
Abstract
Bantang hot spring is natural, geologically formed by the aggregation of both cold and hot spring water. For the first time, this study combined 16S rRNA genes high-throughput sequencing and bioinformatics analyses to determine the prokaryotic microbe diversity and community structure in the water. A total of 5266 OTUs were obtained, and the community of water was found to comprise 32 bacterial phyla with the most abundant phylum of members belonging to Proteobacteria, and six archaeal phyla with the most abundant phylum of members belonging to Woesearchaeota. Alphaproteobacteria, Burkholderiales, and Comamonadaceae were the highest represented classified species at the level of class, order, and family, respectively. The most abundant bacterial and archaeal genus were Subdivision3 genera incertae sedis and Pacearchaeota Incertae Sedis AR13, and more than a quarter of microorganisms were unclassified genus. Among the top 50 most abundant OTUs, 18 OTUs including the earliest and latest species in evolution have not yet been classified. In the COG analysis, ‘General function prediction only’ was found to be the highest represented category, whereas among predicted KEGG pathways, ‘Membrane Transport’ was the most abundant. Thus, findings from this study greatly improve our understanding to explore novel microorganisms in future.
Introduction
Microorganisms are the most ancient inhabitants, found in various environmental conditions on our earth. They have adapted to almost all extreme environmental niches due to their high variability, and some new compounds for drug research and development have been explored under different extreme environmental conditions, such as high-temperature conditions (Miroshnichenko and Bonch-Osmolovskaya Citation2006; Mahajan and Balachandran Citation2017). Hot springs are one of the extreme environments that estuary population of microbes and are especially suitable for the colonization of thermophiles (Wagner and Wiegel Citation2008; Menzel et al. Citation2015). Bacteria and archaea are the prokaryotic microbe that can thrive well in this environment of hot springs (Huang et al. Citation2013). Microbial communities are good model systems for ecological and evolutionary analysis of microbial communities (Rozanov et al. Citation2017). The diversity analysis of such extreme environments has got sufficient attention due to their diverse and unique ecology, chemistry, and opportunity they provide to identify rare compounds and genes (Kuddus and Ramteke Citation2012). Since the 1950s, people have shown great interests in hot spring microorganisms that grow in the temperature of thermophilic (>55°C) and hyperthermophilic (>80°C) (Selvarajan et al. Citation2014). The reason is that the thermal stability of enzymes such as protease, lipase, amylase, cellulase, phosphatase, asperginase, esters, carboxylase from thermophiles is gaining importance owing to a number of commercial and industry applications (López-López et al. Citation2013). One famous example is the Taq polymerase named after Thermus aquaticus, a thermostable DNA polymerase which was isolated by Brock et al (Chien et al. Citation1976). It is frequently used in polymerase chain reaction, which is a method for amplifying DNA.
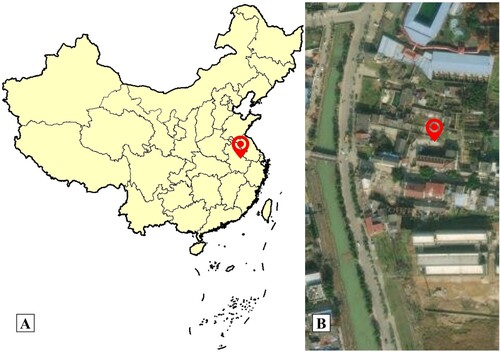
The Bantang hot spring is in the town of Bantang, which is one of the four major hot springs in China. It is located at the foot of Tangshan mountain in the northeast of Chaohu, Hefei City, Anhui Province (Figure ). The Bantang hot spring is composed of two springs, one thermal and the other cold, with half thermal and half cold, which is different from other hot springs in China. The Bantang hot spring has a long history of tourism, medical, and other resources exploitation. However, there are few studies on its microorganisms, and the microbial diversity and microbe community structure of it has not been reported so far.
Figure 1. A: A map that shows the hot spring from Anhui Province, China; B: Sampling location in the town of Bantang, satellite photo. Red marks the location of the Bantang hot spring (31°38′ 50.05″N, 117°54′50.21″E).
Cultivation-dependent studies are valuable for isolating novel organisms and exploring their properties. But some microorganism exists in a viable but nonculturable state, making it difficult to determine the entire microbial composition using culture-dependent methods (Cowan et al. Citation2005). Less than 10% of bacterial diversity, present in an environmental sample can be cultured in vitro with known cultivation techniques in the laboratory (Bull et al. Citation2000). Cultivation-independent methods offer a more comprehensive assessment of microbial diversity (Azimirad et al. Citation2019). Metagenomics is an effective way to identify the microorganisms present in samples, which involves analysis of the total DNA from all microbes present in a given environment, including those in the viable but nonculturable state, and is independent of cultivation (De Filippis et al. Citation2017). 16S rRNA gene is the preferred gene for the metagenomics analysis of microbial diversity to compare microbial diversity among habitats rather than using whole metagenomic DNA (Yang et al. Citation2016). The method of metagenomics analysis has been used to reveal the microbial diversity in many hot springs in different places, such as Tengchong (Pagaling et al. Citation2012; Hou et al. Citation2013), Tibet (Song et al. Citation2013), Yellowstone National Park (Jackson et al. Citation2001; Macur et al. Citation2013), Philippines (Huang et al. Citation2013), Colombian Andes (Bohorquez et al. Citation2012), Eritrea (Ghilamicael et al. Citation2017), and Kenya (Kambura et al. Citation2016).
In this study, the variable V3 and V4 regions of the 16S rRNA genes were used to determine the microbial community in hot spring water of Bantang. The study investigated the overall prokaryotic microbial diversity present in hot spring water using high-throughput sequencing. High-throughput sequencing could produce more sequencing information in comparison with clone library and denaturing gradient gel electrophoresis, which was helpful to explore novel bacteria and archaea using non-culture techniques in future (Song et al. Citation2013).
