
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Unlike cytotoxic chemotherapy, cancer immunotherapy offers targeted therapies that exploit the effector mechanisms of the immune system to combat cancer. However, most therapeutic strategies have so far focused predominantly on the orchestration of the adaptive immune responses to anti-cancer immunotherapies. Unfortunately, the emergence of resistance and associated severe toxicities rendered this modality of treatment imperfect. Because of their complex nature and the late ability to selectively separate distinct innate immune responses, the enormous potential of innate immunity as an immunotherapy was largely neglected. Recently, the growing demand to find alternatives to adaptive immunity-based immunotherapy concurred with growing appreciation of the innate immune effectors contributions to anti-tumor immunity. In particular, the innate immunity anti-infective responses overlap with those that target cancer indicating that these responses can readily be manipulated to design new therapeutic approaches. The paradigm of lectin pathway in recognition of distinct ‘non-self’ (antigenic) glycans on the surface of pathogenic microbes in concert with cancer’s indigenous aberrant (antigenic) glycans render lectin pathway a canonical component of innate immune system that can be extrapolated to cancer immunotherapy. By virtue of recent advances in lectin engineering, the encouraging results of using engineered lectins as anti-viral agents can be replicated in cancer immunotherapy.
Introduction
The inception of cancer immunotherapy roots back to the nineteenth century. In 1893, the observation of spontaneous regression of tumors following the development of erysipelas, a superficial skin infection most commonly caused by Streptococcus pyogenes, led William Coley, often called the ‘Father of Cancer Immunotherapy’ to inject patients with cancer with ‘Coley’s toxins’ made of extracts of heat-inactivated Streptococcus pyogenes and Serratia marcescens (Waldman et al. Citation2020). The immune response associated with Coley’s toxin demonstrated favorable outcome in various cancers especially sarcomas (Coley Citation1893; Decker and Safdar Citation2009). Later, 1909 marked another milestone in the history of cancer immunotherapy when Paul Ehrlich hypothesized that immune system could protect the host from neoplastic disease (Decker et al. Citation2017). However, this hypothesis was only formally introduced as the cancer immunosurveillance concept by Lewis Thomas and Sir Frank Macfarlane Burnet in 1959 (Oiseth and Aziz Citation2017). Admirably, numerous advances in cancer immunotherapy energized by very enthusiastic research spanning decades were needed to convince skeptics that tumors, being derived from self-tissues, would be visible to the immune system. By the year 2013, the unprecedented clinical successes of cancer immunotherapies designed to strengthen cancer immunosurveillance had finally earned recognition by the journal Science as the 2013 Scientific Breakthrough of the year (Finn Citation2018).
Cancer immunosurveillance was initially thought to be a host-protective function carried out by the adaptive immune system only at the earliest stages of cellular transformation (Dunn et al. Citation2004b). However, the role of the immune system in prompting the evolution of more aggressive tumors unveiled the ugly face of the immune system (Dunn et al. Citation2002). Nevertheless, a significant body of evidence has sustained the validity of the cancer immunosurveillance hypothesis which led to its refinement and broadening into ‘the cancer immunoediting’ hypothesis depicting the dual and opposing host-protective and tumor-sculpting effects of the immunue system on developing tumors. These double-faceted actions were conceived as a continuum of three distinct phases, the three Es of cancer immunoediting (elimination, equilibrium, and escape), but linear progressesion can be terminated at the elimination phase if it was successful at destroying a developing tumor. The three Es of cancer immunoediting correspond to the spectrum from cancer immunosurveillance (elimination) to a long period of latency that produces tumor cell variants with increasing capacity to survive immune attacks (equilibrium) leading to eventually the emergence of clinically detectable malignant disease (escape) (Dunn et al. Citation2004a). It has also become clear that cancer immunosurveillance involves the contributions of both innate and adaptive immunity working together to protect the host against neoplastic disease. Compelling evidence indicated that innate immune cells comprise a critical arm of the immunosurveillance circuit. Recently, innate immune cell-based therapy has shown potent anti-tumor activity against hematologic malignancies and some solid tumors (Wang et al. Citation2019). Therefore, priming anti-tumor innate immune responses can provide appealing targets for therapeutic interventions especially in the context of convergent innate and adaptive anti-tumor immunotherapies (Moynihan and Irvine Citation2017). Since complement-dependent cytotoxicity (CDC) has an important contribution to the therapeutic effects of mAb-based therapies in the control and eradication of malignant cells (Wang et al. Citation2020), innate immune cell-based therapies that focus on triggering the CDC by antibody-independent mechanisms are predicted to have enhanced safety and efficacy. How novel immunogenic neo-antigens created by alterations in glycan biosynthesis can substaitianly rejuvenate the landscape of innate immune cell-based cancer immunotherapy is the focus of this article.
Glycans (carbohydrate chains) are integral part of the immune system’s ability to distinguish self from danger. Like pathogens, cancer cells have adapted to the selection pressure exerted by the immune system through harnessing their glycome (the total set of glycans in a biological species) to programme immune evasion. This is called aberrant glycosylation (an enzymatic process that links glycan sugars to other glyans, lipids or proteins), a universal feature of cancer cells found in essentially all tumor cells (Hakomori Citation2001), that give rise to immunogenic glycans that are collectively referred as tumor-associated carbohydrate antigens (TACAs) or cancer-associated glycans. The tumor ‘glyco-code’ denotes the specific glycan signature of tumor cells. TACAs function as immune targets that bridge innate immunosurveillance and adaptive anti-tumor immunity in clinical applications by allowing both attack on tumor cells and interference with the tumor-generated immunosuppressive factors. Furthermore, TACA expression on cancer cells is associated with organ tropism (the specificity of metastatic sites for each tumor entity) underlying extravasation and metastases (Ono and Hakomori Citation2004). Since they are barely expressed on normal tissues, TACAs have the advantage of tumor-specificity. TACAs have a powerful advantage of being pan tumor antigens on malignantly transformed cells which can be harnessed for development of novel cancer immunotherapies (Kieber-Emmons et al. Citation2017). In fact, both anti-TACA natural antibodies and vaccine-induced antibodies have demonstrated oncolytic properties (Brändlein et al. Citation2003; Varambally et al. Citation2004; Vollmers and Brändlein Citation2009; Makhoul et al. Citation2015). Several TACA-targeting approaches for cancer immunotherapy are being investigated in preclinical and clinical trials. These approaches include antibody-drug conjugates (ADCs), chimeric antigen receptor (CAR)-expressing immune cells and inhibiting anti-cancer immune responses by directly engagement of immune receptors on leukocytes (Chua and Durrant Citation2017; Rodrigues Mantuano et al. Citation2020). As an example, the anti- ganglioside GD2 (a tumor-specific marker for neuroblastoma) mAb dinutuximab is currently approved for the treatment of high-risk neuroblastoma in pediatric patients (Keyel and Reynolds Citation2018).
In reality, most of these TACA-targeting approaches are basically artificial analogs of lectins, efficient multivalent glycan receptors that are able to decipher the glyco-code on cell surfaces. In simple terms, lectins are natural glycan-binding proteins that closely mimic antibodies in binding specifically with certain antigens (glycans) but are themselves not produced in response to those antigens. As part of innate immunosurveillance, lectins recognize these antigens as ‘non-self’ which activates the lectin pathway (LP) to trigger CDC and they may also act as opsonins for phagocytosis (Singrang et al. Citation2021). This feature was utilized for many therapeutic applications. Notably, several plant lectins have been reported to have anti-viral properties and have been investigated as potential anti-viral agents (Balzarini Citation2007a). Similarly, the lectins’ inherent selectivity to bind distinctive glycans in tandem with identification and characterization of glycans as potential tumor markers enables coupling tumor recognition to innate immunity through glycan-dependent mechanisms (He et al. Citation2010; Wang et al. Citation2017). On the other hand, malignant transformation also results in altered expression of glycan-binding lectins which are usually absent in normal tissues. The immune lectin families relevant in cancer include selectins, sialic acid binding immunoglobulin-type lectins (Siglecs), galectins (Gal), macrophage galactose-type lectin (MGL) and dendritic cell specific ICAM-3-grabbing non-integrin 1 (DC-SIGN). Recent advances in chemical tools to study the specificity and dynamics of mammalian lectins have transformed our understanding of lectin-glycan interactions in vivo; and it has also become clear that individual lectin families have their own unique properties and peculiarities (Belardi and Bertozzi Citation2015). The cross-talk between immune lectins and cancer-associated glycans has immune-inhibitory properties that are exploited by tumor cells to drive immune suppression within the tumor microenvironment (TME) and facilitate immune evasion. Furthermore, targeting these inhibitory glycan-lectin interactions can be envisioned as a novel type of cancer immunotherapies (RodrÍguez et al. Citation2018). In support of the therapeutic potential of engineered TACA-binding lectins, by using several specific glycosphingolipid globotriaosyl ceramide (Gb3)-binding lectins of different origins (bacteria, fungi, plants, and animals), Siukstaite and colleagues confirmed that the variety of fold and binding site of Gb3-binding lectins results in differences in affinity and specificity, and inferred that the structural diversity of lectins was inspiring for designing a toolbox of artificial lectins that can be used in diagnostic or therapeutical strategies for cancer (Siukstaite et al. Citation2021). Recently, Imberty and colleagues demonstrated that SaroL-1, a novel pore-forming β-trefoil lectin from Salpingoeca rosetta, can exert cytotoxic activity on H1299 non-small cell lung cancer (NSCLC) cells characterized by increased cell surface Gb3 expression, only upon binding to the tumor-associated Gb3 (Notova et al. Citation2022).
