleus formation was more rigid and more regular, but 19D-treated cells showed a similar extent of multinuclear aberration with varied polymorphonuclear structures. We found that this 19D treatment caused the accumulation of cells with multi-lobed nuclei, suggesting a failure of cytokinesis (Figure (c)).
To better evaluate multi-nuclei cell or multi-lobed nuclei, multicolor immunofluorescence staining with nuclear envelope specific lamin antibody, beta-tubulin antibody and DAPI was performed. Results proved that 19D-treated cells showed both multi-nuclei and multi-lobed nucleus formation. Lamin expression was intact in the nuclear envelopes within single cells. We hypothesize that 19D-treated cell nucleus shifts to multiple-nuclei form while the nuclear envelope starts to degrade in 24 h incubation with 19D (Figure (d)). In flow cytometry cell cycle analysis, we showed that 19D caused DNA fragmentations; these small nuclear envelop fragments can actually be represented as the SubG1 population. Additionally, 19D caused an accumulation of tubulin around the nucleus of cells, whereas tubulin was uniformly distributed in the control group. Collectively, 19D treatment caused membrane blebbing, multi-lobed nuclei formation, nuclear envelope degradation and possible tubulin instability in cells.
19D activated apoptosis via caspase 3/7 molecule
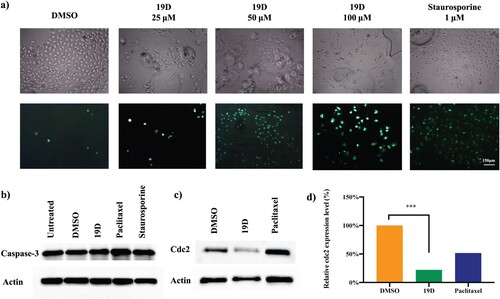
In order to gain further insight into molecular mechanisms underlying 19D-dependent G2/M arrest and cellular and nuclear morphological changes, caspase 3/7 activation in different cells were investigated. Immunofluorescence staining showed a significant increase in caspase 3/7 expression in 19D-treated cells in a dose-dependent manner (Figure (a)). To discriminate the mediated apoptosis by caspase 3 or caspase 7, we further checked the level of caspase 3 with western blot. However, caspase 3 level did not significantly change in 19D-treated cells, leading to profound apoptosis in the absence of caspase 3 activity (Figure (b)). This finding further suggests that 19D activated the apoptosis mechanism through activation of terminal effector in the apoptotic cascade.
Figure 5. Caspase activation and CDC2 inhibition in 19D treated and control cells. (a) Caspase-3/7 activation shown by light microscopy and live cell staining by four short peptide-based fluorogenic substrate DEVD of HUVEC cells treated with DMSO (control), 25, 50 and 100 µM 19D and staurosporine as a positive control. (b) Western blot image of HUVEC cells treated with DMSO, 19D, Paclitaxel and staurosporine showing Caspase-3 expression relative to actin expression. (c) Western blot image of HUVEC cells treated with DMSO, 19D and Paclitaxel showing CDC2 expression relative to actin expression. (d) Relative CDC2 expression percentage level according to western blot bands.
19D induced significant reduction in CDC2 protein level
Further exploration of cell cycle arrest caused by 19D treatment was investigated via evaluation of mitotic regulatory protein CDC2, which plays a key role in the control of G2/M checkpoint of the cell cycle (Sancar et al. Citation2004). The level of CDC2 was significantly reduced in 24 h treatment (Figure (c)). Similar to the 19D treatment, Paclitaxel-treated cells showed reduced CDC2 expression level after 24 h treatment. However, when compared to Paclitaxel, 19D showed a more evident reduction in CDC2 expression. Therefore, we concluded that 19D promoted G2/M phase arrest in an irreversible manner by downregulation of CDC2 levels in cells.