Materials and methods
Sample collection
A total of 10-L hot spring water in Bantang was collected in a sterile jar using a water pump nearby the central of hot spring in the town of Bantang. The geographical position of the sampling site in terms of latitude and longitude was taken using the ArcGIS Earth Android (version 1.4.2) (Figure ), and the temperature and pH value of water were also measured at the site of sampling. Then, the water sample in jar was brought to the laboratory using a thermos bag for maintaining the habitat temperature. The microorganisms in the water of hot spring were collected through 5 of 0.22-μm microporous barrel-type filter. After being collected, these filter membranes were removed and plated onto a 90-mm sterile petri dish, sealed, and stored in the dark at −20°C less than 24 h.
DNA extraction
The filter membrane in the petri dish was taken from −20°C, placed at room temperature for 30 min,15 mL sterile normal saline was added, and shaken vigorously by ultrasonic. Then, the microorganisms in the filter membrane were dissolved in sterile normal saline by buffeting the filter membrane with a sterile dropper, and were collected at the bottom of the centrifuge tube by centrifuging at 1000 rpm for 5 min. Good-quality metagenomic DNA is essential for an effective analysis of microbial diversity. The total DNA from the water sample was extracted according to the manufacturer’s instructions of the E.Z.N.A.® Soil DNA kit produced by Omega Bio-tek (Norcross, GA, USA). A microspectrophotometer was used to detect the concentration and purity of the metagenomic DNA. The size of the metagenomics DNA was verified on a 0.8% agarose gel electrophoresis. The resulted DNA was diluted to a DNA concentration of 1 ng/μL and subsequently used as a template for PCR to amplify 16S rRNA genes.
PCR amplification for 16S rRNA gene
Two universal primers 341F (5′-CCCTACACGACGCTCTTCCGATCTG(barcode)CCTACGGGNGGCWGCAG-3′) and 805R (5′-GACTGGAGTTCCTTGGCACCCGAGAATTCCAGACTACHVGGGTATCTAATCC-3′) with a set of 6-nucleotide barcodes (AGAGAC) were used in PCR to amplify the V3–V4 region of the bacterial and archaeal 16S rRNA genes. To obtain good-quality PCR products, two experiments of PCR amplification were used in this study. The first PCR amplification was carried out in 30 μL volumes with 15 ng of template DNA, 15 μL of 2 × Taq master Mix, and 1 μL of each 10 μM primer. The first PCR amplification was run under following conditions: (1) pre-denature at 94°C for 3 min, (2) followed by five cycles of denaturing at 94°C for 30 s, annealing at 45°C for 20 s, and extension at 65°C for 30 s, (3) followed by 20 cycles of denaturing at 94°C for 20 s, annealing at 55°C for 20 s, and extension at 72°C for 30 s, and (4) a final extension at 72°C for 5 min. The second PCR amplification was carried out in 30 μL volumes with 20 ng of first PCR products, 15 μL of 2 × Taq master Mix, and 1 μL of each 10 μM primer. The second PCR amplification was run under following conditions: (1) pre-denature at 95°C for 3 min, (2) followed by 5 cycles of denaturing at 94°C for 20 s, annealing at 55°C for 20 s, and extension at 72°C for 30 s, and (3) a final extension at 72°C for 5 min. The second PCR products were detected by 2% denaturing agarose gels, then the 16S rRNA gene amplicons were purified using the Agencourt AMPure XP kit (Beckman, USA) and quantified with the Qubit DNA test kit (Life Invitrogen, USA). Finally, the purified 16S rRNA gene amplicons were to construct DNA library using the TruSeq DNA PCR-free sample preparation kit (Illumina, USA) and pair-end sequenced on an Illumina HiSeq2500 PE250 platform at Sangon Biotech Company (Shanghai, China).
Bioinformatics analysis of the sequence data
The original image data file obtained by high-throughput sequencing was transformed into the original sequence by the CASAVA base recognition analysis, which was called raw reads. The obtained raw reads removed primer adapter using the cutadapt (version 1.2.1) (He et al. Citation2020), merged sequences using the PEAR (version 0.9.6) (Zhang et al. Citation2014), quality-filtered sequences using the PRINSEQ (version 0.20.4) (Schmieder and Edwards Citation2011), and identified chimeras sequences using the UCHIME (version 4.2.40) (Edgar et al. Citation2011). Chimeras and non-specific amplification sequences were removed and the remaining sequences were then assigned to OTUs (operational taxonomic units) at a 97% similarity cutoff using the USEARCH (version 5.2.236) (Edgar Citation2010).
The standard of the representative sequence of each OTU was to choose the sequence with high frequency of occurrence that has the largest number of hits to other sequences in the OTU. The taxonomy of each OTU representative sequence was assigned using the Ribosomal Database Project [http://rdp.cme.msu.edu/misc/resources.jsp] classifier with a minimum bootstrap threshold of 80%. Subsequently, taxonomic information at the six-level classification level domain, phylum, class, order, family, and genus was obtained (Balvočiūtė and Huson Citation2017).
Five kinds of the alpha diversity index, such as Shannon, ACE, Chao1, Coverage, and Simpson were calculated using the mothur (version 1.30.1) (Schloss et al. Citation2009). The curves of 16S rRNA gene sequence, such as rarefaction and rank-abundance were analyzed using the R Package (version 2.15.3) (Dessau and Pipper Citation2008). The taxonomic and phylogenetic tree of microorganism in the hot spring water were visualized using the GraPhlAn (version 0.9.7) (Asnicar et al. Citation2015). The phylogenetic tree of the top 50 sequences with high frequency of occurrence and the longest sequence in the database with the same species information were constructed using the Maximum-likelihood method and Tamura 3-parameter model provided in the software of the MEGA (version 11.0.10) (Tamura et al. Citation2021). Bootstrap confidence values were obtained with 1000 replicates. Metagenome functional features of 16S rRNA genes from the hot spring water were predicted using the PICRUSt (version 1.0.0) (Langille et al. Citation2013).