Pitfalls of current immunotherapies for acute leukemias
Acute leukemia types are acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML). Acute lymphoblastic leukemia (ALL), monoclonal and/or oligoclonal proliferation of lymphoid progenitor cells, is the most common pediatric cancer (Mohseni et al. Citation2018). Despite successful cure of most children with B-cell precursor ALL (BCP-ALL) via modern chemotherapy regimens, relapsed/refractory (R/R) BCP-ALL remains a major source of childhood cancer-associated mortality (Tasian and Hunger Citation2017). Although ALL is less frequent in adults, it peaks again after the age of 50 years (Jemal et al. Citation2006) and imposes a tremendous challenge to treat (Faderl et al. Citation2010). Adult patients with BCP-ALL treated with the same multi-agent chemotherapy regimens used in pediatric BCP-ALL have higher rates of treatment-related toxicity as well as lower overall survival (OS) (Leonard and Stock Citation2017), and those patients with R/R BCP-ALL have dismal outcomes, with survival of less than 6 months when treated with conventional cytotoxic chemotherapy (Paul et al. Citation2019). The outcome for T-cell ALL is even worse than that for BCP-ALL. Most T-ALL disease recurrences occur within 2 years of diagnosis, and relapsed disease remains very difficult to salvage, with survival rates lower than 25% (Reismüller et al. Citation2009). On the other hand, AML is an intractable malignancy that if not completely eliminated at first attempt, becomes resistant to further treatments. Relapse after conventional chemotherapy remains the major cause of death after diagnosis of AML. AML is stigmatized with lack of leukemia-specific antigens and poor treatment outcomes despite reaching the ceiling of treatment intensification with conventional chemotherapy (Rubnitz and Kaspers Citation2021). Moreover, relapse remains the major cause of treatment failure after allogeneic stem cell transplantation (allo-SCT) in patients with ALL (Willasch et al. Citation2017). It carries a poor prognosis and is a common cause of death (Bajwa et al. Citation2013). The 5-year OS after relapse in adult patients is only 7% even after allo-SCT (Dinner et al. Citation2014). Again, posttransplant relapse is the greatest obstacle to the success of allo-SCT in AML (Yanada et al. Citation2021). Posttransplant relapse occurs in a considerable proportion of patients with AML (Yanada et al. Citation2020), and the prognosis remains dismal despite treatment with chemotherapy, donor lymphocyte infusion (DLI), and 2nd allo-SCT (Schmid et al. Citation2012; Bejanyan et al. Citation2015; Lim et al. Citation2018).
It is undeniable that immunotherapies have brought hope to patients with R/R acute leukemias (Shang and Zhou Citation2019). Instead of relying on the cytotoxic mechanisms of conventional therapy, cancer immunotherapies harness components of the immune system to induce long-term cancer remission (Ragoonanan et al. Citation2021). Antibodies have been viewed as ideal candidates or ‘magic bullets’ for the use in cancer immunotherapy that offers less off-target toxicities. The success of the first generation anti-CD20 monoclonal antibody (mAb) rituximab for the treatment of B-cell lymphomas inspired the development of more advanced antibody formats which invigorated the field of antibody-based immunotherapy. However, mAbs (also called naked antibodies) have limited efficacy. Strikingly, mAbs have historically been largely ineffective in AML (Short et al. Citation2020). Therefore, efforts shifted to improve their efficacy through developing several approaches including conjugating mAbs to potent cytotoxic drugs to generate antibody-drug conjugates (ADCs) (e.g. inotuzumab), or linking a pair of mAbs designed to harness cytotoxic T-cells to construct bispecific T-cell receptor-engaging (BITE) antibodies (e.g. blinatumomab), or fusing an extracellular antigen recognition moiety derived from a mAb (scFv) with intracellular signaling endo-domains to generate CAR- engineered T cells (i.e. CAR-T cells) (Caruana et al. Citation2014). ADCs, BITE and CAR-T cells represent the three major categories of immunotherapies currently approved for treatment of R/R acute leukemias. The main challenges that restrict the widespread use of these immunotherapies are resistance, toxicity and applicability. Primary and secondary/acquired resistances are key elements responsible for curtailing overall survival rates in patients treated with immunotherapy. Factors driving primary therapy resistance can be either tumor cell-intrinsic, determined by the traits of the tumor cell itself, or tumor cell-extrinsic, involving the cells in the stroma of the local TME (van Elsas et al. Citation2020). TME is a crucial determinant of metastatic dissemination as well as a detrimental factor limiting benefit from cancer immunotherapies. T-cell exhaustion is the classic tumor cell-extrinsic primary resistance mechanism that unequivocally facilitates the initial steps of the metastatic cascade (Asiry et al. Citation2021).
Another major flaw with cancer immunotherapies is their inability to eradicate cancer stem cells (CSCs). Substantial evidence from prior studies supports the cancer stem cell (CSC) hypothesis that proposes the existence within many tumors of a subpopulation of cells with stem cell characteristics (the CSC or cancer-initiating cells). The CSC concept was provoked by studies on leukemias and postulates that many tumors are hierarchically organized with these putative CSCs being at the top of the hierarchy (Reya et al. Citation2001; Cho and Clarke Citation2008). The CSC population, distinct from the bulk of the tumor, composed of highly resistant tumor cells, is thought to be responsible for relapse and explains why cancer immunotherapies can only improve the survival/ quality of life but cannot completely cure cancer patients. Although, the CSC concept also implies that targeted elimination of CSCs would be more effective than therapies targeting only the bulk of the tumor mass, the lack of CSC-specific markers incurs many therapeutic challenges (Santamaria et al. Citation2017). CSCs were initially discovered in AML where it is referred to as leukemic stem cells (LSCs) or leukemia-initiating cells (LICs) (Dick Citation2008). Although LSCs were best characterized in AML, stem cells have also been discovered in various other leukemias including, ALL, chronic myelogenous leukemia (CML), and chronic lymphocytic leukemia (CLL) (Misaghian et al. Citation2009). Later, the existence of CSC has been identified in many solid tumors, including brain, breast, and colon (Ailles and Weissman Citation2007). Recent works revealed that AML/ALL cells hijack HSC niches in the bone marrow (BM) and become LSCs at the expense of normal hematopoietic stem cells (HSCs) (Hira et al. Citation2017). Normally, HSCs reside in the bone marrow (BM) in a specialized microenvironment that harbor, protect, and support HSCs, known as BM microenvironment (BMM), or simply as BM niche. Niche hijack by AML/ALL cells and their transformation into LSCs is considered to be the most prominent cause of tumor recurrence (Azizidoost et al. Citation2017). Niche hijack is critical for cancer development as LSCs are quiescent and resistant to conventional chemotherapy. Niche hijack also implies that LSCs manipulate BM niche in their favor to create their own BM TME which acts as a protective niche for leukemic cells. TME mediates therapy resistance by promoting tumor cell growth/survival and contributing to the phenomenon of minimal residual disease (MRD). MRD is best viewed as a sanctuary of drug-resistant cancer that eventually leads to disease relapse (Meads et al. Citation2008). This highlights the need for TME-targeted therapies so that the subpopulation of LSCs can be eliminated by either targeting of LSCs in BM niches and/or preventing sequestration of leukemic cells in the BM. Furthermore, ‘by preventing the ways a tumour cell acts to modify normal microenvironments can stop cancer progression or at least make cancers more sensitive to treatment,’ Dr. Ingrid G. Winkler says.
