
Abstract
BMPR2 encodes the bone morphogenetic protein receptor type 2. Most of heritable pulmonary arterial hypertension is caused by mutations of BMPR2. Pulmonary arterial hypertension is characterized by increased pulmonary vascular resistance and sustained elevation of mean pulmonary arterial pressure. Here we sought to identify novel mutations in a family with pulmonary arterial hypertension. Whole-exome sequencing obtained variants data from the patient's blood Genomic DNA who was diagnosed with pulmonary arterial hypertension. Sanger sequencing was used to confirm potential causative variants in the patient. A novel frame-shift mutation in BMPR2 (NM_001204:c.453dupA, p.I152Nfs*29) was identified in the patient with pulmonary arterial hypertension, she also had subclinical hypothyroidism and hyperuricemia. This frame-shift mutation will cause the BMPR2 protein loss of function, leading to the obstruction of BMPs signaling pathway, further affect the growth, proliferation and differentiation of vascular cells. Our study expands the spectrum of BMPR2 mutations and enriches the clinical features.
Introduction
Pulmonary arterial hypertension is a rare dominant disease characterized by elevated pulmonary artery pressure, pulmonary arteriolar hypertrophy and pulmonary vessels stenosis (Gordeuk et al. Citation2016; Dodson et al. Citation2018). The diagnostic criterion for pulmonary arterial hypertension is the patient’s right heart catheterization performed mean pulmonary arterial pressure >25 mmHg at rest (Hayes et al. Citation2014). The symptoms of pulmonary arterial hypertension can occur at all ages and the mean age at diagnosis is 36 years. Common onset symptoms include dyspnea, fatigue and syncope. With the disease worsen, most victims died of right heart failure. According to previous epidemiological survey, the prevalence of pulmonary arterial hypertension is likely to occur in 7 cases per million populations (Peacock et al. Citation2007).
Pulmonary arterial hypertension is an autosomal dominant disease, which mutations of many genes are responsible for it, including bone morphogenetic protein receptor type 2 (BMPR2), activin receptor-like type 1, caveolin 1 (CAV1), endoglin (ENG), potassium two-pore-domain channel subfamily K member 3 (KCNK3), SMAD family member 9 (SMAD9), SMAD family member 4 (SMAD4), T-box 4 (TBX4) and so on, over 75% of heritable pulmonary arterial hypertension cases are caused by mutations of BMPR2, and 20% of idiopathic pulmonary arterial hypertension patients carry a heterozygous BMPR2 mutation (Ma and Chung Citation2017).
In this study, we applied whole-exome sequencing to identify the genetic lesion of a Chinese family with pulmonary arterial hypertension. In combination with bioinformatics analysis and Sanger sequencing validation, a novel mutation (c.453dupA, p.I152Nfs*29) of BMPR2 was identified.
Case presentation
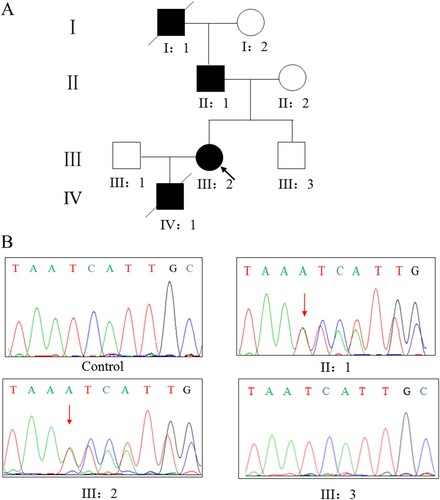
This research was approved by the Review Board of Xiangya Hospital of the Central South University. Written informed consent for publication of their details was obtained from the patient. We identified a pulmonary arterial hypertension family from Hunan province, China (Figure A). The proband was a female patient with pulmonary arterial hypertension without other diseases beforehand. After long time (8 months) chronic dyspnea at age 30, the proband’s condition aggravated and activity tolerance decreased significantly. Therefore she checked in the Department of Cardiology, Xiangya Hospital of the Central South University.
Figure 1. (A) Pedigree of the family affected with pulmonary arterial hypertension. Squares,male family members; circles, female members; closed symbols,affected members; open symbols, unaffected members; arrow, proband. (B) Sanger DNA sequencing chromatogram demonstrating the heterozygosity for a BMPR2 mutation (c.453dupA, p.I152Nfs*29).