Nucleotide sequence accession numbers
The metagenome sequence data of the 16S rRNA genes for this study were publicly available in the NCBI Sequence Read Archive (SRA) under the accession number SRR13684275.
Results
Water quality parameters
The temperature and pH of the hot spring water were detected as 60°C and 6.5 (acidic) at the site of sampling. The chemical components contained in the hot spring water of Bantang have been reported in our previous study, which are mainly HCO3–, Ca2+, SO42-, Mg2+, H2SiO3, Cl–, Na+, NO3–, and Sr, belonging to weak acid medical hot mineral water (Fang et al. Citation2015).
16S rRNA gene sequence processing
A total of 59994 raw reads with an average read length of 456.67 bp were obtained using high-throughput sequencing. The bases with minimum base call accuracy of 99% in effective tags (Q20) for each sequence amplicon were all greater than 98%, and the rarefaction curve (Figure ) was tended to be flat, indicating high levels of accuracy in the sequencing data and indicating that they were reliable for further study. After the quality control, the average length of quality control reads was 415.63 bp, and the quality control reads were preprocessed, chimera removed, and non-specific amplified sequences removed. The sequence information of microorganism in the hot spring water was shown in Table .
Figure 2. Rank-abundance curve of microbiome diversity in the hot spring water.
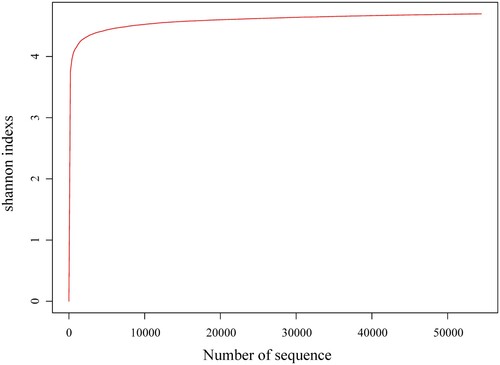
Table 1. Sample sequencing information and alpha diversity of microbiome in the hot spring water.
OTU clustering and taxonomic annotation of microbiome
To better understand the OTU information and their taxonomic annotation, the tag and OTUs were calculated and summarized. Finally, a total of 5266 OTUs were clustered (Table ). The taxonomic annotation for the OTUs of the hot spring water was assigned to bacteria and archaea domains, and the predominant phyla and genera identified (Table ; Figures ). At 80% confidence threshold in the RDP classifier, these 5266 OTUs spanned a wide range within the domain bacteria and archaea, occupying 32 phyla. The results showed that the phylum Proteobacteria (39.01%), Verrucomicrobia (17.75%), Bacteroidetes (10.36%), Actinobacteria (9.00%), Parcubacteria (6.31%), Planctomycetes (3.92%), Chlamydiae (2.45%), Nitrospirae (2.14%), and Firmicutes (1.04%) (in order from high to low abundance) were all present at levels of more than 0.5% of all microbiome; Proteobacteria was the predominant bacteria phylum, representing more than 39% of all microbiome present. Unclassified bacteria phylum was more than 6% of all microbiome present. At the class level taxonomic distribution, members like Alphaproteobacteria (23.29%), Subdivision3 (17.58%), Betaproteobacteria (11.24%), Actinobacteria (8.99%), unclassified (8.22%), Parcubacteria unclassified (6.31%), Cytophagia (5.83%), Planctomycetia (3.85%), Sphingobacteriia (2.72%), Chlamydiia (2.45%), Deltaproteobacteria (2.31%), Nitrospira (2.14%), and Gammaproteobacteria (1.5%) were present in majority (more than 1%) whereas class Flavobacteriia, Clostridia, Gemmatimonadetes, Thaumarchaeota unclassified, Anaerolineae, Armatimonadetes unclassified were present in minority (low than 1%). At the order level taxonomic distribution, members like Subdivision3 unclassified (17.58%), unclassified (11.52%), Burkholderiales (10.58%), Sphingomonadales (9.62%), Actinomycetales (8.15%), Parcubacteria unclassified (6.31%), Cytophagales (5.83%), Rhodospirillales (5.76%), Rhodobacterales (5.44%), Planctomycetales (3.58%), Sphingobacteriales (2.72%), Chlamydiales (2.45%), and Nitrospirales (2.14%) were present in majority (more than 1%), whereas order Flavobacteriales, Bdellovibrionales, Gaiellales, Rhizobiales, Myxococcales, Methylococcales, Methylophilales, and so on were present in minority (low than 1%). Prominent families in the metagenome were Subdivision3 unclassified (17.58%), unclassified (13.29%), Comamonadaceae (9.89%), Sphingomonadaceae (9.58%), Microbacteriaceae (7.94%), Parcubacteria unclassified (6.31%), Cytophagaceae (5.82%), and Rhodobacteraceae (5.44%) (Figure ). At the genus level, Subdivision3 genera incertae sedis (16.09%), Sphingorhabdus (9.39%), Rhodoluna (7.82%), Parcubacteria genera incertae sedis (6.31%), Pseudarcicella (5.76%), Rhodobacter (5.14%), Reyranella (3.05%), Curvibacter (2.67%), Sediminibacterium (2.50%), Neochlamydia (2.31%), Limnohabitans (2.14%), Nitrospira (2.11%), Vampirovibrio (0.76%), Gaiella (0.75%), Flavobacterium (0.61%), Limisphaera (0.53%), and Methylothermus (0.51%) (in order from high to low abundance) were present at levels of more than 0.5% of all microbiome present; Subdivision3 genera incertae sedis was the predominant genus belonging to Verrucomicrobia, representing more than 16% of all microbiome present. Unclassified genus was more than 25% of all microbiome present. In addition, archaea were also found in the hot spring water, the phylum Woesearchaeota (0.30%), Pacearchaeota (0.19%), Thaumarchaeota (0.11%), Euryarchaeota (0.11%), Crenarchaeota (0.09%), and Diapherotrites (0.08%) were all present of all microbiome. Woesearchaeota was the predominant phylum, representing 0.30% of all microbiome present. At the genus level, Pacearchaeota Incertae Sedis AR13 (0.19%), Nitrosopumilus (0.09%), Woesearchaeota Incertae Sedis AR20 (0.08%), Woesearchaeota Incertae Sedis AR16 (0.06%), Candidatus Iainarchaeum (0.06%), Diapherotrites Incertae Sedis AR10 (0.02%), Nitrososphaera (0.02%), Woesearchaeota Incertae Sedis AR17 (0.02%), and Woesearchaeota Incertae Sedis AR18 (0.02%) were present of all microbiome; Pacearchaeota Incertae Sedis AR13 was the predominant genus, representing 0.19% of all microbiome present. In this study, some thermophiles were also found in the hot spring water, such as Methylothermus and Meiothermus. The presence of many novel species was indicated by more than 25% unclassified OTUs.