In addition, cancer immmunotherapies had brought a new wave of unique toxicity profiles that are distinct from the toxicities of other cancer therapies (Kennedy and Salama Citation2020). Unfortunately, paucity of truly tumor-specific antigens compelled targeting tumor antigens that are co-expressed on normal tissues. The resulting direct attack on normal tissues that have the shared expression of the targeted antigen is called ‘the on-target off-tumor toxicity.’ The ensuing damage can be significant as is the case with T-cell mediated/engaging therapies (BITE and CAR-T cells) which, due to their potency and toxicity, ideally should either target tumor-specific targets or targets that are vastly over-expressed in tumors as compared with normal tissues (Strohl and Naso Citation2019). The fact that ‘on-target off-tumor’ toxicity resulting from the almost universal B-cell aplasia is the most striking toxicity associated with the anti-CD19 CAR-T cells can infer that the brisk immune response can be a double-edged weapon (Sun et al. Citation2018). B-cell aplasia occurs secondary to eradicating the entire B cell compartment as CD19 is also expressed on normal B lymphocytes. This scenario is even worse in AML with significant potential for on-target/off-tumor toxicity due to target antigen expression on normal hematopoietic stem/progenitor cells (HSPCs) that may lead to prolonged or permanent marrow aplasia (Lamble and Tasian Citation2019). This may spark contention around listing these therapies as targeted therapies in the first place and emphasizes the need for search for novel cancer neo-antigens escepcially that the success of any antigen-specific immunotherapy depends critically on the choice of target antigen (Cheever et al. Citation2009). Moreover, cost poses hindrances to the widespread availability of these immunotherapies, being among most expensive drugs on the planet!. In the United States, blinatumomab amounts to around $89 000 per cycle, the cost of inotuzumab is around $89 760 for the first cycle, $67 320 for subsequent cycles for patients in complete remission, and $89 760 for subsequent cycles for patients not in complete remission after the first cycle, and the price of tisagenlecleucel (an anti-CD19 CAR-T cell product) is set at $475 000. These prices do not include the hospital cost incurred for management of the infusion or related side effects, e.g. long-term (potentially lifelong) use of intravenous immunoglobulins or other costs related to these drugs. The cost-effectiveness of the current immunotherapies remains speculative given the limitations of the financial analyses done to date (Jain and Litzow Citation2018).
The crippling effect of T-cell exhaustion on T-cell mediated immunotherapies
T-cells destroy tumor cells by recognizing and reacting to tumor-associated antigens through their T-cell receptors (TCRs). An important stumbling block for the T-cell mediated immunotherapies is a biological self-defense mechanism often referred to T-cell exhaustion that occurs in chronic infections and cancer where T-cells are exposed to persistent antigen and/or inflammatory signals. This scenario is often associated with the deterioration of T-cell function: a state called ‘exhaustion’ (Wherry and Kurachi Citation2015). Exhausted T- cells have deprived T-cell effector function by losing their proliferative potential and cytotoxicity along with sustained expression of inhibitory receptors (exhaustion markers) and a distinct transcriptional state (Patil et al. Citation2015). Persistent tumor antigen stimulation, the presence of inhibitory immune cells and cytokines in TME, upregulated expression of multiple inhibitory receptors/immune checkpoints (ICs) or ‘don’t eat me’ signals, changes in T-cell-related transcription factors, and metabolic factors can all result in T-cell exhaustion (Tang et al. Citation2021).
Tumor expression of inhibitory ICs leads to tumor immune escape which is a hallmark of tumor growth (Prendergast Citation2008). The upregulation of ICs, also termed surface exhaustion markers, is one of the most important mechanisms of T-cell exhaustion in hematological malignancies. T-cell activation, through antigen recognition by the TCR, is tightly regulated by the balance between co-stimulatory and co-inhibitory signals ICs. Several co-stimulatory or co-inhibitory molecules on T-cells with their respective ligands are collectively known as B7-CD28 family. The main co-inhibitory ICs are the programmed cell death protein 1 (PD-1), T-cell immunoglobulin and mucin domain 3 (TIM-3) and Cytotoxic T-lymphocyte antigen-4 (CTLA-4). Many cancers exploit co-inhibitory ICs to escape immune surveillance (Ok and Young Citation2017), and blocking of such molecules to treat cancer has shown promising results in clinical trials (Ferris et al. Citation2016; Nghiem et al. Citation2016; Hao et al. Citation2017). PD-1 (CD279), a member of the CD28/CTLA4 family, and its ligand PD-L1 (CD274) are the most important T-cell exhaustion markers in hematological malignancies. The increased expression of PD-L1 can engage the PD-1 receptor on activated T cells inhibiting their function (Gianchecchi et al. Citation2013). TIM-3, a specific surface glycoprotein expressed on T-cells, is another co-inhibitory IC. As galectin-9 (Gal-9) receptor, TIM-3 induces T-cell apoptosis when combined with Gal-9. TIM-3-mediated interaction between bone marrow T-cells (as tumor-infiltrating lymphocytes) and leukemia cells was shown as a strong risk factor for relapse in pediatric BCP-ALL (Blaeschke et al. Citation2020). Although their functions are opposite, CLTA-4 and CD28 share the same ligands, B7-1 (CD80) and B7-2 (CD86), both of which are expressed on antigen-presenting cells (APCs). CTLA-4 is a co-inhibitory molecule with the ability to directly inhibit T-cell activation, as it counteracts CD28 co-stimulation by outcompeting its binding to their mutual ligands, CD80 and CD86 (Linsley et al. Citation1994; Riley et al. Citation2002). CTLA-4 expression is upregulated in patients with peripheral T-cell lymphoma, mycosis fungoides, and Sézary syndrome, but not seen in B-cell lymphoma (Xerri et al. Citation1997; Wong et al. Citation2006; Gibson et al. Citation2013).
Researchers are looking for strategies to revive exhausted T-cells. In 2018, both James P. Allison and Tasuku Honjo received Nobel Prize in Medicine and Physiology for their discovery of immunologic checkpoint blockade, a novel cancer therapy likened to taking the brakes off immune cells, freeing them to fight cancer (Wolchok Citation2018). However, reversing exhaustion is neither always desirable nor risk-free (Dolgin Citation2020). To demonstrate, as PD-1 and PD-L1 play a crucial role in peripheral immune tolerance, PD-1/PD-L1 inhibitors could lead to novel immune-related adverse events (irAEs) including colitis, hepatitis, myositis, pneumonitis, endocrinopathies, kidney injury, and skin toxicities (Wang et al. Citation2018). irAEs complications are more frequent with the use of CTLA-4-targeting agents compared to PD-1/PD-L1 inhibitors (Roth et al. Citation2021). In addition, resistance to PD-1/PD-L1 inhibitors is observed in some cancer patients, where T-cells may no longer be reinvigorated via anti-PD-1 treatment (Qin et al. Citation2019). Resistance to immune checkpoint blockers (ICB) can be either primary when patients do not respond to initial IC blockade or secondary/acquired which occurs in approximately one fourth to one third of patients who initially responeded to ICB but relapse over time despite receiving continued therapy (Sharma et al. Citation2017). In clinical practice, most patients still show limited efficacy with either a poor response or a transient reinvigoration soon to be resistant to ICB (Jiang et al. Citation2021). This is clearly manifested in solid tumors where the response rate of ICB therapy is less than 30% (Zhang et al. Citation2020). Counterintuitive results from the Andrea Schietinger Lab led the researchers to a shocking conclusion: ‘The brakes are there for a reason – to prevent the cells from getting overstimulated and dying,’ Dr. Schietinger says. Furthermore, as exhausted T-cells develop as a consequence of persistent antigen exposure, it can be inferred that the best way to avert T-cell exhaustion is to use the T-cell mediated immunotherapies in upfront therapy protocols.
B-cell precursor acute lymphoblastic leukemia
Blinatumomab
The CD3/CD19 BiTE mAb blinatumomab has shown promising results in patients with R/R BCP-ALL. Blinatumomab is an antibody-based T-cell-mediated immunotherapy that transiently engages CD3-positive cytotoxic T-cells to CD19-positive target B-cells inducing perforin-mediated cytotoxicity via granzyme entry into the BCP-ALL blasts, resulting in apoptosis and cell lysis. However, blinatumomab is fraught with many setbacks:
Unique toxicities. Neurological side effects are one of the most feared toxicities leading to both treatment interruption and discontinuation. Blinatumomab is also associated with the cytokine release syndrome (CRS), a severe systemic inflammatory response characterized by markedly elevated interleukin (IL)-6, IL-10, and interferon-γ (INF- γ) that can be life-threatening or even fatal (Teachey et al. Citation2013). CRS and neurotoxicity are the main adverse effects occurring coincidentally with T-cell activation. Infusion-related reactions (IRRs) are common and may not be distinguishable from mild/moderate cytokine release syndrome (low grade 1/2) (Topp et al. Citation2015). Hypogammaglobulinemia, cytopenias, and infections were also reported in patients treated with blinatumomab. Owing to such severe toxicities and in order to prevent the risks of the preparation and administration errors associated with the use of blinatumomab, the US Food and Drug Administration (FDA) requested specific warnings on the product label as well as the implementation of a Risk Evaluation and Mitigation Strategy Exit Disclaimer (REMS) (Yamazaki and Galluzzi Citation2017).
Resistance. Although around half of R/R BCP-ALL patients treated with blinatumomab achieve remission, early relapses after initial response to blinatumomab was the main hurdle that prevented responders from receiving allogeneic stem cell transplant (alloSCT) consolidation. Relapse of disease at extramedullary (EM) sites and loss of CD19 antigen expression are potential mechanisms of resistance which were common during blinatumomab failure in R/R BCP-ALL. CD19-negative relapses occurred in almost 40% of patients who responded to blinatumomab and subsequently relapsed. Sustained CD19-antibody pressure can result in lineage switches as described in KMT2A- and ZNF384-rearranged BCP-ALL (Jacoby et al. Citation2016; Oberley et al. Citation2018). EM disease progression can occur during blinatumomab therapy even when remission is attained in the BM (Aldoss et al. Citation2017). As blinatumomab rely on T-cell–mediated clearance of leukemic blasts, immune-based resistance mechanisms include increased expression of T-cell exhaustion markers. Increased expression of PD-L1 on leukemic blasts and its ligand PD-1 on patient T-cells strongly reduced the efficacy of blinatumomab-mediated lysis of target cells both in ex vivo and in vivo and occurs particularly in relapsed disease and can be increased further with blinatumomab treatment. The expression levels of PD-1 and TIM-3 are also higher among leukemia patients as compared to healthy controls and are upregulated during T-cell attack against leukemia (Köhnke et al. Citation2015; Feucht et al. Citation2016).