Right cardiac catheterization confirmed that the patient had pulmonary hypertension, estimated pulmonary arterial pressure was 68 mm Hg. The patient’s right atrium and right ventricle were significantly enlarged, moderate tricuspid regurgitation, and pulmonary artery widened. The patient had no pulmonary embolism or congenital heart disease. Laboratory examination showed high levels in three indicators include uric acid, N-terminal pro-brain natriuretic peptide, and thyroid-stimulating hormone (Table ). In conclusion, the proband was diagnosed as pulmonary arterial hypertension with cardiac function grade 2–3, subclinical hypothyroidism and hyperuricemia.
Table 1. The clinical data of the patient.
After treatment, condition of the patient was controlled and improved, but two years later, the patient appeared edema of lower limbs and hands for unknown reasons, pain in anterior cardiac area after exercise, the frequency of chest pain increasing and lasting for about half an hour each time, and significantly decreased activity tolerance (climb one floor or a little activity would feel dyspnea). Then she checked again in our hospital. Her family history related to disease as follow: (1) Her grandfather died in 20 years old with a history of repeated edema in both lower limbs; (2) Her father had a history of pulmonary arterial hypertension; (3) Her son suffered from congenital ventricular septal defect, and died of pulmonary arterial hypertension; (4) Others are unaffected.
Genetic analysis
Genomic DNA was obtained from patient’s peripheral blood lymphocytes by DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA, USA). The Novogene Bioinformatics Institute (Beijing, China) performed exome capture, high-throughput sequencing. The proband exome were captured by means of Agilent SureSelect Human All Exon V5 kits and sequenced with the Illumina HiSeq2000 platform. The whole-exome sequencing data was filtered as follow: first, we exclude the variation outside the coding region and the synonym variation, and then include variants with MAF < 1.0% in dbSNP132, 1000 Genomes Project, ExAC, and GnomAD databases; By searching “pulmonary arterial hypertension” in GeneCards (https://www.genecards.org) we obtained a list of genes associated with the pulmonary arterial hypertension, then we selected genes with a score greater than 20 in GeneCards to match with the proband's whole-exome sequencing data; MutationTaster and SIFT bioinformatics programs were used to predict the functional effects of mutations. Sanger sequencing was used to confirm potential causative variants in the patient. Primers used to amplify fragments encompassing individual variants were designed by Integrated DNA Technologies (https://sg.idtdna.com/pages, F: 5′-AAAGGGCAGTCTGTCAGTATTT, R: 5′- ACTATTGAGGCTGGGTGTATTT). The target fragment was amplified by polymerase chain reaction (PCR) and analyzed using the ABI 3100 Genetic Analyzer (ABI, Foster City, CA, USA).
Identification of BMPR2 mutation
Whole-exome sequencing yielded 9.75 Gb of data with 99.6% coverage of the target region and 99.0% of the target covered over 10×. A total of 57, 214 variants were detected in the patient. After filtering the whole-exome sequencing data (Table ), we finally identified a heterozygous mutation (c.453dupA, p.I152Nfs*29) in BMPR2 and further confirmed it by Sanger sequencing (Figure B). The mutation (c.453dupA, p.I152Nfs*29) of BMPR2 was highly suspected to be the genetic lesions of the family. Sanger sequencing showed that the novel mutation in the patient was inherited from her father. And this mutation was not detected in the cohort consisting of 200 controls previously studied by our group.
Table 2. The gene list after whole-exome sequencing data filtration of the patient.
Discussion
BMPR2 plays an important role in heritable pulmonary arterial hypertension pathological processes. To today, hundreds mutations of BMPR2 have been detected (Evans et al. Citation2016). The BMPR2 gene encodes the bone morphogenetic protein receptor type 2, a serine threonine kinase receptor, belonging to TGF-β receptor superfamily and widely expressing in various tissues (Ma and Chung Citation2017). BMPR2 consists of 4 domain include extracellular domain, trans-membrane domain, kinase domain and cytoplasmic tail domain (Machado et al. Citation2001). BMPR2 mediated activation of the BMPs signaling pathway is crucial to control vascular smooth muscle growth and differentiation (Andruska and Spiekerkoetter Citation2018).
