
Abstract
Interstitial lung disease (ILD) presents as progressive scarring of lung tissue, which may affect respiratory function and restrict oxygen-exchange into the bloodstream, finally leading to dyspnea, dry cough and decreased quality of life. Increasing evidence indicates that genetic factors may be crucial in the development of ILD. At present, the main pathogenic genes of ILD include genes vital for surfactant metabolism and function and telomere biology associated genes. Mutations in both sets of genes may induce the apoptosis of alveolar epithelial cells and finally result in remodeling of the lung interstitium. In this study, one Han Chinese family with interstitial pneumonia and lung cancer was recruited. The proband was selected for whole exome sequencing analysis. After data filtering, only a novel mutation (c.404G > A /p.G135E) of SFTPA2 (NM_001098668) was identified. No other meaningful mutations were detected. The mutation co-segregated with the affected patients in the pedigree and was absent in the healthy controls. Bioinformatics analysis predicted that this variant is pathogenic and located in an evolutionarily conserved site of the SP-A2 protein. This study further expands the variant spectrum of the SFTPA2 gene and may be helpful in the genetic counseling of interstitial pneumonia and lung cancer patients.
Introduction
Interstitial lung disease (ILD) is a series of disorders that may present as progressive scarring of lung tissue (Antoniou et al. Citation2014; Cottin et al. Citation2018), such as idiopathic pulmonary fibrosis, interstitial lung pneumonia, hypersensitivity pneumonitis, sarcoidosis and asbestosis. The primary signs and symptoms of ILD include shortness of breath and increasing cough caused by impaired gas-exchange (Azadeh et al. Citation2018). The prevalence of ILD is estimated to be 5–60 in 100,000 people and the average survival time for some ILD patients (idiopathic pulmonary fibrosis) is only 3–5 years after diagnosis (Antoniou et al. Citation2014; Cottin et al. Citation2018).
Recently, many studies have suggested that genetic factors may play a crucial role in the occurrence and development of ILD (Devine and Garcia Citation2012; du Bois Citation2002; Newton et al. Citation2018; van Moorsel et al. Citation2015). Several investigations have revealed that up to 20% of people with ILD have a family history of ILD or lung cancer (Antoniou et al. Citation2014; Newton et al. Citation2018). At present, at least two groups of genes have been identified as ILD disease-causing genes (Newton et al. Citation2018; Wolters et al. Citation2014): genes participating in surfactant metabolism and function, including surfactant protein C (SFTPC), surfactant protein A2 (SFTPA2), surfactant protein A1 (SFTPA1) and ATP-binding cassette transporter A3 (ABCA3); and telomere related genes, such as telomerase reverse transcriptase (TERT), telomerase RNA component (TERC), poly(A)-specific ribonuclease (PARN) and regulator of telomere elongation helicase 1 (RTEL1). Mutations in dyskerin pseudouridine synthase 1 (DKC1), and filamin A (FLNA) have also been found in ILD patients (Antoniou et al. Citation2014; Cottin et al. Citation2018).
In this study, a novel heterozygous mutation (NM_001098668: c.404G > A /p.G135E) of SFTPA2 was identified via employing whole exome sequencing (WES) and Sanger sequencing in a Han Chinese family with interstitial pneumonia and lung cancer. This mutation may have critical implications for genetic monitoring.
Material and methods
Subjects
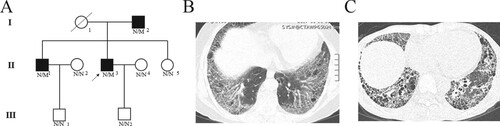
In total of six family members (I-2, II-1, II-3, II-5, III-1 and III-2) participated in this study (Figure (a)). Blood was collected from the proband and their family members and all the subjects accepted test for pulmonary function and HRCT. Simultaneously, 200 unrelated local healthy people were also enrolled to serve as normal controls (Liu and Luo Citation2018). This study was approved by the Ethics Committee of the Second Xiangya Hospital of Central South University and performed in accordance with the principles enshrined in the Declaration of Helsinki. Written informed consent was obtained from each participant.