Questionable benefit in isolated CNS relapses. Because there is no evidence that blinatumomab crosses the blood-brain barrier or can treat CNS disease (Curran and Stock Citation2019); the benefit of blinatumomab in isolated CNS relapse is not known. This was redemonstrated in 2 international randomized clinical trials conducted by Brown et al. (Citation2021) and Locatelli et al. (Citation2021) between 2015 and 2019 on the effect of blinatumomab administered in combination with conventional chemotherapy compared with conventional chemotherapy alone in children with first relapse of BCP-ALL. The incidence of EM relapse in both studies was around 10% to 15% of patients but the role of blinatumomab for EM relapse was also unclear in both studies (Shukla and Sulis Citation2021).
Uncertain benefit in multiply relapsed leukemia. As inhibiting perforin activation abrogates blinatumomab-induced cytotoxicity, the efficacy of blinatumomab in patients with multiply relapsed leukemia whose cytotoxic T-cells are probably dysfunctional and likely perforin-deficient remains a speculation that requires further research (Löffler et al. Citation2000; Portell et al. Citation2013; Qin et al. Citation2018).
Administration. Blinatumomab requires continuous intravenous infusion due to its short half-life which can be cumbersome for many patients and their families.
Post-trasplant immune-related complications. Immune-related toxicities are theoretically increased in blinatumomab-bridged transplanted patients, plausibly because of immune alteration triggered by the treatment (Badar et al. Citation2021).
Anti-CD19 CAR-T cells
The adoptive transfer of patient-derived T-cells modified to express chimeric antigen receptors is another T-cell-mediated CD19-targeting immunotherapy that has demonstrated dramatic success in R/R BCP-ALL, albeit the response and durability of remission requires both exponential CAR-T cell expansion and persistence. Both CAR-T cell therapy and blinatumomab share unique toxicities attributable to utilization of T-cells for cytotoxicity to tumor cells. Hypogammaglobulinemia, cytopenias and increased risk of infections are also associated with CAR-T cell therapy. As with blinatumomab, CRS and neurotoxicity are commonly seen with CAR T-cell therapy. The most feared complication of CAR-T therapy is the immune effector cell-associated neurotoxicity syndrome (ICANS) that can occur either during CRS, after CRS has subsided, or in the absence of CRS. CRS is the most well-described cytokine-mediated toxicity that typically occurs during initial CAR-T cell expansion. However, the spectrum of cytokine-mediated toxicities also include late-onset life-threatening systemic inflammatory toxicities resembling hemophagocytic lymphohistiocytosis (HLH) or macrophage activation syndrome (MAS) occurring after the infusion of either the anti-CD19 CAR-T cell or blinatumomab. CD19/CD22 CAR-T cell therapy can causepersistent elevation of inflammatory cytokines along with pronounced natural killer (NK) cell lymphopenia leading to HLH-like toxicities (Lichtenstein et al. Citation2021; Masih et al. Citation2021). The severity and high incidence rate of these toxicities represents a therapeutic challenge.
Growing experience revealed that CAR-T cell-induced remissions are short-lived in a substantial number of patients owing to either poor CAR-T cell persistence associated with antigen-positive relapses and/or cancer cell resistance resulting from antigen loss or modulation associated with antigen-negative relapses. Antigen loss as a sequel to the eventual adaptation of tumor cells to the selective immune pressure of CAR-T cells is a major mechanism of CAR-T cell therapy failure. The type of co-stimulatory molecule (e.g. 4-1BB vs. CD28), rejection due to the murine component in tisagenlecleucel (an anti-CD19 CAR-T cell product), and T-cell exhaustion are considered important factors for poor CAR-T cell persistence (Shah and Fry Citation2019). The PD-1/PD-L1 axis known to limit response to blinatumomab may also limit CAR-T cells, more often in CD28-containing CAR-T cells (Long et al. Citation2015; Rupp et al. Citation2017; Jacoby Citation2019). As the currently available CAR-T cells are only CD19-directed, flow detectable CD19-positive disease must be present in the BM at relapse. Unfortunately, the increasing incidence of CD19-negative relapses not only confers resistance to therapy but is also difficult to treat (Shah et al. Citation2019). Even diminution of antigen expression can be sufficient for the development of resistance to initially effective CAR-T cell therapy. Likewise, the antecedent blinatumomab therapy that depletes CD19-positive cells renders the anti-CD19 CAR-T cell therapy ineffective (Pillai et al. Citation2019). Therefore, receipt of prior therapy with blinatumomab is an exclusion criterion of the pivotal registration trial of tisagenlecleucel in patients with ALL (Maude Citation2018). In addition to antigen loss and exhaustion of CAR-T cells, microenvironment-mediated upregulation of antiapoptotic pathways (as the case in many hematological cancers) have also been identifed as major mode of tumor escape from CAR-T cell therapy (Rasche et al. Citation2022).
A link between poor CAR-T cell granule-mediated cytotoxicity and subsequent secondary inflammation was demonstrated in a murine model of perforin-deficient CAR-T cells. This is a significant finding since perforin-deficient CAR-T cells may more accurately reflect the human CAR-T cells derived from patients with multiply relapsed leukemia. Unlike blinatumomab, perforin-deficient CAR-T cells could mediate anti-leukemia activity despite the absence of perforin, although with reduced potency (Ishii et al. Citation2020). The full scope of interactions intertwining the adoptively transferred CAR-T cells with recipient’s immune system remains elusive. Given the extremely high marketed prices of CAR-T cell therapy, serious concerns are raised over the cost-effectiveness of this therapy.
Inotuzumab
The ability of mAbs acting as vehicles for therapeutic delivery is typified by the CD22-directed-ADC inotuzumab ozogamicin (InO) which consists of calicheamicin (a cytotoxic anti-tumor antibiotic that causes double-strand DNA breaks) linked to a humanized antibody directed against CD22. Despite the high expression of CD22 on malignant B cells (>90% of BCP-ALL blasts), naked mAbs that target CD22 alone have had little activity in clinical trials. InO has demonstrated efficacy in patients with R/R CD22-positive BCP-ALL and ongoing studies are evaluating the value of InO as part of upfront treatment in combination with chemotherapy (Yurkiewicz et al. Citation2018). Unlike Blinatumomab or the anti-CD19 CAR-T cells which both need a debulking regimen if used for patients with high tumor load, Ino holds the advantage of not needing any prior debulking therapy.
InO-related adverse events are neutropenia, thrombocytopenia, infusion-related reactions, tumor lysis syndrome, and prolonged QT syndrome (Jain and Litzow Citation2020). The calicheamicin component of the drug is associated with the hepatotoxicity, especially the potentially life-threatening veno-occlusive disease (VOD), which can occur either with the use of InO alone or with alloSCT. This toxicity is especially worrisome since the goal of treatment is to proceed to alloSCT after achieving complete response (Thomas Citation2014). Although InO efficacy is independent of CD22 positivity as even low CD22 expression levels can result in high intracellular calicheamicin levels (Zwaan et al. Citation2003; Dijoseph et al. Citation2007; DiJoseph et al. Citation2011; de Vries et al. Citation2012; Lanza et al. Citation2020), low baseline CD22 expression has been identified as a potential biomarker of poor InO response and; vice versa, patients with leukemic blast CD22 positivity ≥90% had improved outcomes (Shah et al. Citation2020; Kantarjian et al. Citation2021).
Acute myeloid leukemia
AML is an aggressive disease with unfavorable OS due to inevitable chemotherapy resistance and high relapse rate due to mainly the persistent existence of LSCs. AML LSC populations consist of clonally diverse LSC populations that likely form the basis of emergence and selection of resistant subclones under therapies (Morita et al. Citation2020). To reiterate, LSCs home to and engraft within the osteoblast-rich area of the BM where they are protected from chemotherapy-induced apoptosis (Ishikawa et al. Citation2007). These residual LSCs originating from AML cells that anchor in the BM niche following chemotherapy are thought to mediate treatment refractoriness and relapse in AML (Ho et al. Citation2016; Yao et al. Citation2021). Furthermore, transient or long-term quiescence, the latter referred to as dormancy, is likely the main reason for LSC (and normal HSCs) resistance to cell-cycle targeting chemotherapeutic agents, such as cytarabine (Ara-C), azacitidine and decitabine (Essers and Trumpp Citation2010; Cogle et al. Citation2016).