19D inhibited CDC2 kinase
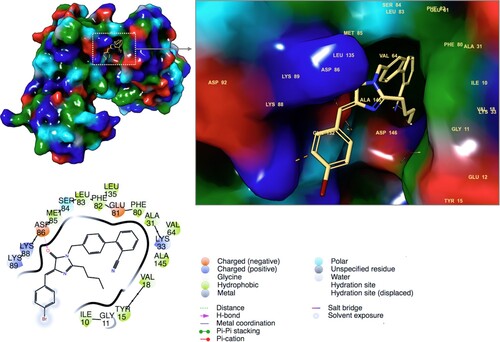
To confirm that 19D is also targeted the CDC2 kinase protein, molecular docking studies were performed. Ligands and target protein are prepared using Maestro molecular modeling package. For this aim, LigPrep and Protein Preparation modules were used, respectively. OPLS3 force field was used for geometry optimization of ligand and refinement of atom positions of target protein. LigPrep generates accurate, energy minimized 3D molecular ligand structures. It also applies sophisticated rules to correct Lewis structures and eliminate mistakes in ligands in order to reduce downstream computational errors. Even high-resolution crystallographic target protein structures may need proper preparation, such as correction of missing hydrogen atoms, incomplete side chains and loops, ambiguous protonation states of the residues and flipped residues. Thus, the protein preparation module of Maestro was used for this aim. Docking score of the 19D at the binding pocket of the target protein was calculated as −5.40 kcal/mol. Figure represented 2D and 3D ligand interactions diagram of the 19D at the binding site. Compound is able to form strong interactions with the target protein. While Asp86 constructs a hydrogen bond with the imidazolone ring of the ligand, most of the other crucial contacts were hydrophobic nonpolar interactions (Figure ). Three positive charged Lys residues at the position of 33, 88 and 89 and two negative charged residues Glu81, and Asp86 are crucial for polar interactions. Nonpolar interactions were formed with the following residues: Ile10, Tyr15, Val18, Ala31, Val64, Phe80, Phe82, Leu83, Met85 and Ala145. Various imidazolone derivative and CDC2 protein interactions were also highlighted for the known inhibitors in previous studies (Raffa et al. Citation2009). In the study of Raffa et al. purvanolol A is used as a positive control. We also docked purvanolol A using same docking protocol with 19D and it was found that positive control has a similar docking score (−5.61 kcal/mol) with 19D. Its 2D ligand interaction diagram shows similar residue contacts (Figure S1). Main contacts were formed via hydrophobic interactions such as Ile10, Tyr15, Val18, Ala31, Val64, Phe80, Phe82, Leu83, Leu135 and Ala145. Lys33, Asp146 and Leu83 (from its backbone) construct hydrogen bonds with the positive control molecule (Supplementary Figure S1).
Figure 6. 2D and 3D ligand interactions diagram of 19D at the binding pocket of CDC2 (CDK1).
Discussion
Despite the recent advances in cancer drug development, the major treatment strategy used still targets the cell proliferation potential of cancer cells. Since cancer cells have unique characteristics in rapid cell division and proliferation, targeting proliferation-associated molecules in different cellular pathways and inducing cell death are considered the ultimate goals in anti-cancer drug discovery (Evan and Vousden Citation2001). Angiotensin II and its receptor type I (AT1R) are the main effector peptides of the renin angiotensin system and they have a role in a variety of processes including cell proliferation in various tissue types including cancer cells (Ray et al. Citation1991; Bataller et al. Citation2000; Fujimoto et al. Citation2001; Du et al. Citation2012). Therefore, AT1R antagonists have recently been recognized as potential molecules to be used against different types of cancer (Deshayes and Nahmias Citation2005; Kosaka et al. Citation2007; Chen et al. Citation2013).
In the present study, an imidazolone derivate, a small molecule (compound 19D), which causes G2/M phase arrest in cell division was discovered. 19D significantly reduced cell proliferation and accelerated apoptotic cell death in a concentration-dependent manner in all tested cell lines. Further, 19D triggered the cancer cells’ deterioration by triggering nuclear aberrations. It is worth to notice that 19D molecule triggered CDC2 checkpoint marker reduction and cell cycle checkpoint failure. Depending on the cell type, the maximum cell proliferation inhibition was achieved within the rapidly dividing cells tested like HUVEC and HCT compared to U-87 MG cells.
Previous reports of AT1R antagonists provided both in vitro and in vivo efficacy of inhibitors for cancer treatment. Koyama et al. (Citation2014) showed that among the different sartan family of AT1R blockers, only telmisartan demonstrated cell proliferation blockage and apoptosis induction, whereas irbesartan and valsartan did not possess the same activity. They also showed the in vivo tumor weight loss in human endometrial cancers (Koyama et al. Citation2014). Kozako et al. (Citation2016) further confirmed the results in T cell leukemia by showing in vitro efficacy of telmisartan but not the other sartan family molecules (Kozako et al. Citation2016). Nevertheless, Liu et al. (Citation2005) showed that losartan, another sartan family molecule, caused apoptotic induction and cell proliferation blocking activity on human pancreatic cells (Liu et al. Citation2005). Previous results suggest that telmisartan- or losartan-like AT1R blockers might be a new therapeutic option for the treatment of different cancers (Fujihara et al. Citation2017). Our results also support the same idea. Compound 19D might be a promising alternative for the treatment of various cancer types including colon cancer, breast cancer, glial tumors andothers.