Figure 3. Graphlan chart representation of taxonomic classification and phylogenetic tree of the hot spring water metagenome.
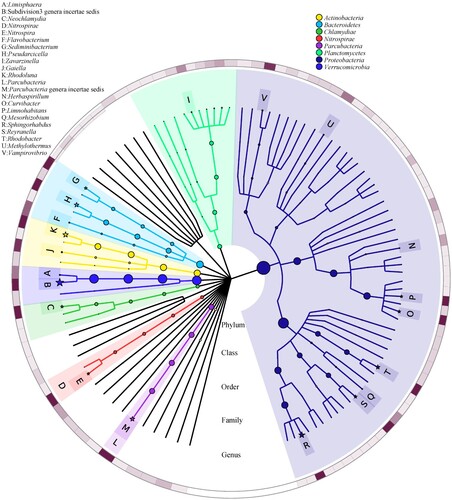
Figure 4. Phylum level distribution of the microbiome population present in the hot spring water.

Figure 5. Genus level distribution of microbiome population present in the hot spring water.

Figure 6. Family level distribution of microbiome population present in the hot spring water.
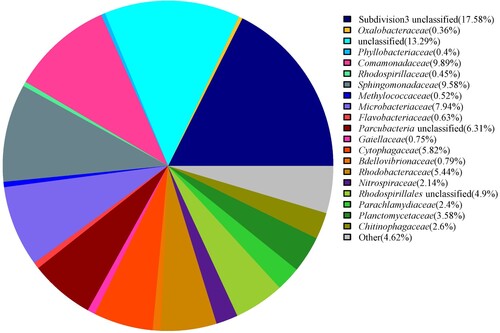
Table 2. Relative abundance of the bacterial (more than 0.5%) and archaea phylum and genus in the hot spring water.
Alpha diversity and phylogenetic analysis of microbiome
The alpha diversity analysis revealed that >99% of the species were estimated, while high values of Chao1 richness indices (46578.58) and Shannon diversity indices (4.69825) indicated that microbial communities in Bantang hot spring are highly rich and diverse (Table ). The rarefaction analysis derived from the observed OTUs showed a near saturation of OTUs to a plateau at 3% genetic distance for the hot spring water, taken as an example of Shannon (Figure ), demonstrating that our sequencing depth was sufficient and the majority of the microbe diversity was included.
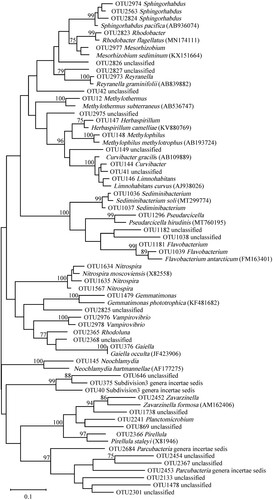
The representative sequences of top 50 most abundant OTUs and 18 longest classified database sequences with the same genus were selected for phylogenetic analyses (Figure ). Among the top 50 OTUs, 18 OTUs of microorganism that could not be clustered into any recognized microbe divisions or candidate divisions. The phylogenetic tree revealed that OTU2368 unclassified microorganism was the earliest species in evolution and has the closest sequence similarity to OTU376 Gaiella. OTU2367 unclassified microorganism was the latest species in evolution and has the closest sequence similarity to OTU2454 unclassified microorganism. Among 18 unclassified OTUs, OTU41, 646, and 869 had the closest sequence similarity to any classified species.
Figure 7. Phylogenetic tree based on 16S rRNA gene sequences of microbiome constructed by Maximum-likelihood method and Tamura 3-parameter model, numbers on the nodes are the bootstrap values (percentages) based on 1000 replicates, bootstrap values lower than 70% are deleted. Scale bar indicates substitutions per nucleotide position.