Recently, targeting of LSCs has become a prime issue for AML therapy (Anguille et al. Citation2012). Moreover, most antigens of potential immunotherapeutic interest in AML are also expressed on HSPCs. Therefore, the challenge inherent in developing immunotherapies for AML has been to identify targetable cell surface proteins on AML blasts that spare the normal HSPCs. As both CD33 and CD123 are almost ubiquitously expressed on AML blasts, they were contemplated as appealing target antigens. However, they are not ideal targets as they are also expressed on healthy HSPCs (i.e. non-leukemia specific antigens) (Ehninger et al. Citation2014).
Gemtuzumab
Given its low cell surface density, slow antibody-induced internalization, and expression on hematopoietic progenitor cells, CD33 is a challenging target that is if not precisely targeted would cause refractory cytopenias (Walter Citation2014; Stokke and Bhojwani Citation2021). Several CD33-targeted therapeutics has been destined to failure with the exception of gemtuzumab ozogamicin (GO). GO and InO are ADCs with distinct molecular targets whose cytotoxicity is dependent on the highly potent chemotherapeutic agent calicheamicin. Nonetheless, several factors contribute to resistance to GO and the prognosis of children with R/R AML remains poor (Zwaan et al. Citation2010; Hoffman et al. Citation2021). AML blasts have higher intrinsic resistance to calicheamicin leading to defective calicheamicin-induced cell-kill. Moreover, the lack of CD33 expression on LSCs is another mechanism of resistance to GO. There is lack of benefit of GO in AML patients with lower CD33 expression (wild type FLT3/NPM1 and the core-binding factor (CBF)- AML). CD33 expression on blasts is independently prognostic for outcomes regardless of genetic associations in pediatric AML. In younger and older adults, a positive correlation also exists between the percentage of CD33-positivity and GO benefit in non-CBF AML (Pollard et al. Citation2012; Khan et al. Citation2017). Efflux mediated by P-glycoprotein (Pgp) is the third mechanism of resistance. The efficacy of GO depends on the expression of Pgp which correlates with the multidrug resistance (MDR) phenotype. Actually, the cytotoxic effects of both GO and InO are inversely proportional to the amount of Pgp which indicates that Pgp contributes to their clinical resistance (Linenberger et al. Citation2001; Walter et al. Citation2007). As an ADC that employ calicheamicin, prior treatment with GO has also been shown to increase the risk of VOD (Richardson and Corbacioglu Citation2020).
Flotetuzumab
The high expression of CD123 in patients with R/R AML correlates with poor outcomes which make CD123 another attractive therapeutic target in AML (Testa et al. Citation2002; Vergez et al. Citation2011; Kandeel et al. Citation2020). Although CD123 expression intensity is higher on AML blasts and LSCs, CD123 is also expressed in normal HSPCs resulting in the risk of long-lasting or even permanent myelosuppression (Haubner et al. Citation2019). Similar to blinatumomab, flotetuzumab is an investigational bispecific antibody-based molecule to CD3ϵ and CD123 engineered in a Dual Affinity Re-Targeting (DART) format that is currently being evaluated in an ongoing phase 1/2 clinical trial in adult patients with R/R AML (NCT02152956), and in another ongoing phase 1 clinical trial in pediatric patients with R/R AML (NCT04158739). The published findings from the adult trial indicate that flotetuzumab provides encouraging evidence of clinical activity in patients with R/R AML. IRRs/CRS remain the most frequent and significant toxicities observed following treatment with flotetuzumab; albeit with increased frequency and severity as compared with T-cell mediated B-cell immunotherapies (Uy et al. Citation2021). As has been reported for BCP-ALL, T-cell exhaustion hallmarked by reduced T-cell function might contribute to primary or secondary resistance to bispecific antibodies in AML patients (Knaus et al. Citation2018; Daver et al. Citation2021). PD-L1 upregulation on AML cells is a common adaptive immune escape strategy that resulted in reduced efficacy of flotetuzumab in vitro. Moreover, patients with R/R AML who progressed early (within 2 weeks) on flotetuzumab treatment had higher baseline levels of PD-L1 on AML cells indicating that AML blasts expressing high levels of PD-L1 are less susceptible to flotetuzumab-mediated killing in vivo as well (Rettig et al. Citation2017).
CAR-T cells
The primary challenge limiting the use of CAR-T cells in myeloid malignancies is the absence of AML-specific antigens; as myeloid antigens are often co-expressed on normal HSPCs. AML cells share the same cell surface antigens with healthy HSPCs and their myeloid and/or lymphoid progenitors including CD123, CD34, CD33, and many others (Cummins and Gill Citation2019). To illustrate, CAR-T cells directed against CD33 and CD123 have both shown highly potent anti-tumor activity in pre-clinical models (Gill et al. Citation2014; Kim et al. Citation2018; Petrov et al. Citation2018) but sustained complete responses have not yet been reported (Mardiros et al. Citation2015; Wang et al. Citation2015b).
As CAR-T cells were also unable to differentiate between normal and cancerous cells, targeting CD33 and CD123 has the potential to elicit long-term myelosuppression. Depletion of normal myeloid progenitors leads to intolerable BM failure and is ultimately fatal due to neutropenic infections and bleeding complications. The manufacture of CAR-T cells in patients with active AML is another challenge since AML cells secrete soluble factors that inhibit T-cell proliferation (Orleans-Lindsay et al. Citation2001). Inhibition of T-cell expansion can occur also by prior exposure to T-cell-damaging chemotherapy. While many barriers limit the full therapeutic potential of CAR-T cells, substantial efforts have been invested in developing and testing a variety of solutions (Mardiana and Gill Citation2020). Currently there are 26 ongoing clinical trials to assess the use of CAR-T cell therapies in AML, including 12 that are specifically for anti-CD123 CAR-T, 5 for anti-CD33 CAR-T, and 3 that are investigating CAR-T cells that target multiple antigens (clinicaltrials.gov) (Carter et al. Citation2020).
T-cell precursor acute lymphoblastic leukemia
T-cell acute lymphoblastic leukemia (T-ALL) is biologically distinct from its B lymphoblastic (B-ALL) counterpart (Raetz and Teachey Citation2016), rendering T-cell ALL blasts more resistant to conventional chemotherapeutic drugs used to treat BCP-ALL blasts (Pieters et al. Citation1993). T-cell and B-ALL also differ clinically and even the National Cancer Institute (NCI) risk stratification criteria for BCP-ALL do not apply on T-cell ALL (Teachey and Pui Citation2019). Molecular biomarkers are not yet included in the risk stratification of newly diagnosed patients with T-ALL and lack of predictive molecular biomarkers at diagnosis remains an unmet need. There has been remarkably little progress in the treatment of R/R T-ALL associated with dismal prognosis. Unlike other leukemias, targeted therapies for patients with R/R T-ALL remain limited. Ironically, if immunotherapy for AML is still in infancy, immunotherapy for T-cell ALL has not yet been born.
Daratumumab
Recently, the anti-CD38 mAb daratumumab (approved for treatment of R/R multiple myeloma) had demonstrated efficacy in T-ALL in pre-clinical animal studies (Bride et al. Citation2018). Currently, an ongoing early phase 2 trial (NCT03384654; EudraCT 2017-003377-34) seeks to evaluate the efficacy and safety of daratumumab in pediatric and young adult patients with R/R T-ALL or R/R BCP-ALL.
CAR-T cells
The adaptation of CAR-T cells to T-cell malignancies has been slower than those targeting B-cell malignancies. The use of CAR-T cells in T-cell malignancies was hampered by many challenges implicit in the design and implementation of immunotherapies for T-cell malignancies. The three main challenges seen in adapting CAR technology for T-cell malignancies are: the fratricide effect (CAR-T cells share the same surface markers with their malignant T-cell targets resulting in rapid self-extinguishing and limited expansion of the CAR-T cells), T-cell aplasia (CAR-T cells would also target healthy T-cells resulting in profound immunosuppression and deadly infections), and product contamination (failure to isolate normal T-cells from malignant T-cells; even single malignant cell contamination can result in antigen-positive relapse). Despite the complex manufacturing processes, several preclinical and clinical trials designed to test CAR-T cell products are currently underway (Fleischer et al. Citation2019; Diorio and Teachey Citation2020; Cordo et al. Citation2021).
Complement-dependent cytotoxicity for cancer immunotherapy
The complement system sits at the center of the interplay between the adaptive and innate immune responses. It aids in the destruction of microorganisms and damaged cells, including cancer cells. Antibodies (including mAbs) trigger CDC through activation of the classic complement pathway (CP) which is the major effector mechanism of the antibody-mediated immunity. The alternative (AP) and LP are the effector mechanisms of the innate immunity and can also effectuate CDC by antibody-independent immune responses. Initiation of the three complement pathways converge at the point of C3 activation (the central and most abundant component of the complement system), and subsequently result in activation of the common terminal lytic pathway that culminates with the formation of the multiprotein complex (C5b, C6, C7, C8, and C9) known as the membrane attack complex (MAC). MAC is inserted as pores of up to 11 nm into the cell membrane inducing loss of membrane integrity and disruption of the proton gradient across the membrane resulting in cell lysis and death. Activated complement components act also as opsonins, and thus contribute to phagocytosis.