Figure 1. Clinical features of the family. (a) Pedigree figure. Family members are identified by generations and numbers. Squares indicate male family members; circles, female members; closed symbols, the affected members; open symbols, unaffected members; arrow, proband, N, normal, M, mutation. HRTC testing of II-3 (b) and II-1(c).
Whole exome sequencing
Genomic DNA was extracted from peripheral blood lymphocytes of all the participants with DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA). We selected the proband’s DNA to perform the whole exome sequencing. Whole exome sequencing services were provided by the Novogene Bioinformatics Institute (Beijing, China). Exomes were captured by Agilent SureSelect Human All Exon V6 kits, and next-generation sequencing was conducted with an Illumina HiSeq X-10 system.
The strategies of data filtering are as follows: (a) variants in intergenic, intronic, and UTR regions as well as synonymous variants were excluded; (b) variants with MAF >0.01 in the NHLBI Exome Sequencing Project Exome Variant Server, 1000 Genomes project, dbSNP147 and the ExAC database were excluded; (c) variants with MAF<0.05 in in-house exome databases of Novogene (2500 exomes) and genomAD database were remained; (d) SIFT, Polyphen-2 and MutationTaster were applied to predict the effects of variants. (e) Co-segregation analysis was conducted in the family.
Variant validation and co-segregation analysis
Sanger sequencing was applied to validate the candidate variants found in whole exome sequencing. Co-segregation analysis was performed on each family members. The candidate variants were also examined in 200 healthy adults of both sexes, who were enrolled and used as an internal control for genetic variants potentially specific for the Han Chinese.
Results
Case presentation
The family comes from the central southern region of China (Jiangxi province) (Figure (a)). The proband (II-3), a 50-year-old man, suffered from repeated cough for eight years and dyspnea for one year without smoking history. Pulmonary function indicated mild obstructive pulmonary ventilation dysfunction and severely damaged diffuse capacity. HRCT showed ground-glass opacity with honeycombing in the subpleural, and basal lungs, which was in accordance with the usual interstitial pneumonia (UIP) pattern and suggested idiopathic pulmonary fibrosis (Table , Figure (b)). A family history investigation demonstrated that his brother (II-1) also showed similar symptoms five years ago. HRCT of his brother (II-1) showed a reticular pattern with honeycombing in the subpleural and basal lungs with traction bronchiectasis (Figure (c)). Patient II-1 also presented a weakly positive SSA antibody and strong positive Ro-52 profile but showed no symptoms of rheumatic diseases. After the consultation of two rheumatologists the patient was finally diagnosed with IPF. In addition, the proband’s father (I-2) was diagnosed with non-small cell lung cancer two years ago (76-years-old) but refused medical testing due to being far away from our hospital. We could only visit to collect blood samples after informed consent was obtained. Other clinical data is shown in Table .
Table 1. The clinical data of the family with ILD.
Genetic analysis
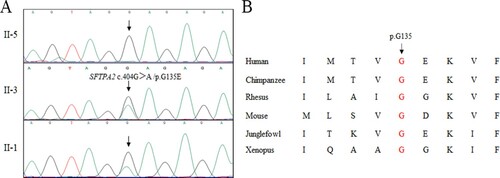
The gDNA of II-3 was selected for whole exome sequencing. The mean coverage was 99.6%, and the average sequencing depth was 90.10×. In total, 14530 SNPs and indels were detected in the patient (II-3). According to the above filtering strategy, a novel heterozygous variant (c.404G>A /p.G135E) in exon six of SFTPA2 (NM_001098668) was detected. No other meaningful variants were found.