Microbial functional gene diversity
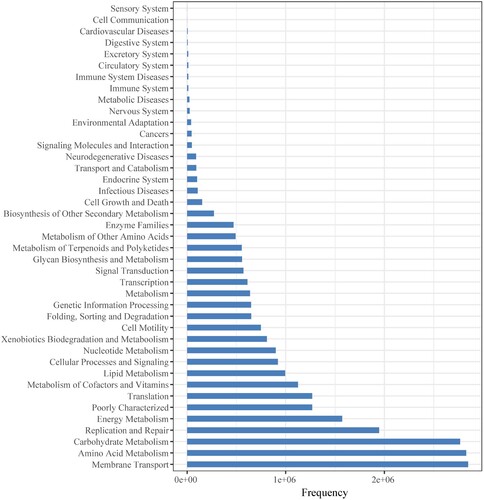
The PICRUSt software was used to predict abundance of gene categories (COGs) (Figure ) and metabolic pathways (KEGG) (Figure ) from 16S rRNA gene sequencing data of microorganisms present in the hot spring water. Among predicted gene categories, ‘General function prediction only’ was the most abundant category at first tier followed by ‘Amino acid transport and metabolism’, ‘Transcription’ (Figure ). Genes involved in ‘General function prediction only’ predominated, accounting for 3252407 copies of all genes. Among predicted metabolic pathways, ‘Membrane Transport’ was the most abundant category at first tier followed by ‘Amino Acid Metabolism’, ‘Carbohydrate Metabolism’, ‘Replication and Repair’ (Figure ). Genes involved in ‘Membrane Transport’ predominated, accounting for 2848275 copies of all genes.
Figure 8. COGs metagenome functional predictions of OTUs from microbiome in the hot spring water.
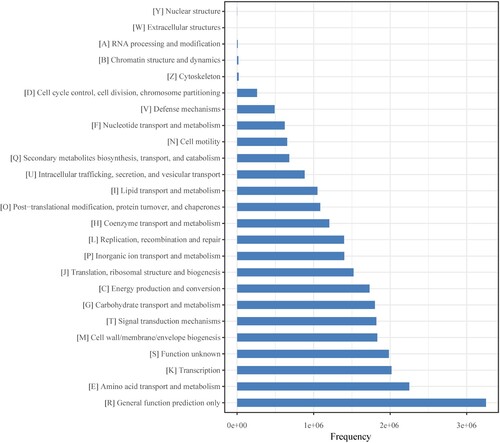
Figure 9. KEGG metagenome functional predictions of OTUs from microbiome in the hot spring water.
Discussion
In this study, prokaryotic microbe diversity in the hot spring water of Bantang from the Anhui province of China was revealed using high-throughput sequencing of the V3 and V4 regions of the 16S rRNA gene. More details of microbial diversity in hot springs have been obtained with the use of the high-throughput sequencing technique instead of conventional clone library and DGGE approaches. Shannon indices showed that microbial diversity in hot spring water of China was much richer in Bantang than in Tengchong (Hou et al. Citation2013). The maximum representation of members from the bacteria phyla found in the hot spring water were Proteobacteria, Verrucomicrobia, Bacteroidetes, Actinobacteria, and Parcubacteria. Part of them (e.g. Proteobacteria, Bacteroidetes) are also the dominant phyla in several hot springs such as Yunnan hot spring, Tibetan hot spring (Song et al. Citation2013), Tengchong hot spring (Pagaling et al. Citation2012), Tapovan hot spring (Rawat and Joshi Citation2019), Y-shaped Sungai Klah hot spring (Lee et al. Citation2018), and Trans-Himalayas hot spring (Roy et al. Citation2020), but the relative abundance of bacteria phyla in these above hot springs are found to be dissimilar. Notably, unclassified bacterial phylum (6.41%) was also found among top five bacterial phyla. In addition, some bacterial phyla were detected with relative low abundance from the Bantang hot spring, but are the preponderant bacterial phyla in other hot springs, such as Firmicutes, Chloroflexi, Aquificae, Deinococcus-Thermus, and Acidobacteria (López-López et al. Citation2015; Rawat and Joshi Citation2019). The reason is probably the physical and chemical structure or local environment such as geochemical structure, temperature, dissolved oxygen level, and the quantity of plant litter in the hot spring is different. It is well known that abiotic factors work together on the dynamics of microbial populations (Chan et al. Citation2017). Bacteria in Proteobacteria are Gram-negative and known for their industrial and agricultural value, and extreme metabolic diversity, such as chemorganotropic, chemolithotropic, and phototropic microorganisms (Marin Citation2011). In the process of decomposing bagasse at 50°C, Proteobacteria were the predominant phylum (Mhuantong et al. Citation2015), which is in accordance with the similar Bantang hot spring.
At the genus level, Subdivision3 genera incertae sedis, Sphingorhabdus, Rhodoluna, Parcubacteria genera incertae sedis, Pseudarcicella, and Rhodobacter were the maximum representation of microbe. The genus Sphingorhabdus belonging to the phylum Proteobacteria was only found in the Bantang hot spring none in others of China (Jogler et al. Citation2013). The genus Rhodoluna belonging to the phylum Actinobacteria is a free-living planktonic freshwater aerobic chemo-organoheterotrophs bacterium and first obtained from Lake Taihu, China, can synthesize actinorhodopsin (Hahn et al. Citation2014). The genus Pseudarcicella belonging to the phylum Bacteroidetes is first isolated from the skin of the medical leech Hirudo medicinalis in Germany (Kampfer et al. Citation2012). The genus Rhodobacter also belonging to the phylum Proteobacteria have vesicular photosynthetic membranes, require thiamine for growth factor (Imhoff et al. Citation1984). Notably, unclassified genus (25.05%) dominated the hot spring water, indicated that we can explore some novel bacteria in Bantang hot spring using culturable techniques in future. Unclassified genus dominate the hot spring sample are also reported in Jakrem hot spring (63.50%) and Deulajhari hot spring (46.29%), India, respectively (Panda et al. Citation2015; Singh and Subudhi Citation2015), which are similar with this study in Bantang hot spring.