Antibody-mediated complement-dependent cytotoxicity
mAbs acquire the cell killing effects by antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and CDC (Hendriks et al. Citation2017). The importance of CDC for the action of mAbs in the control and eradication of malignant cells has been best illustrated by the mechanism of B-cell depletion with rituximab (Golay et al. Citation2000; van Meerten et al. Citation2006; Zhou et al. Citation2008; Weiner Citation2010). One of the mechanisms for rituximab resistance involves the upregulation of membrane complement regulatory proteins (mCRPs). mCRPs are complement-inhibitory proteins that confer protection against the potentially deleterious uncontrollable CDC on normal cells and include CD35, CD46, CD55, and CD59 (O’Brien et al. Citation2021). This protection is a two-edged sword as it can also over-regulate the complement system to the extent that enables tumor cells evade CDC. Many strategies have been adopted in an attempt to overcome rituximab resistance induced by deficient CDC. The addition of CD46 depleter (Ad35K++ protein) to rituximab has demonstrated increased rituximab-mediated killing in preclinical studies (Richter et al. Citation2016). Adding fresh frozen plasma (FFP) with rituximab to overcome complement depletion resulted in a rapid and dramatic clinical response in all rituximab-resistant CLL patients (Rezvani and Maloney Citation2011).
mCRPs also may function as biomarkers of malignant transformation and derive the therapeutic resistance in numerous cancer types which highlights the critical role of the clinical application of cancer-specific mCRP blockade in cancer treatment (Geller and Yan Citation2019). Both downmodulation and the use of mAbs blocking CD55 and CD59 sensitized tumor cells to CDC in rituximab-resistant patients (Ziller et al. Citation2005; Mamidi et al. Citation2015). Moreover, inhibition of mCRPs by neutralizing antibodies or posttranscriptional gene silencing increases CDC of various tumor cells (Jurianz et al. Citation1999; Zell et al. Citation2007; Geis et al. Citation2010; Bellone et al. Citation2012). The dependence of rituximab on CDC for its in vivo efficacy had resulted in the clinical approval of the next-generation anti-CD20 antibody Ofatumumab for the treatment of patients with CLL in 2009. Ofatumumab generates greater CDC activity leading to more potent tumor cell lysis than rituximab (Weiner et al. Citation2010).
Antibody-independent complement-dependent cytotoxicity
The model of paroxysmal nocturnal hemoglobinuria (PNH) is intriguing to be imitated by cancer immunotherapy. PNH is caused by defective synthesis of the glycosylphosphatidylinositol-anchored proteins (GPI-APs) CD55 or CD59, mCRPs that normally inhibit complement-mediated cell lysis by the AP, leading to chronic intravascular hemolysis (Brodsky Citation2014). The uncontrolled complement-mediated lysis of erythrocytes is mediated by the uninhibited MAC formation. The mechanism of CDC in PNH occur independent of antibodies. Triggering CDC in an antibody-independent fashion can also be initiated by the LP. The LP plays a critical role in host defense against variety of infections (Ali et al. Citation2012; Mazalovska and Kouokam Citation2018; Tang et al. Citation2018; Mnich et al. Citation2020; Świerzko and Cedzyński Citation2020) through recognizing ‘foreign’ particles and macromolecules and promoting their elimination either by opsonization (enhancing their uptake by phagocytic cells) or lysis (via formation of the MAC).
Activation of the LP is similar to the CP except that it bypasses antibodies to target activation on pathogens. Instead, the LP is activated cascadically upon recognition of glycans by 2 distinct major lectin groups: collectins (mannose-binding lectin (MBL), collectin-10, collectin-11) and ficolins (ficolin-1, ficolin-2, ficolin-3) through association with proteins of the MBL-associated serine protease (MASP) family. Collectins and ficolins are equivalent to C1q in the CP and act as pattern recognition receptors (PRRs) reactive against pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) (Cedzyński and Świerzko Citation2020). The MASPs (−1, −2, and −3) are structurally and functionally similar to the proteolytic enzymes, C1r and C1s, of the CP (Carroll and Sim Citation2011). The lectin-MASP complex results in activation of the proenzyme forms of MASPs. MASP-1 and -2 are active proteases, but only MASP-2 cleaves both C4 and C2 to generate the same C3 convertase (C4b2a) as the CP. In fact, MASP-2 has intrinsic ability to self-activate, binds individually to MBL with high affinity and cleaves C4 and C2 (and hence fulfills the roles of both C1r and C1s in C1) indicating that reconstitution of MBL with MASP-2 alone is sufficient to trigger complement activation (Vorup-Jensen et al. Citation2000; Thielens et al. Citation2001). Subsequent steps in the LP are essentially the same as in the CP and completion of the complement cascade ensues with the generation of the MAC.
Glycobiology
The biological significance of glycans
Most of human proteome (the total set of proteins in a biological species) do not remain ‘naked’ but undergo extensive post-translational modifications. As a matter of fact, the most common post-translational modification of proteins is glycosylation (Opdenakker et al. Citation1993). Around 70% of the human proteome (including 80% of membrane proteins) is glycosylated (Apweiler et al. Citation1999; Arnaud et al. Citation2013). Golgi apparatus is the main site of glycosylation but some glycosylation also takes place in other organelles (endoplasmic reticulum and the plasma membrane) (Stanley Citation2011; Moremen et al. Citation2012). Glycan biosynthesis runs in an orderly stepwise process orchestrated by glycosyltransferases, special enzymes that add specific sugar residues to growing glycan chains, and glycosidases responsible for the fragmentation of glycoconjugates by catalyzing the hydrolysis of glycosidic linkages; however, they can also be used for their synthesis (Flitsch Citation2012). The addition of N- and O-glycans affects intracellular processes like the folding and trafficking of most glycoproteins. The linkage of distinct glycan sets to cell surface proteins and lipids form the glycocalyx (the pericellular matrix) where glycans are densely expressed on the outermost layer of the cell surface which can extend more than 30 nm from the plasma membrane on some cells (Martinez-Palomo et al. Citation1969). Glycocalyx can promote or hinder the binding of canonical protein ligands to their cell-surface receptors, as well as mediate ligand-independent receptor clustering and activation (Griffin and Hsieh-Wilson Citation2016). In cancer, glycocalyx is vital for cancer cell adhesion, migration and invasion; and therapeutic manipulation of glycocalyx definitely impedes its invasive potential.
The diverse structures of glycans underline their varied functions (Table ). Glycans play fundamental role in numerous biological processes, including cell adhesion, immune regulation, infection, inflammation, and cancer metastasis. The glycans’ potential as a therapeutic target led to the development of glycomimetics. Glycomimetics, a recent trend of glycan engineering, are ‘drug-like’ compounds which mimic the structure and function of native carbohydrates (Hevey Citation2019). The paradigm of a glycomimetic drug in the classical sense is oseltamivir (Ernst and Magnani Citation2009). Both influenza medications oseltamivir and zanamivir are sialic acid mimetics that function as inhibitors of the influenza sialic acid releasing enzyme sialidase (neuraminidase, the ‘N’ in ‘H1N1’) to prevent virion release from infected cells (von Itzstein Citation2007). Nevertheless, paucity of good data has undermined previous findings for oseltamivir’s prevention of complications from influenza (Jefferson et al. Citation2014a). A cochrane review published in 2009 revealed that the company behind oseltamivir funded research into the drug, and much of that research failed to emphasize the likelihood of ‘serious adverse events, especially neuropsychiatric events’ associated with taking the drug (Cohen Citation2009). In treatment trials, oseltamivir reduced the time to first alleviation of symptoms by only 16.8 hours in healthy adults and by 29 hours in healthy children (Jefferson et al. Citation2014b). The side effects included nausea, diarrhea, insomnia, abdominal pain, and headache (Moscona Citation2005). However, the most important serious adverse events were neuropsychiatric events such as depressed mood, behavior disturbance, panic attack, suicidal ideation, delusion, delirium, convulsion, and encephalitis (Gupta et al. Citation2015). Another similar systematic review concluded that the combination of diagnostic uncertainty, risk for virus strain resistance, possible side effects and financial cost outweigh the small benefits of oseltamivir or zanamivir for the prophylaxis and treatment of healthy individuals (Michiels et al. Citation2013). The questionable risk-benefit ratio of neuraminidase inhibitors should incite shifting the focus from glycan engineering (i.e, glycomimetics) to a new landscape of lectin-based therapeutics.
Table 1. Structure and classification of glycans.
Glycosaminoglycans and proteoglycans
Rather than existing in a free state, glycans usually exist as glycoconjugates either conjugated to proteins (glycoproteins, proteoglycans and GPI-APs) or conjugated to lipids (glycolipids). However, glycans can also be secreted without conjugation to other macromolecules in the form of glycosaminoglycans (GAGs). GAGs are crucial to the formation of the glycocalyx. They are a family of highly sulfated, complex, unbranched (linear), often long, polysaccharides with a repeating disaccharide unit. GAG chains, the essential functional parts, are often highly sulfated, with resulting capability to bind cytokines, chemokines, or growth factors. Based on the difference of repeating disaccharide units comprising GAGs, they can be categorized into four main groups: heparin/heparan sulfate (HS), chondroitin sulfate (CS)/dermatan sulfate (DS), keratan sulfate (KS), and hyaluronan (also known as hyaluronic acid, HA).