Sanger sequencing validated this mutation (Figure (a)) and confirmed that this mutation also existed in the affected patients (I-2, II-1 and II-3) but was absent in our 200 control individuals. Moreover, the family members II-5, III-1 and III-2 benefited from pre-symptomatic diagnosis in the context of the familial mutation and they were found to be non-carriers. Bioinformatics programs predicted the pathogenesis of this novel variant(c.404G>A /p.G135E), which is located in an evolutionarily conserved site of the SP-A2 protein (Figure (b)). According to ACMG guidelines (Richards et al. Citation2015), this mutation was located in a hot spot and well-established functional domain (PM1, PM: pathogenic moderate), was absent from controls (PM2), co-segregated with affected family members (PP1, PP: pathogenic supporting) and predicted to be deleterious by different programs (SIFT, Polyphen2 and mutation taster) (PP3). Hence, this mutation is likely pathogenic.
Figure 2. Genetic analysis of the family (a) Sequencing results of the SFTPA2 mutation. Sequence chromatogram indicates a G to A transition of nucleotide 404. (b) Conservation analysis of the alanine residue at position 135 (p.G135).
Discussion
The human SFTPA2 gene encoding surfactant protein A2 protein is located on chromosome 10q22.3, and consists of six exons, spanning approximately 4.5 kilobases (kb). Previous reports found that SP-A2, a hydrophilic calcium-dependent lectin, played a crucial role in pulmonary innate immunity (Heinrich et al. Citation2006). The expression of SFTPA2 is restricted to type II alveolar epithelial cells and club cells of the lung (Madsen et al. Citation2003). These cell types are essential for the pathogenesis of lung cancer and ILD (Guillot et al. Citation2013). In this study, we identified a novel mutation (c.404G>A /p.G135E) in a Han-Chinese family with interstitial pneumonia and lung cancer. Our study is consistent with previous studies showing that pathogenic variations in SFTPA2 may lead to interstitial pneumonia and lung cancer.
In 2009, two heterozygous missense mutations of the SFTPA2 gene were identified in two families with idiopathic pulmonary fibrosis and lung cancer (Wang et al. Citation2009). Since then, an increasing number of SFTPA2 variants have been detected in familial and sporadic interstitial lung diseases including interstitial pneumonia and lung cancer (Legendre et al. Citation2020; Liu et al. Citation2020; van Moorsel et al. Citation2015). At present, 15 mutations of SFTPA2 have been identified in different types of lung disease including idiopathic pulmonary fibrosis and interstitial pneumonia and lung cancer (Coghlan et al. Citation2014; Legendre et al. Citation2020; Liu et al. Citation2020; van Moorsel et al. Citation2015; Wang et al. Citation2009; Yang et al. Citation2014). Previous studies also showed a poor genotype-phenotype correlation in ILD patients, and the same mutation in SFTPA2 can be found in different types of lung disease. For example, the mutation p.F198S of SFTPA2 was detected in patients with lung cancer, IPF and undefined lung disease (Song et al. Citation2012; van Moorsel et al. Citation2015; Wang et al. Citation2009; Yang, Li, Wang, Xu, Zhao, Ma, Li and Chen Citation2014). In this study, we identified a novel mutation (c.404G>A /p.G135E) of SFTPA2 in patients with interstitial pneumonia and lung cancer. Our study expanded the spectrum of SFTPA2 mutations and provided more insights into the SFTPA2 gene.
SP-A2 can bind, aggregate, opsonize, and permeabilize microorganisms (McCormack and Whitsett Citation2002). The binding of pathogens and lipids to SP-A2 is mediated by the highly conserved carbohydrate binding domain (CRD, 134aa-248aa) (Kishore et al. Citation2006). The second amino acid in the CRD, the functional domain of the protein, is highly conserved. To date, all the described mutations have been located in this domain. Previous studies have demonstrated that mutations in the CRD may lead to abnormal protein formation (Wang et al. Citation2009). Aberrant protein accumulation in the cytoplasm and endoplasmic reticulum (ER) can cause ER stress, induce the activation of the unfolded protein response and lead to type II alveolar epithelial cell apoptosis in cases of long-standing or severe activation (Maitra et al. Citation2010). Meanwhile, mutations of surfactant related genes may also lead to the accumulation of surfactant in pulmonary alveolus, which may induce the auto-immune pulmonary alveolar proteinosis, one rare pulmonary disease with anti- granulocyte macrophage-colony stimulating factor antibodies blocking activation of alveolar macrophages (Borie et al. Citation2011). In our study, the mutation (c.404G>A /p.G135E) is located in the CRD domain of SP-A2. We speculated that the mutation may disrupt the stability of the SP-A2 protein and finally induce interstitial pneumonia and lung cancer under the influence of microorganisms.