In addition to bacteria, archaea were also identified in the hot spring water, only 0.08%–0.30% of the OTUs identified in the hot spring water were affiliated with archaea phyla, including the predominant phyla of Woesearchaeota and the predominant genera of Pacearchaeota Incertae Sedis AR13. Woesearchaeota and Pacearchaeota have been frequently reported in the literature since 1990s but rarely discussed. Pacearchaeota is rarely found in hot springs but detected in Bantang hot spring water. Woesearchaeota and Pacearchaeota are the dominating archaea phyla in the surface water of oligotrophic lakes, which is consistent with this study (Ortiz-Alvarez and Casamayor Citation2016). Thaumarchaeota play an important role in the nitrogen cycle of marine environment through oxidizing ammonia to nitrite. Methanogenic Euryarchaeota was detected in the hot spring but with only in the acidic environment (Wemheuer et al. Citation2013), which is consistent with the study in Tengchong hot spring (Pagaling et al. Citation2012). Hyperthermophilic Crenarchaetoa was detected in this study, which always dominates hot springs in our earth, such as hot springs in Yellowstone, Iceland, Kamchatka, and Thailand (Song et al. Citation2013), but Woesearchaeota dominates in Bantang hot spring. The archaeal genes especially Crenarchaetoa participated in ammonium oxide, which is reported in hot springs like Russia, Iceland, the Great Basin and YNP (USA), and China (Reigstad et al. Citation2008; Zhang et al. Citation2008; Zhang et al. Citation2008). Crenarchaeota, Diapherotrites, Thaumarchaeota, and Euryarchaeota are also found in the Lirima hot springs located in the Chilean Andean highlands, but Diapherotrites is detected only at the hot spring with 42°C (Pérez et al. Citation2020). These archaea are present in a relatively small portion in the hot spring water, perhaps the archaea use more energy to maintain metabolism in extreme environments and are thus less able to diversify (Aller and Kemp Citation2008). In addition, the pH and temperature also influence the abundance of archaea, the Bantang hot spring water is weak acid (pH 6.5) and moderate temperature (60°C). Menzel et al. (2015) found that the abundance of archaea is higher in hot springs with low pH and high-temperature habitat (Menzel et al. Citation2015). More than quarter of the unclassified genus and the phylogenetic analysis (Figure ) strongly suggest that the Bantang hot spring hides the wealth of uncultivable and novel microorganism for exploring.
Predicted abundance of gene categories revealed that in COG analysis, ‘General function prediction only’ was the highest represented category at first tier, which is in agreement with the study in Aguas Calientes (Peru) hot spring (Paul et al. Citation2016), and ‘Amino acid transport and metabolism’ was the highest represented category at second tier, followed by ‘Transcription’ and ‘Function unknown’. In KEGG pathways prediction, ‘Amino Acid Metabolism’ was the highest represented category at second tier, which is in agreement with the COG analysis above and very similar to the study in Sungai Klah (Malaysia) hot spring (Chan et al. Citation2015), whose ‘Amino Acid Metabolism’ is at first tier. In addition, it is reported that more than 30% of genes in metagenome could not be identified using bioinformatics methods, while the metagenome functional features prediction provides a good chance to find some new activities that still exist about the function a large part of the microbial proteome (López-López et al. Citation2015). Otherwise, microbial functional genes are also greatly influenced by the environmental conditions, like oxygen, temperature, light, nutrient content, and so on (Shapleigh Citation2009).
Conclusion
This study has documented, for the first time, the prokaryotic microbial community in the hot spring water from Bantang of China using 16S rRNA gene high-throughput sequencing. The majority of the classified OTUs observed in this study belong to bacterial phyla suggesting that bacteria are the most dominant taxa in Bantang hot spring. The prokaryotic microbe diversity was richer compared to other hot spring in China, like Tengchong hot spring. Among the top 50 most abundant OTUs, 18 OTUs (36%) including the earliest and latest species in evolution have not yet been classified which strongly suggested that the Bantang hot spring is a good source to explore novel bacteria and archaea in future.
Authors’ contributions
Shu Fang participated in the experiments design, data analysis and drafted a manuscript. Juan Yan participated in the experiment operation. Both authors have read and approved the final manuscript.
Availability of data and materials
Raw data supporting the findings of this study are in the OSFHOME repository at https://osf.io/s38uk. In addition, the metagenome sequence data of the 16S rRNA genes for this study are publicly available in the NCBI Sequence Read Archive (SRA) repository [https://www.ncbi.nlm.nih.gov/sra/] under accession number SRR13684275.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Aller JY, Kemp PF. 2008. Are archaea inherently less diverse than Bacteria in the same environments? FEMS Microbiol Ecol. 65(1):74–87.
- Asnicar F, Weingart G, Tickle TL, Huttenhower C, Segata N. 2015. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ. 3:e1029.
- Azimirad M, Tajeddin E, Hasani Z, Mirjalali H, Alebouyeh M, Zali MR. 2019. Development of a culture-independent method for rapid monitoring of microbial indicators in water samples. Int J Environ Sci Technol. 16:3165–3170.
- Balvočiūtė M, Huson DH. 2017. SILVA, RDP, Greengenes, NCBI and OTT – how do these taxonomies compare? BMC Genomics. 18(Suppl 2):114.
- Bohorquez LC, Delgado-Serrano L, López G, Osorio-Forero C, Klepac-Ceraj V, Kolter R, Junca H, Baena S, Zambrano MM. 2012. In-depth characterization via complementing culture-independent approaches of the microbial community in an acidic hot spring of the Colombian Andes. Microb Ecol. 63(1):103–115.