As an alternative AT1R blocker, 19D compound may be considered a more potent option in comparison to the other sartan family antagonists, due to (i) being a small molecule, (ii) being an imidazolone derivative and (iii) for its specific inhibition on G2/M phase of the cell cycle. Firstly, small-molecule inhibitors are organic molecules with low molecular weight. Their small size makes their use in vivo even more practical, and possibly more cost-efficient, compared to small peptides or antibody-based inhibitors (Azmi and Mohammad Citation2009). The common fact about small molecule inhibitors is that the smaller the molecule, the more soluble, cost-efficient and diffusive it is to be used by systemic delivery. Especially for the brain tumors, small-molecule inhibitors are more promising due to their potential passage across the blood-brain barrier. To date, several small-molecule inhibitors against AT1R were developed and tested (Mavromoustakos et al. Citation2013; Smith, Woodruff, et al. Citation2013; Smith, Wyse, et al. Citation2013). To our knowledge, none of them showed anti-cancer activity or they may have not been tested – this excludes the sartan family molecules. Therefore, 19D is a novel small molecule that shows anti-cancer activity by targeting AT1R. It can easily be modified to increase the binding affinity and tested in further cancer types with AT1R expression such as brain tumors. Secondly, the form of 19D as an imidazolone derivative makes it more flexible to be modified according to the desirable soluble form, low-cost production and purification, high yield and ease of workup (Fozooni et al. Citation2017). Therefore, we can speculate that 19D molecule can easily be modified to increase its solubility in DMSO or other solvents. Further, by increasing the solubility, the minimum inhibitory concentration (IC50) can be decreased down to a nanomolar level with suitable solvents. Finally, as a G2/M phase blocker, 19D molecule has several advantages. Compared to G1 phase blockers, G2/M blockers are more selective and more efficient in anti-cancer activity (Kastan and Bartek Citation2004). G1/S checkpoint is an important checkpoint, triggered by the Chk2-p53-MDM2-p21 pathway in non-cancer cells. Yet, most cancer cells have a deficient or ineffectively working G1 checkpoint due to mutations in the tumor suppressor genes or an imbalance in Cdks and cyclins. Thus, in many cancer cells, the most important checkpoint in response to DNA damage is the G2/M checkpoint which is associated with the CHK1-CDC25 phosphatase pathway (Swift and Golsteyn Citation2014). Compound 19D can be considered as a more potent and more effective cell cycle inhibitor as it specifically causes G2/M phase arrest phase via a reduction in the CDC2 checkpoint marker.
As a conclusion; controlling the cell division in different cancers at a specific phase of the cell cycle is considered as a main strategy for anti-cancer activity of a novel molecule. 19D compound as an AT1R antagonist has great potential in targeting cancer as its effect in arresting G2/M phase was as strong as the currently used chemotherapeutic agent, Paclitaxel. Moreover, 19D directs the arrested cells into apoptotic cell death. Nevertheless, there are some limitations of this molecule that requires further studies. For example, in vivo activity of 19D compound is not known yet. The effects on liver and kidney should be extensively tested on in vivo tumor models. Further, the stability of the molecule should be improved and inhibition concentration should be decreased for safer and more efficient anti-cancer activity. In subsequent studies, we aim to modify the chemical structure of 19D compound to increase its binding activity and decrease its inhibitory concentrations. Further, we will also observe in vivo efficacy of the 19D compound in cancer models in order to provide a more solid understanding of its activity against cancer.
Acknowledgments
The authors have no personal, financial or institutional interest in any of the methods, materials, or devices described in this article. All authors state that the manuscript has been read and approved by all the authors, and each author believes that the manuscript represents honest work not published in any form and not submitted to any other journal.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data Availability
All necessary experimental raw data including cell proliferation assays, cell cycle inhibition assays, cell images and western blot assays were uploaded to Zenodo Database via link below: https://doi.org/10.5281/zenodo.4748912
Additional information
Funding
References
- Azmi AS, Mohammad RM. 2009. Non-peptidic small molecule inhibitors against Bcl-2 for cancer therapy. J Cell Physiol. 218(1):13–21.
- Barreca M, Spanò V, Montalbano A, Cueto M, Díaz marrero AR, Deniz I, Erdoğan A, Lukić Bilela L, Moulin C, Taffin-de-givenchy E, et al. 2020. Marine anticancer agents: an overview with a particular focus on their chemical classes. Mar Drugs. 18(12):619.
- Bataller R, Ginès P, Nicolás JM, Görbig MN, Garcia-ramallo E, Gasull X, Bosch J, Arroyo V, Rodés J. 2000. Angiotensin II induces contraction and proliferation of human hepatic stellate cells. Gastroenterology. 118(6):1149–1156.