We observed that several mutations are associated with cardiomyopathy in Table , so how could further screening be performed? To solve this problem, MAF in GnomAD, MutationTaster and SIFT analysis were used in the Table analysis, and we made a systematic evaluation of each potential gene in Table according to the ACMG evaluation method and obtained corresponding evaluation results. The results showed that only the mutation of BMPR2 gene was highly pathogenic, while the variation of other genes was benign or uncertain significance. Mutations of uncertain significance include TTN and FLNC. According to existing literature reports (Taylor et al. Citation2011; Ader et al. Citation2019; Chauveau et al. Citation2014; Brun et al. Citation2014), mutations of TTN and FLNC are genetic pathogenic factors of cardiomyopathy, which mainly lead to hypertrophic cardiomyopathy and dilated cardiomyopathy. Both types of cardiomyopathy are characterized by marked enlargement or hypertrophy of the left ventricle and arrhythmia. However, in our patient, there was no abnormal left ventricle and the rhythm was tune. In conclusion, we conclude that BMPR2 gene mutation is a genetic risk factor for our patient with pulmonary hypertension.
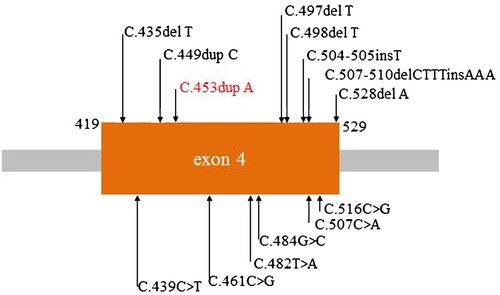
In this study a heterozygous mutation (c.453dupA, p.I152Nfs*29) of BMPR2 was identified in a Chinese family with pulmonary arterial hypertension by whole-exome sequencing. This mutation is located on exon 4 of BMPR2, exon 4 will be transcribed and translated into the trans-membrane domain. Previous studies had demonstrated that mutations in this domain could lead to pulmonary arterial hypertension, for example c.435delT, p.F145Lfs*7 and c.497delT, p.A167Pfs*9 (Figure ) (Machado et al. Citation2001; Deng et al. Citation2000; Machado et al. Citation2015; Morisaki et al. Citation2004; Kataoka et al. Citation2013; Momose et al. Citation2015; Girerd et al. Citation2010; Pfarr et al. Citation2011; Baloira Villar et al. Citation2015; Kabata et al. Citation2013; Machado et al. Citation2006; Cogan et al. Citation2006). Our patient also had subclinical hypothyroidism and hyperuricemia.
Figure 2. Reported mutations of BMPR2 exon 4, red words stands for present study.
Subclinical hypothyroidism and hyperuricemia are often present in patients with heart failure. High uric acid levels are a risk factor for pulmonary arterial hypertension patients. Uric acid could inhibit acetylcholine-mediated vasodilation by acting on the vascular endothelium (Khosla et al. Citation2005). Uric acid also can trigger inflammatory responses, production and release of various profibrotic and pro-inflammatory chemokines such as interleukin 1, interleukin 6 and tumor necrosis factor alpha (Bao et al. Citation2018). These factors can accelerate the pathologic process of pulmonary arterial hypertension. The incidence of subclinical hypothyroidism was in patients with idiopathic pulmonary arterial hypertension is higher than general population (Richter et al. Citation2016; Hollowell et al. Citation2002). Both excessive and insufficient thyroid hormones can induce or aggravate cardiovascular diseases. Treatment with thyroid hormones improves cardiovascular risk factors in patients with hypothyroidism (Jabbar et al. Citation2017). But in a pulmonary arterial hypertension rat models, experimental hypothyroidism prevented and reversed the development of pulmonary arterial hypertension (Husseini et al. Citation2013). Further trials are needed to evaluate whether treatment of subclinical hypothyroidism can improve pulmonary hypertension symptoms. The presence of subclinical hypothyroidism and hyperuricemia in our patients may be due to the patient's severity heart failure.