- Bull AT, Ward AC, Goodfellow M. 2000. Search and discovery strategies for biotechnology: the paradigm shift. Microbiol Mol Biol Rev. 64(3):573–606.
- Chan CS, Chan KG, Ee R, Hong KW, Urbieta MS, Donati ER, Shamsir MS, Goh KM. 2017. Effects of physiochemical factors on prokaryotic biodiversity in Malaysian circumneutral hot springs. Front Microbiol. 8:1252.
- Chan CS, Chan KG, Tay YL, Chua YH, Goh KM. 2015. Diversity of thermophiles in a Malaysian hot spring determined using 16S rRNA and shotgun metagenome sequencing. Front Microbiol. 6:177.
- Chien A, Edgar DB, Trela JM. 1976. Deoxyribonucleic acid polymerase from the extreme thermophile Thermus aquaticus. J Bacteriol. 127(3):1550–1557.
- Cowan D, Meyer Q, Stafford W, Muyanga S, Cameron R, Wittwer P. 2005. Metagenomic gene discovery: past, present and future. Trends Biotechnol. 23(6):321–329.
- De Filippis F, Parente E, Ercolini D. 2017. Metagenomics insights into food fermentations. Microb Biotechnol. 10(1):91–102.
- Dessau RB, Pipper CB. 2008. [“R”–project for statistical computing]. Ugeskr Laeger. 170(5):328–330.
- Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 26(19):2460–2461.
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 27(16):2194–2200.
- Fang S, Dai C, Yan J. 2015. Phylogenetic analysis of strain CHBT-1721 with other thermophile bacteria isolated from hot springs of China. Journal of Zhejiang University (Agriculture and Life Sciences). 41(1):15–24.
- Ghilamicael AM, Budambula NLM, Anami SE, Mehari T, Boga HI. 2017. Evaluation of prokaryotic diversity of five hot springs in Eritrea. BMC Microbiol. 17(1):203.
- Hahn MW, Schmidt J, Taipale SJ, Doolittle WF, Koll U. 2014. Rhodoluna lacicola gen. nov., sp. nov., a planktonic freshwater bacterium with stream-lined genome. Int J Syst Evol Microbiol. 64(Pt 9):3254–3263.
- He B, Zhu R, Yang H, Lu Q, Wang W, Song L, Sun X, Zhang G, Li S, Yang J, et al. 2020. Assessing the impact of data preprocessing on analyzing next generation sequencing data. Front Bioeng Biotechnol. 8:817.
- Hou W, Wang S, Dong H, Jiang H, Briggs BR, Peacock JP, Huang Q, Huang L, Wu G, Zhi X, et al. 2013. A comprehensive census of microbial diversity in hot springs of Tengchong, Yunnan Province China using 16S rRNA gene pyrosequencing. PLoS One. 8(1):e53350.
- Huang Q, Jiang H, Briggs BR, Wang S, Hou W, Li G, Wu G, Solis R, Arcilla CA, Abrajano T, et al. 2013. Archaeal and bacterial diversity in acidic to circumneutral hot springs in the Philippines. FEMS Microbiol Ecol. 85(3):452–464.
- Imhoff JF, TRüPER HG, Pfennig N. 1984. Rearrangement of the species and genera of the phototrophic “purple nonsulfur bacteria”. Int J Syst Bacteriol. 34(3):340–343.
- Jackson CR, Langner HW, Donahoe-Christiansen J, Inskeep WP, McDermott TR. 2001. Molecular analysis of microbial community structure in an arsenite-oxidizing acidic thermal spring. Environ Microbiol. 3(8):532–542.
- Jogler M, Chen H, Simon J, Rohde M, Busse HJ, Klenk HP, Tindall BJ, Overmann J. 2013. Description of Sphingorhabdus planktonica gen. nov., sp. nov. and reclassification of three related members of the genus Sphingopyxis in the genus Sphingorhabdus gen. nov. Int J Syst Evol Microbiol. 63(Pt 4):1342–1349.
- Kambura AK, Mwirichia RK, Kasili RW, Karanja EN, Makonde HM, Boga HI. 2016. Bacteria and Archaea diversity within the hot springs of lake Magadi and Little Magadi in Kenya. BMC Microbiol. 16(1):136.
- Kampfer P, Busse HJ, Longaric I, Rossello-Mora R, Galatis H, Lodders N. 2012. Pseudarcicella hirudinis gen. nov., sp. nov., isolated from the skin of the medical leech Hirudo medicinalis. Int J Syst Evol Microbiol. 62(Pt 9):2247–2251.
- Kuddus M, Ramteke PW. 2012. Recent developments in production and biotechnological applications of cold-active microbial proteases. Crit Rev Microbiol. 38(4):330–338.
- Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al. 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 31(9):814–821.
- Lee LS, Goh KM, Chan CS, Annie Tan GY, Yin WF, Chong CS, Chan KG. 2018. Microbial diversity of thermophiles with biomass deconstruction potential in a foliage-rich hot spring. Microbiologyopen. 7(6):e00615.
- López-López O, Cerdán ME, González-Siso MI. 2013. Hot spring metagenomics. Life (Basel). 3(2):308–320.
- López-López O, Knapik K, Cerdán ME, González-Siso MI. 2015. Metagenomics of an alkaline hot spring in Galicia (Spain): microbial diversity analysis and screening for novel lipolytic enzymes. Front Microbiol. 6:1291.
- Macur RE, Jay ZJ, Taylor WP, Kozubal MA, Kocar BD, Inskeep WP. 2013. Microbial community structure and sulfur biogeochemistry in mildly-acidic sulfidic geothermal springs in Yellowstone National Park. Geobiology. 11(1):86–99.
- Mahajan GB, Balachandran L. 2017. Sources of antibiotics: Hot springs. Biochem Pharmacol. 134:35–41.