A mouse model of pneumonia with microorganisms, such as Klebsiella pneumoniae, Streptococcus pneumoniae, Pseudomonas aeruginosa, and others, has revealed that SP-A-/- (ortholog of SFTPA2 in mice) mice show increased vulnerability to infection and injury (Korfhagen et al. Citation1998; Mikerov et al. Citation2008). Bronchoalveolar lavage results showed that the baseline levels of host defense molecules in knockout mice were different from those in WT mice (Ali et al. Citation2010). SP-A can promote alveolar macrophage competence, resulting in a successful innate host defense response to various injurious agents (Phelps et al. Citation2011), which provides a new therapeutic target for patients with ILD. Simultaneously, our study also revealed that genetic testing for damaging mutations in surfactant protein-related genes, telomere-related genes and other ILD-susceptibility genes is strongly recommended for people with a family history of ILD (Newton et al. Citation2018).
In short, we identified a novel heterozygous variant (c.404G>A /p.G135E) of SFTPA2 in a Han-Chinese families with interstitial pneumonia and lung cancer via whole exome sequencing. Our study not only expands the spectrum of SFTPA2 mutations but helps the family members to mitigate ILD risk factors. The study also supplements and improves genetic testing strategies and ILD risk estimation methodologies for China.
Acknowledgments
We thank all subjects for participating in this study. We thank Dr. Shuai Guo from University of Texas MD Anderson Cancer Center, the USA for editing the language.
Consent for publication statement
Written informed consent was obtained from the proband and family members for the publication of any potentially identifiable images or data included in this article.
Data availability statement
Due to the nature of this research, participants of this study did not agree for their data to be shared publicly, so supporting data is not available.
Disclosure statement
The authors declared they have no conflicts of interest.
Additional information
Funding
References
- Ali M, Umstead TM, Haque R, Mikerov AN, Freeman WM, Floros J, Phelps DS. 2010. Differences in the BAL proteome after Klebsiella pneumoniae infection in wild type and SP-A-/- mice. Proteome Sci. 17(8):34.
- Antoniou KM, Margaritopoulos GA, Tomassetti S, Bonella F, Costabel U, Poletti V. 2014. Interstitial lung disease. Eur Respir Rev. 1(23):40–54.
- Azadeh N, Moua T, Baqir M, Ryu JH. 2018. Treatment of acute exacerbations of interstitial lung disease. Expert Rev Respir Med. 12:309–313.
- Borie R, Danel C, Debray MP, Taille C, Dombret MC, Aubier M, Epaud R, Crestani B. 2011. Pulmonary alveolar proteinosis. Eur Respir Rev. 20:98–107.
- Coghlan MA, Shifren A, Huang HJ, Russell TD, Mitra RD, Zhang Q, Wegner DJ, Cole FS, Hamvas A. 2014. Sequencing of idiopathic pulmonary fibrosis-related genes reveals independent single gene associations. BMJ Open Respir Res. 1:e000057.
- Cottin V, Hirani NA, Hotchkin DL, Nambiar AM, Ogura T, Otaola M, Skowasch D, Park JS, Poonyagariyagorn HK, Wuyts W, et al. 2018. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 31:27.
- Devine MS, Garcia CK. 2012. Genetic interstitial lung disease. Clin Chest Med. 33:95–110.
- du Bois RM. 2002. The genetic predisposition to interstitial lung disease: functional relevance. Chest. 121:14S–20S.
- Guillot L, Nathan N, Tabary O, Thouvenin G, Le Rouzic P, Corvol H, Amselem S, Clement A. 2013. Alveolar epithelial cells: master regulators of lung homeostasis. Int J Biochem Cell Biol. 45:2568–2573.