GAGs can either found free in the extracellular matrix (ECM) or occur in proteoglycans. Proteoglycans are highly glycosylated glycoproteins found in high concentrations within the ECM of connective tissues. They consist of a core protein that, in addition to containing canonical N-glycans and O-glycans, one or more GAG chains are covalently attached to serine (Ser) or threonine (Thr) residues (O-linked glycosylation). The negatively charged carboxyl and sulfate groups of the GAG component are important for attracting water into tissues (Hellicar et al. Citation2022). The highly sulfated motifs of HS of syndecan-1 (CD138), a cell surface proteoglycan highly expressed in multiple myeloma cells, play crucial roles in multiple myeloma cell survival, proliferation, and metastasis (Delcommenne and Klingemann Citation2012). High serglycin (the major secretory-vesicle proteoglycan in multiple myeloma) levels are present in BM aspirates of >30% of newly diagnosed multiple myeloma patients, and are required for adhesion, in vivo growth, and vascularization of multiple myeloma cell (Skliris et al. Citation2013; Purushothaman and Toole Citation2014). Serglycin level is correlated with drug resistance in hematological malignancy cell lines (Beyer-Sehlmeyer et al. Citation1999). Serglycin expression was found to distinguish AML from ALL. In contrast to myeloperoxidase, serglycin was found to be a selective marker for immature myeloid cells, distinguishing AML from Philadelphia chromosome-negative chronic myeloproliferative disorders (Niemann et al. Citation2007).
Sialic acids: the decorative masks of cancer
Biochemistry
Glycans are primarily constructed from ten monosaccharides in humans: glucose (Glc), galactose (Gal), N- acetylglucosamine (GlcNAc), N-acetylgalactosamine (GalNAc), fucose (Fuc), xylose (Xyl), sialic acid (Neu5Ac), glucuronic acid (GlcA), mannose (Man) and iduronic acid (IdoA). The generic term sialic acids (Sias), also called neuraminic acids, defines a large family of various N- and O-derivatives of the basic neuraminic acid molecule (Neu), 5-amino-3,5-dideoxy-2- nonulosonic acid (Schauer et al. Citation2011). Sialic acids are α-keto acids with a nine-carbon backbone that usually do not exist in free sugar forms in nature (Lih and Wu Citation2017). Instead, sialic acids are often found in all cell types as the terminal (outermost) saccharide on branches of N- and O-linked glycoproteins, proteoglycans, glycolipids, and occasionally capping side chains of GPI anchors. They play important roles in various biological processes largely in two ways, one related to their hydrophilic and acidic properties exerting physicochemical effects on glycoconjugates to which they are attached, and the other as recognition sites or in an opposing fashion as masking sites (Schauer Citation2000; Miyagi and Yamaguchi Citation2007). The biological versatility of sialic acids is due to their large structural diversity that arises from the natural modifications and the different linkages (α2-3/6/8) to underlying glycans and glycoconjugates. The terms sialoglycans and sialoglycoconjugates indicate sialylated glycans and glycoconjugates respectively. The sum total of the diversity of sialoglycans in a cell or organism is dubbed its ‘sialome.’
N-acetyl neuraminic acid (Neu5Ac or NANA) is the most common form of sialic acids in humans. Neu5Ac is synthesized from Glc through uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) as a key intermediate for sialic acid metabolism. The Neu5Ac synthesized in the cytosol is activated in the nucleus by conjugation to cytidine 5-monophosphate (CMP) to form CMP-Sia by CMP-sialic acid synthetase (CMAS). CMP-Sia is then transported into the Golgi apparatus where various sialyltransferases use the activated sugar nucleotides, CMP-Sia, as donor to incorporate the negatively charged Sia residues into glycans via distinct glycosidic linkages (α2-3/6/8) to form glycoconjugates which are secreted or expressed on cell surfaces.
The sialic acids found in mammalian organisms vary in their substituent at C5, which in Neu5Ac is an acetylated amino group, in N-glycolylneuraminic acid (Neu5Gc) a glycolylated amino group and in 2-keto-3-deoxy-nonulosonic acid (Kdn) a hydroxyl group (D’Addio et al. Citation2020). The 5-amino group can also exist in nature in non-N-acylated form, giving rise to neuraminic acid (Neu). Neu5Ac, Neu5Gc, Kdn, and Neu collectively comprise the four ‘core’ Sia molecules. Neu5Gc is formed in the cytosol from CMP-Neu5Ac by CMP-Neu5Ac hydroxylase (CMAH)-catalyzed reaction (Varki Citation2001). CMAH gene is inactive in humans, and although Neu5Gc is common in mammals, it is not synthesized by our species (Varki et al. Citation2017). Although humans are incapable of synthesizing Neu5Gc due to an inactivated CMAH gene, the accumulation of Neu5Gc from dietary sources (especially red meat of mammalian origin) in human tissues can be attributed to the irreversibility of the CMAH reaction (CMP-Neu5Ac → CMP-Neu5Gc) (Bergfeld et al. Citation2012). As a result, Neu5Gc can be metabolically incorporated and displayed on the glycocalyx of the human epithelia and associated carcinomas (Pearce and Varki Citation2010). Neu5Gc-containing glycoconjugates have been found on human cancer cells and in various human tissues (Kooner et al. Citation2019). For example, the ganglioside GM3 containing the non-human Neu5Gc (Neu5Gc-GM3) is relatively specific for different types of cancer (Samraj et al. Citation2014). Neu5Gc is a foreign antigen (xeno-autoantigen) against which varying levels of anti-Neu5Gc antibodies (xeno-autoantibodies) are elicited (Leviatan Ben-Arye et al. Citation2017; Dhar et al. Citation2019). These xeno-autoantibodies are implicated in ‘serum sickness’ and chronic inflammation (‘xenosialitis’) which can progress into various diseases including cancer, type 2 diabetes and atherosclerosis (Samraj et al. Citation2018; Dhar et al. Citation2019).
The four ‘core’ Sia molecules can also undergo various natural modifications that impart a second level of diversity as they sometimes carry one or more additional substitutions on the hydroxyl groups at C-4, C-7, C-8, and C-9 (O-acetyl, O-methyl, O-sulfate, or O-lactyl groups). The most common modification of sialic acids is O-acetylation preferentially occuring at the C-4, C-7, C-8, and C-9 hydroxyl groups of the nonulosonic acid and sialic acid backbone. However, because O-acetyl esters from C-7 and C-8 positions are known to spontaneously migrate to C-9 even under physiological conditions, O-acetylation at C-9 is considered the most common biologically occurring modification (Vandamme-Feldhaus and Schauer Citation1998). O-acetylated sialic acids (O-Ac-Sias) derivative of Neu5Ac include 5-N-acetyl-4-O-acetyl neuraminic acid (Neu4,5Ac2), 5-N-Acetyl-7-O-acetyl neuraminic acid (Neu5,7Ac2), 5-N-Acetyl-8-O-acetyl neuraminic acid (Neu5,8Ac2) and 5-N-Acetyl-9-O-acetyl neuraminic acid (Neu5,9Ac2) (Varki et al. Citation2015a). Sialyltransferases – the enzymes responsible for the addition of sialic acid to growing glycoconjugate chains – conjugate O-Ac-Sias (with CMP as leaving group) to the glycans of glycoproteins and glycolipids that exit the Golgi system via the secretory pathway. Humans possess 20 sialyltransferase isoenzymes (Bowles and Gloster Citation2021). ST3Gal1 and ST6Gal1 are the most widely studied sialyltransferases. Studies with cell lines and mice suggest that the sialyltransferase ST6GAL1 installs Neu5,9Ac2 on N-glycans, ST3GAL1 is the enzyme responsible for attaching Sia residues to T antigens in many types of cancer (adds O-Ac-Sia to O-glycans), and ST3GAL5 and ST8SIA1 can transfer O-Ac-Sia to gangliosides that can carry O-Ac-Sia on the terminal and possibly on the inner sialic acid (Visser et al. Citation2021). ST6GAL1 is the primary enzyme responsible for α2-6 sialylation of N-glycans on selected glycoproteins. Upregulated ST6Gal1 is associated with pro-survival pathways, and its sialylated products can inhibit tumor cell apoptosis and activate growth factor pathways (Park and Lee Citation2013; Chakraborty et al. Citation2018). High ST6GAL1 has been linked with increased metastatic potential and poor survival in multiple types of cancer (Gessner et al. Citation1993; Recchi et al. Citation1998; Petretti et al. Citation1999).