- Marin I. 2011. Proteobacteria. In: Gargaud M, et al. (eds) Encyclopedia of Astrobiology, vol 1. Springer, Berlin, Heidelberg. p. 1350.
- Menzel P, Gudbergsdóttir SR, Rike AG, Lin L, Zhang Q, Contursi P, Moracci M, Kristjansson JK, Bolduc B, Gavrilov S, et al. 2015. Comparative metagenomics of eight geographically remote terrestrial hot springs. Microb Ecol. 70(2):411–424.
- Mhuantong W, Charoensawan V, Kanokratana P, Tangphatsornruang S, Champreda V. 2015. Comparative analysis of sugarcane bagasse metagenome reveals unique and conserved biomass-degrading enzymes among lignocellulolytic microbial communities. Biotechnol Biofuels. 8:16.
- Miroshnichenko ML, Bonch-Osmolovskaya EA. 2006. Recent developments in the thermophilic microbiology of deep-sea hydrothermal vents. Extremophiles. 10(2):85–96.
- Ortiz-Alvarez R, Casamayor EO. 2016. High occurrence of Pacearchaeota and Woesearchaeota (Archaea superphylum DPANN) in the surface waters of oligotrophic high-altitude lakes. Environ Microbiol Rep. 8(2):210–217.
- Pagaling E, Grant WD, Cowan DA, Jones BE, Ma Y, Ventosa A, Heaphy S. 2012. Bacterial and archaeal diversity in two hot spring microbial mats from the geothermal region of Tengchong, China. Extremophiles. 16(4):607–618.
- Panda AK, Bisht SS, Kumar NS, De Mandal S. 2015. Investigations on microbial diversity of Jakrem hot spring, Meghalaya, India using cultivation-independent approach. Genom Data. 4:156–157.
- Paul S, Cortez Y, Vera N, Villena GK, Gutiérrez-Correa M. 2016. Metagenomic analysis of microbial community of an Amazonian geothermal spring in Peru. Genom Data. 9:63–66.
- Pérez V, Cortés J, Marchant F, Dorador C, Molina V, Cornejo-D'Ottone M, Hernández K, Jeffrey W, Barahona S, Hengst MB. 2020. Aquatic thermal reservoirs of microbial life in a remote and extreme high Andean hydrothermal system. Microorganisms. 8(2):208.
- Rawat N, Joshi GK. 2019. Bacterial community structure analysis of a hot spring soil by next generation sequencing of ribosomal RNA. Genomics. 111(5):1053–1058.
- Reigstad LJ, Richter A, Daims H, Urich T, Schwark L, Schleper C. 2008. Nitrification in terrestrial hot springs of Iceland and Kamchatka. FEMS Microbiol Ecol. 64(2):167–174.
- Roy C, Rameez MJ, Haldar PK, Peketi A, Mondal N, Bakshi U, Mapder T, Pyne P, Fernandes S, Bhattacharya S, et al. 2020. Microbiome and ecology of a hot spring-microbialite system on the trans-Himalayan plateau. Sci Rep. 10(1):5917.
- Rozanov AS, Bryanskaya AV, Ivanisenko TV, Malup TK, Peltek SE. 2017. Biodiversity of the microbial mat of the Garga hot spring. BMC Evol Biol. 17(Suppl 2):254.
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 75(23):7537–7541.
- Schmieder R, Edwards R. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 27(6):863–864.
- Selvarajan R, Maredza A, Tekere M. 2014. Microbial exploration in extreme conditions: Metagenomic analysis and future perspectives. Nova Science Publishers, Inc, New York, Hauppauge. p. 157–181.
- Shapleigh JP. 2009. Dissimilatory and assimilatory nitrate reduction in the purple photosynthetic bacteria. In: Hunter CN, et al. (eds) The Purple Phototrophic Bacteria. Advances in Photosynthesis and Respiration, vol 28. Springer, Dordrecht. p. 623–642.
- Singh A, Subudhi E. 2015. Structural insights of microbial community of Deulajhari (India) hot spring using 16s-rRNA based metagenomic sequencing. Genom Data. 7:101–102.
- Song ZQ, Wang FP, Zhi XY, Chen JQ, Zhou EM, Liang F, Xiao X, Tang SK, Jiang HC, Zhang CL, et al. 2013. Bacterial and archaeal diversities in Yunnan and Tibetan hot springs, China. Environ Microbiol. 15(4):1160–1175.
- Tamura K, Stecher G, Kumar S. 2021. MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol. 38(7):3022–3027.
- Wagner ID, Wiegel J. 2008. Diversity of thermophilic anaerobes. Ann N Y Acad Sci. 1125:1–43.
- Wemheuer B, Taube R, Akyol P, Wemheuer F, Daniel R. 2013. Microbial diversity and biochemical potential encoded by thermal spring metagenomes derived from the Kamchatka Peninsula. Archaea. 2013:136714.
- Yang B, Wang Y, Qian PY. 2016. Sensitivity and correlation of hypervariable regions in 16S rRNA genes in phylogenetic analysis. BMC Bioinformatics. 17:135.
- Zhang CL, Ye Q, Huang Z, Li W, Chen J, Song Z, Zhao W, Bagwell C, Inskeep WP, Ross C, et al. 2008. Global occurrence of archaeal amoA genes in terrestrial hot springs. Appl Environ Microbiol. 74(20):6417–6426.
- Zhang G, Liu CQ, Liu H, Jin Z, Han G, Li L. 2008. Geochemistry of the Rehai and Ruidian geothermal waters, Yunnan Province, China. Geothermics. 37(1):73–83.
- Zhang J, Kobert K, Flouri T, Stamatakis A. 2014. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 30(5):614–620.