- Bauer JH, Reams GP. 1995. The angiotensin II type 1 receptor antagonists: a new class of antihypertensive drugs. Arch Intern Med. 155(13):1361–1368.
- Chen X, Meng Q, Zhao Y, Liu M, Li D, Yang Y, Sun L, Sui G, Cai L, Dong X. 2013. Angiotensin II type 1 receptor antagonists inhibit cell proliferation and angiogenesis in breast cancer. Cancer Lett. 328(2):318–324.
- Debatin K-M. 2000. Activation of apoptosis pathways by anticancer treatment. Toxicol Lett. 112-113:41–48.
- De Paepe B, Verstraeten VM, De Potter CR, Bullock GR. 2002. Increased angiotensin II type-2 receptor density in hyperplasia, DCIS and invasive carcinoma of the breast is paralleled with increased iNOS expression. Histochem Cell Biol. 117(1):13–19.
- Deshayes F, Nahmias C. 2005. Angiotensin receptors: a new role in cancer? Trends Endocrinol Metab. 16(7):293–299.
- Du N, Feng J, Hu LJ, Sun X, Sun HB, Zhao Y, Yang YP, Ren H. 2012. Angiotensin II receptor type 1 blockers suppress the cell proliferation effects of angiotensin II in breast cancer cells by inhibiting AT1R signaling. Oncol Rep. 27:1893–1903.
- Durdagi S, Aksoydan B, Erol I, Kantarcioglu I, Ergun Y, Bulut G, Acar M, Avsar T, Liapakis G, Karageorgos V, et al. 2018. Integration of multi-scale molecular modeling approaches with experiments for the in silico guided design and discovery of novel hERG-neutral antihypertensive oxazalone and imidazolone derivatives and analysis of their potential restrictive effects on cell proliferation. Eur J Med Chem. 145:273–290.
- Evan GI, Vousden KH. 2001. Proliferation, cell cycle and apoptosis in cancer. Nature. 411(6835):342–348.
- Fogarty DJ, Sánchez-gómez MV, Matute C. 2002. Multiple angiotensin receptor subtypes in normal and tumor astrocytes in vitro. Glia. 39(3):304–313.
- Fozooni S, Khoshdast H, Hassani H, Hamidian H. 2017. Synthesis of oxazolone and imidazolone derivatives in presence of H2O2 promoted fly ash as a novel and efficient catalyst. J Sci Islamic Repub Iran. 28:221–230.
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. 2006. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 49(21):6177–6196.
- Fujihara S, Morishita A, Ogawa K, Tadokoro T, Chiyo T, Kato K, Kobara H, Mori H, Iwama H, Masaki T. 2017. The angiotensin II type 1 receptor antagonist telmisartan inhibits cell proliferation and tumor growth of esophageal adenocarcinoma via the AMPKα/mTOR pathway in vitro and in vivo. Oncotarget. 8(5):8536–8549.
- Fujimoto Y, Sasaki T, Tsuchida A, Chayama K. 2001. Angiotensin II type 1 receptor expression in human pancreatic cancer and growth inhibition by angiotensin II type 1 receptor antagonist. FEBS Lett. 495(3):197–200.
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell. 144(5):646–674.
- Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL. 2016. OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J Chem Theory Comput. 12(1):281–296.
- Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, Friesner RA. 2004. A hierarchical approach to all-atom protein loop prediction. Proteins Struct Funct Bioinf. 55(2):351–367.
- Jordan MA, Wendell K, Gardiner S, Derry WB, Copp H, Wilson L. 1996. Mitotic block induced in HeLa cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res. 56:816–825.
- Juillerat-Jeanneret L, Celerier J, Chapuis bernasconi C, Nguyen G, Wostl W, Maerki HP, Janzer RC, Corvol P, Gasc JM. 2004. Renin and angiotensinogen expression and functions in growth and apoptosis of human glioblastoma. Br J Cancer. 90(5):1059–1068.
- Kastan MB, Bartek J. 2004. Cell-cycle checkpoints and cancer. Nature. 432(7015):316–323.
- Kosaka T, Miyajima A, Takayama E, Kikuchi E, Nakashima J, Ohigashi T, Asano T, Sakamoto M, Okita H, Murai M, Hayakawa M. 2007. Angiotensin II type 1 receptor antagonist as an angiogenic inhibitor in prostate cancer. Prostate. 67(1):41–49.
- Koyama N, Nishida Y, Ishii T, Yoshida T, Furukawa Y, Narahara H. 2014. Telmisartan induces growth inhibition, DNA double-strand breaks and apoptosis in human endometrial cancer cells. PLoS One. 9(3):e93050.