Bronchial arterial hypertrophy and bronchial microvessel density were significantly increased in pulmonary arterial hypertension patients with BMPR2 mutation compared with pulmonary arterial hypertension patients without the mutation (Ghigna et al. Citation2016), and carriers of BMPR2 mutations have more severe heart failure than non-carriers BMPR2 in pulmonary arterial hypertension patients (Bruggen et al. Citation2016). Although BMPR2 is the most common genetic cause of pulmonary arterial hypertension, its penetrance is very low, only about 20% (Ma and Chung Citation2017). It may be haploid mutations still have enough BMPR2 to maintain normal function, however, under the influence of other factors, such as inflammatory factor tumor necrosis factor-α which could directly suppresses BMPR2 mRNA and protein expression (Morrell et al. Citation2019; Sánchez-Duffhues et al. Citation2019), induces endothelial-to-mesenchymal transition and accelerate the pathological progression of pulmonary arterial hypertension. When vascular lesions develop to end stage, leading to heart failure and show other obvious symptoms, it is very difficult to cure.
Currently, the most commonly used drug therapy for pulmonary arterial hypertension in clinical practice includes oral anticoagulants and diuretics, prostanoids (epoprostenol, trepoprostenil, iloprost), endothelin receptor antagonists (bosentan, ambrisentan) and phosphodiesterase type 5 inhibitors (sildenafil, tadalafil) (Montani et al. Citation2013). In this study, our patient was first diagnosed with pulmonary arterial hypertension and then treated with anticoagulants, diuretics and endothelin receptor antagonists, but the patient’s disease was not effectively resolved. Two years later, she was hospitalized again because of her worsening condition. It suggests that we need more precise treatment for pulmonary arterial hypertension patients with BMPR2 mutations. Recent studies have shown that BMPR2 plays an important role in the balance between BMP and TGF-β signaling, the reduction of BMPR2 enhances the TGF-β signaling pathway, TGF-β may be a therapeutic target for pulmonary arterial hypertension patients with BMPR2 mutations (Hiepen et al. Citation2019).
Consent for publication
Written informed consent for publication of their details was obtained from the patient.
Acknowledgements
The authors thank all subjects for participating in this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Due to patient privacy, additional supporting data, including genetic data is not available.
Additional information
Funding
References
- Ader F, De Groote P, Réant P, Réant P, Rooryck-Thambo C, Dupin-Deguine D, Rambaud C, Khraiche D, Perret C, Pruny JF, Mathieu-Dramard M, Gérard M, Troadec Y, Gouya L, Jeunemaitre X, Van Maldergem L, Hagège A, Villard E, Charron P, Richard P. 2019. FLNC pathogenic variants in patients with cardiomyopathies: prevalence and genotype-phenotype correlations. Clin Genet. 96(4):317–329.
- Andruska A, Spiekerkoetter E. 2018. Consequences of bmpr2 deficiency in the pulmonary vasculature and beyond: contributions to pulmonary arterial hypertension. Int J Mol Sci. 19:2499.
- Baloira Villar A, Pousada Fernández G, Núñez Fernández M, et al. 2015. Clinical and molecular study of 4 cases of pulmonary hypertension associated with sarcoidosis. Arch de Bronconeumol. 51:E19–E21.
- Bao J, Shi Y, Tao M, et al. 2018. Pharmacological inhibition of autophagy by 3-MA attenuates hyperuricemic nephropathy. Clin Sci (Lond). 132:2299–2322.
- Bruggen CE, Happé CM, Dorfmüller P, et al. 2016. Bone morphogenetic protein receptor type 2 mutation in pulmonary arterial hypertension: a view on the right ventricle. Circulation. 133:1747–1760.
- Brun F, Barnes CV, Sinagra G, et al. 2014. Titin and desmosomal genes in the natural history of arrhythmogenic right ventricular cardiomyopathy. J Med Genet. 51(10):669–676.
- Chauveau C, Rowell J, Ferreiro A. 2014. A rising titan: TTN review and mutation update. Hum Mutat. 35(9):1046–1059.
- Cogan JD, Pauciulo MW, Batchman AP, et al. 2006. High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am J Respir Crit Care Med. 174:590–598.
- Deng Z, Morse JH, Slager SL, et al. 2000. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 67:737–744.
- Dodson MW, Brown LM, Elliott CG. 2018. Pulmonary arterial hypertension. Heart Fail Clin. 14:255–269.
- Evans JD, Girerd B, Montani D, et al. 2016. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis. Lancet Respir Med. 4:129–137.