- Heinrich S, Hartl D, Griese M. 2006. Surfactant protein A–from genes to human lung diseases. Curr Med Chem. 13:3239–3252.
- Kishore U, Greenhough TJ, Waters P, Shrive AK, Ghai R, Kamran MF, Bernal AL, Reid KB, Madan T, Chakraborty T. 2006. Surfactant proteins SP-A and SP-D: structure, function and receptors. Mol Immunol. 43:1293–1315.
- Korfhagen TR, LeVine AM, Whitsett JA. 1998. Surfactant protein A (SP-A) gene targeted mice. Biochim Biophys Acta. 19(1408):296–302.
- Legendre M, Butt A, Borie R, Debray MP, Bouvry D, Filhol-Blin E, Desroziers T, Nau V, Copin B, Dastot-Le Moal F, et al. 2020. Functional assessment and phenotypic heterogeneity of SFTPA1 and SFTPA2 mutations in interstitial lung diseases and lung cancer. Eur Respir J. 56(6):2002806.
- Liu L, Luo H. 2018. Whole-Exome sequencing identified a novel compound heterozygous mutation of LRRC6 in a Chinese primary ciliary dyskinesia patient. Biomed Res Int. 2018:1854269.
- Liu L, Qin J, Guo T, Chen P, Ouyang R, Peng H, Luo H. 2020. Identification and functional characterization of a novel surfactant protein A2 mutation (p.N207Y) in a Chinese family with idiopathic pulmonary fibrosis. Mol Genet Genomic Med. 8:e1393.
- Madsen J, Tornoe I, Nielsen O, Koch C, Steinhilber W, Holmskov U. 2003. Expression and localization of lung surfactant protein A in human tissues. Am J Respir Cell Mol Biol 29:591–597.
- Maitra M, Wang Y, Gerard RD, Mendelson CR, Garcia CK. 2010. Surfactant protein A2 mutations associated with pulmonary fibrosis lead to protein instability and endoplasmic reticulum stress. J Biol Chem. 16(285):22103–22113.
- McCormack FX, Whitsett JA. 2002. The pulmonary collectins, SP-A and SP-D, orchestrate innate immunity in the lung. J Clin Invest. 109:707–712.
- Mikerov AN, Haque R, Gan X, Guo X, Phelps DS, Floros J. 2008. Ablation of SP-A has a negative impact on the susceptibility of mice to Klebsiella pneumoniae infection after ozone exposure: sex differences. Respir Res. 4(9):77.
- Newton CA, Molyneaux PL, Oldham JM. 2018. Clinical Genetics in interstitial lung disease. Front Med (Lausanne). 5:116.
- Phelps DS, Umstead TM, Quintero OA, Yengo CM, Floros J. 2011. In vivo rescue of alveolar macrophages from SP-A knockout mice with exogenous SP-A nearly restores a wild type intracellular proteome; actin involvement. Proteome Sci. 28(9):67.
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 17:405–424.
- Song Y, Fang G, Shen H, Li H, Yang W, Pan B, Huang G, Lin G, Ma L, Willard B, et al. 2012. Human surfactant protein A2 gene mutations impair dimmer/trimer assembly leading to deficiency in protein sialylation and secretion. PLoS One. 7:e46559.
- van Moorsel CH, Ten Klooster L, van Oosterhout MF, de Jong PA, Adams H, van Es H W, Ruven HJ, van der Vis JJ, Grutters JC. 2015. SFTPA2 mutations in familial and sporadic idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 15(192):1249–1252.
- Wang Y, Kuan PJ, Xing C, Cronkhite JT, Torres F, Rosenblatt RL, DiMaio JM, Kinch LN, Grishin NV, Garcia CK. 2009. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 84:52–59.
- Wolters PJ, Collard HR, Jones KD. 2014. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol. 9:157–179.
- Yang HY, Li H, Wang YG, Xu CY, Zhao YL, Ma XG, Li XW, Chen H. 2014. Correlation analysis between single nucleotide polymorphisms of pulmonary surfactant protein A gene and pulmonary tuberculosis in the Han population in China. Int J Infect Dis. 26:31–36.