- Kozako T, Soeda S, Yoshimitsu M, Arima N, Kuroki A, Hirata S, Tanaka H, Imakyure O, Tone N, Honda S, et al. 2016. Angiotensin II type 1 receptor blocker telmisartan induces apoptosis and autophagy in adult T-cell leukemia cells. FEBS Open Bio. 6(5):442–460.
- Li Petri G, Spanò V, Spatola R, Holl R, Raimondi MV, Barraja P, Montalbano A. 2020. Bioactive pyrrole-based compounds with target selectivity. Eur J Med Chem. 208:112783–112783.
- Liu W-B, Wang X-P, Wu K, Zhang R-L. 2005. Effects of angiotensin II receptor antagonist, losartan on the apoptosis, proliferation and migration of the human pancreatic stellate cells. World J Gastroenterol. 11(41):6489–6494.
- Mavromoustakos T, Agelis G, Durdagi S. 2013. AT1 antagonists: a patent review (2008–2012). Expert Opin Ther Pat. 23(11):1483–1494.
- Raffa D, Maggio B, Cascioferro S, Raimondi MV, Daidone G, Plescia S, Schillaci D, Cusimano MG, Titone L, Colomba C. 2009. N-(indazolyl) benzamido derivatives as CDK1 inhibitors: design, synthesis, biological activity, and molecular docking studies. Arch Pharm. 342(5):265–273.
- Ray PE, Aguilera G, Kopp JB, Horikoshi S, Klotman PE. 1991. Angiotensin II receptor-mediated proliferation of cultured human fetal mesangial cells. Kidney Int. 40(4):764–771.
- Sahin Z, Biltekin SN, Yurttas L, Berk B, Özhan Y, Sipahi H, Gao ZG, Jacobson KA, Demirayak Ş. 2021. Novel cyanothiouracil and cyanothiocytosine derivatives as concentration-dependent selective inhibitors of U87MG glioblastomas: adenosine receptor binding and potent PDE4 inhibition. Eur J Med Chem. 212:113125.
- Sancar A, Lindsey-boltz LA, Unsal-kaçmaz K, Linn S. 2004. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 73(1):39–85.
- Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M. 2007. Epik: a software program for pK(a) prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des. 21(12):681–691.
- Smith MT, Woodruff TM, Wyse BD, Muralidharan A, Walther T. 2013. A small molecule angiotensin II type 2 receptor (AT2R) antagonist produces analgesia in a rat model of neuropathic pain by inhibition of p38 mitogen-activated protein kinase (MAPK) and p44/p42 MAPK activation in the dorsal root ganglia. Pain Med. 14(10):1557–1568.
- Smith MT, Wyse BD, Edwards SR. 2013. Small molecule angiotensin II type 2 receptor (AT2R) antagonists as novel analgesics for neuropathic pain: comparative pharmacokinetics, radioligand binding, and efficacy in rats. Pain Med. 14(5):692–705.
- Spanò V, Frasson I, Giallombardo D, Doria F, Parrino B, Carbone A, Montalbano A, Nadai M, Diana P, Cirrincione G, et al. 2016. Synthesis and antiproliferative mechanism of action of pyrrolo[3',2':6,7] cyclohepta[1,2-d]pyrimidin-2-amines as singlet oxygen photosensitizers. Eur J Med Chem. 123:447–461.
- Spanò V, Pennati M, Parrino B, Carbone A, Montalbano A, Cilibrasi V, Zuco V, Lopergolo A, Cominetti D, Diana P, et al. 2016. Preclinical activity of New [1,2]oxazolo[5,4-e]isoindole derivatives in diffuse Malignant peritoneal mesothelioma. J Med Chem. 59(15):7223–7238.
- Spanò V, Rocca R, Barreca M, Giallombardo D, Montalbano A, Carbone A, Raimondi MV, Gaudio E, Bortolozzi R, Bai R, et al. 2020. Pyrrolo[2',3':3,4]cyclohepta[1,2-d][1,2]oxazoles, a new class of antimitotic agents active against multiple malignant cell types. J Med Chem. 63(20):12023–12042.
- Swift LH, Golsteyn RM. 2014. Genotoxic anti-cancer agents and their relationship to DNA damage, mitosis, and checkpoint adaptation in proliferating cancer cells. Int J Mol Sci. 15(3):3403–3431.
- Weaver BA. 2014. How taxol/paclitaxel kills cancer cells. Mol Biol Cell. 25(18):2677–2681.
- Zhang J, Yang PL, Gray NS. 2009. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 9(1):28–39.