- Ghigna MR, Guignabert C, Montani D, et al. 2016. BMPR2 mutation status influences bronchial vascular changes in pulmonary arterial hypertension. Eur Respir J. 48:1668–1681.
- Girerd B, Montani D, Eyries M, et al. 2010. Absence of influence of gender and BMPR2 mutation type on clinical phenotypes of pulmonary arterial hypertension. Respir Res. 11:73.
- Gordeuk VR, Castro OL, Machado RF. 2016. Pathophysiology and treatment of pulmonary hypertension in sickle cell disease. Blood. 127:820–828.
- Hayes DJ, Ramanathan C, Kopp BT. 2014. The BMPR2 missense mutation p.K230N and pulmonary arterial hypertension. Pediatr Pulm. 49:E5–E6.
- Hiepen C, Jatzlau J, Hildebrandt S, et al. 2019. BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFβ responses and altered cell mechanics. PLoS Biol. 17:e3000557.
- Hollowell JG, Staehling NW, Flanders WD, et al. 2002. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): national health and nutrition examination survey (NHANES III). J Clin Endocrinol Metab. 87:489–499.
- Husseini A, Bagnato G, Farkas L, et al. 2013. Thyroid hormone is highly permissive in angioproliferative pulmonary hypertension in rats. Eur Respir J. 41:104–114.
- Jabbar A, Pingitore A, Pearce SH, et al. 2017. Thyroid hormones and cardiovascular disease. Nat Rev Cardiol. 14:39–55.
- Kabata H, Satoh T, Kataoka M, et al. 2013. Bone morphogenetic protein receptor type 2 mutations, clinical phenotypes and outcomes of Japanese patients with sporadic or familial pulmonary hypertension. Respirology. 18:1076–1082.
- Kataoka M, Aimi Y, Yanagisawa R, et al. 2013. Alu-mediated nonallelic homologous and nonhomologous recombination in the BMPR2 gene in heritable pulmonary arterial hypertension. Genet Med. 15:941–947.
- Khosla UM, Zharikov S, Finch JL, et al. 2005. Hyperuricemia induces endothelial dysfunction. Kidney Int. 67:1739–1742.
- Ma L, Chung WK. 2017. The role of genetics in pulmonary arterial hypertension. The J Pathol. 241:273–280.
- Machado RD, Aldred MA, James V, et al. 2006. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum Mutat. 27:121–132.
- Machado RD, Pauciulo MW, Thomson JR, et al. 2001. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet. 68:92–102.
- Machado RD, Southgate L, Eichstaedt CA, et al. 2015. Pulmonary arterial hypertension: a current perspective on established and emerging molecular genetic defects. Hum Mutat. 36:1113–1127.
- Momose Y, Aimi Y, Hirayama T, et al. 2015. De novo mutations in the BMPR2 gene in patients with heritable pulmonary arterial hypertension. Ann Hum Genet. 79:85–91.
- Montani D, Günther S, Dorfmüller P, et al. 2013. Pulmonary arterial hypertension. Orphanet J Rare Dis. 8:97.
- Morisaki H, Nakanishi N, Kyotani S. 2004. BMPR2 mutations found in Japanese patients with familial and sporadic primary pulmonary hypertension. Hum Mutat. 23:632.
- Morrell NW, Aldred MA, Chung WK, et al. 2019. Genetics and genomics of pulmonary arterial hypertension. Eur Respir J. 53:1801899.
- Peacock AJ, Murphy NF, McMurray JJV, et al. 2007. An epidemiological study of pulmonary arterial hypertension. Eur Respir J. 30:104–109.
- Pfarr N, Szamalek-Hoegel J, Fischer C, et al. 2011. Hemodynamic and clinical onset in patients with hereditary pulmonary arterial hypertension and BMPR2 mutations. Respir Res. 12:99.
- Richter MJ, Sommer N, Schermuly R, et al. 2016. The prognostic impact of thyroid function in pulmonary hypertension. J Heart Lung Transplant. 35:1427–1434.
- Sánchez-Duffhues G, García de Vinuesa A, van de Pol V, et al. 2019. Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. J Pathol. 247:333–346.
- Taylor M, Graw S, Sinagra G, et al. 2011. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation. 124(8):876–885.