Sialylation and cancer
Sialic acids are normally expressed on almost all cell types. It is difficult to overstate the importance of biological and pathological roles of sialic acids in humans. By binding to the sugar chains of glycoconjugates and changing their conformation, intermolecular interactions, and/or half-life, sialic acids mediate cell–cell recognition, communication, aggregation, development, carbohydrate–protein interactions, controlling the lifetimes of glycoconjugates in organisms, mediating bacterial and viral infections, tumor growth and metastasis, with role in immunology, microbiome, cell signaling, reproduction, and biology of nervous system (Ghosh Citation2020). Sialic acids facilitate the infection process by either serving as binding sites for human pathogens or are utilized by microorganisms for molecular mimicry (Stencel-Baerenwald et al. Citation2014; Heise et al. Citation2018). Many viruses use sialic acids as receptors for binding to target cells, which is the critical first step for viral infection. Examples of these receptors or viral lectins include the sialic acid-binding hemagglutinin glycoproteins (HAs) of human Influenza A virus (IAV) and the hemagglutinin-esterase-fusion (HEF) protein of Influenza C viruse. Sialic acid-binding proteins of bacteria include bacterial lectins like adhesins and toxins (e.g. Clostridium botulinum toxin) (Ghosh Citation2020). On the other hand, molecular mimicry enables microbial pathogens to evade the host immune system by decorating themselves with sialic acids (Vimr et al. Citation2004; Varki Citation2008; Varki and Schauer Citation2009).
Augmented sialylation of tumor cells (hypersialylation) has been correlated with a metastatic phenotype and poor prognosis in patients with cancer (Amado et al. Citation1998; Baldus et al. Citation1998). Hypersialylation occurs either via the upregulation of sialyltransferases or the downregulation of sialidases (neuraminidases) or a combination of both (Büll et al. Citation2014; Peixoto et al. Citation2019). Hypersialylation is an established hallmark of several cancers, including lung, breast, ovarian, pancreatic and prostate cancer. Like pathogens, hypersialylation decorate cancer cells by ‘self’ antigens which enables tumors evade the host immune system by molecular mimicry. This process is primed by binding between sialoglycans on the hypersialylated cancer cell surface and Siglecs on most white blood cells in the immune system, which promote immunosuppressive signaling to promote continued tumor growth by inhibiting the cytotoxicity of NK cells and the activation of T-cells, and inducing a tumor-associated macrophage (TAM) phenotype (Macauley et al. Citation2014; Rodriguez et al. Citation2021). Moreover, the reduced adhesion between cancer cells and the ECM, and other cells in the same tumor mass is the harbinger of metastatic cancer spread. Upregulation of multiple sialyltransferases causes hypersialylation of integrins and other adhesion molecules, reducing the stability of tumor masses and thereby increasing the spread of tumor cells. Selectins also play a key role in cancer metastasis. The increased expression of selectin ligands (sialofucosylated glycans) on tumors by upregulation of sialyltransferases (and fucosyltransferases) is correlated with enhanced metastasis in many cancers, including melanoma, gastric, pancreatic, colon and lung cancer (Läubli and Borsig Citation2019). In summry, hypersialylation enables tumor spread by metastasis through (1) mimicking host-like cell-surface sialylation leading to evasion of the immunosurveillance system and cell death pathways, (2) stimulating tumor invasion and migration, and (3) playing a cytoprotective role that contributes to chemotherapy and radiotherapy resistance in several cancers.
Aberrant sialylation is also closely linked with the action of sialidases (neuraminidases). Neuraminidases are believed to modulate the function of sialoglycoconjugates by desialylation, the removal of α-glycosidically linked sialic acid residues from the terminal positions of the carbohydrate groups of glycoproteins and glycolipids, which is the initial step in the degradation of these glycoconjugates. The expression levels of specific neuraminidases, such as Neu1 and Neu3, are critical factors in the metastasis and survival of cancer cells, and alteration in sialidase expression may be a defining factor for cancer progression, irrespective of the sialic acid content (Miyagi Citation2008; Yamaguchi et al. Citation2012). The processes by which hypersialylation promote metastasis are complex and the reader is referred to an excellent review (Dobie and Skropeta Citation2021).
Aberrant glycosylation: the sugar sculpting art of cancer
The Warburg effect (aerobic glycolysis)
A key feature of cancer is the energy metabolism shift from mitochondrial oxidative phosphorylation of glucose to CO2 to less efficient glycolysis metabolic pathway. In 1924, Warburg found that unlike most normal tissues, cancer cells tend to ‘ferment’ glucose into lactate even in in the presence of completely functioning mitochondria to support mitochondrial oxidative phosphorylation. Unlike normal differentiated cells which redirect the pyruvate generated by glycolysis away from mitochondrial oxidative phosphorylation by generating lactate only under anaerobic conditions (anaerobic glycolysis), most cancer cells rely primarily on this metabolic pathway to generate the energy needed for cellular processes regardless of the availability of oxygen ‘aerobic glycolysis’ (Vander Heiden et al. Citation2009). Aerobic glycolysis is an inefficient means of generating ATP with nearly 20-fold less ATP generated per unit of glucose as compared to the amount obtained by mitochondrial respiration (Locasale and Cantley Citation2011), but the increased glucose consumption is used as a carbon source for anabolic processes needed to support the uncontrolled proliferation of cancer cells (Koppenol et al. Citation2011; Boroughs and DeBerardinis Citation2015). Although Warburg Effect has been proposed to be an adaptation mechanism to cope with the increased energetic and biosynthetic needs of tumors, its exact functions remains enigmatic (Liberti and Locasale Citation2016).
The increased glucose uptake leads to increased flux through the hexosamine biosynthetic pathway (HBP) to generate the UDP-GlcNAc, which serves as the donor substrate for O-GlcNAcylation. O-GlcNAcylation is a type of post-translational modification by glycosylation where O-linked β-N-acetylglucosamine (O-GlcNAc) moieties are attached to cytoplasmic, nuclear and mitochondrial proteins. Evidence indicates that the HBP, specifically through O-GlcNAcylation, helps fuel cancer cell metabolism, growth, survival, and spread (Akella et al. Citation2019). Elevated O-GlcNAcylation (hyper-O-GlcNAcylation) is a signature of cancer-specific metabolism and linked to various hallmarks of cancer, including cancer cell proliferation, survival, invasion, and metastasis; energy metabolism; and epigenetics (Ma and Vosseller Citation2014). Hyper-O-GlcNAcylation is encountered in various types of cancer and has itself been considered as a hallmark of cancer (Singh et al. Citation2015).
Aberrant glycosylation and cancer hallmarks
As cancer-associated glycans play pivotal roles in cancer cell signaling, tumor cell dissociation and invasion, cell-matrix interactions, angiogenesis, metastasis and immune modulation, aberrant glycosylation heralds the acquisition of all proposed cancer hallmark capabilities. These hallmarks include sustaining proliferative signaling, evading growth suppressors, deregulating cellular energetic, resisting cell death, enabling replicative immortality, activating invasion and metastasis, inducing angiogenesis, genome instability and mutation, tumor promoting inflammation, and avoiding immune destruction (Munkley and Elliott Citation2016). Evidence indicates that aberrant glycosylation is also linked to tumor initiation, progression and metastasis, and can be considered as a new hallmark of cancer development (Vajaria and Patel Citation2017).
Aberrant glycosylation enables the acquisition of cancer hallmark capabilities through 3 main mechanisms:
Interference with receptor tyrosine kinases (RTKs) functions: RTKs and their signaling cascades are the main players in cancer progression. Alterations in glycosylation such as terminal sialylation, fucosylation, or oligosaccharide branching, can affect the conformational arrangements of RTKs’ extracellular domain, causing aberrant activation and signaling, as demonstrated for epidermal growth factor receptor (EGFR) (Contessa et al. Citation2008; Kaszuba et al. Citation2015). Moreover, aberrant glycosylation of RTKs eventually allow galectin recognition and promote receptor retention at the cancer cell surface, facilitating receptor activation leading to increased cancer cell migration, invasion, and proliferation (Lau et al. Citation2007). The interaction with glycosaminoglycans, proteoglycans, gangliosides, and other glycosylated transmembrane proteins can also modulate the activation of RTKs (Shintani et al. Citation2006; Julien et al. Citation2013). For example, HS-containing proteoglycans modulate the activation of RTKs. In this case, growth factors, such as hepatocyte growth factor or vascular endothelial growth factor (VEGF), are tethered to the cancer cell surface by HS chains, facilitating ligand-mediated receptor activation and downstream signal transduction (Shintani et al. Citation2006).
Modulatulation of cell–cell interactions: increased branching of N-glycans on E-cadherin (the principal adhesion molecule of adherens junction in epithelia) impairs cell–cell adhesion and downstream signaling, and contributes to invasion and metastasis.
Modulatulation of cell-matrix interactions: both N- and O-glycosylation directly affect the interaction of integrins with ECM proteins and glycans present in the cancer cell glycocalyx, influencing their biological functions (including cell adhesion), and promotion of cancer migration and invasion (Paszek et al. Citation2014).
The role of glycosylation in immune system evasion
In a similar manner to pathogens’ ability to use their glycocalyx to evade immune recognition, alterations in the glycocalyx of cancer cells facilitate immune escape. These changes can change the TME and metastatic process with significant effects on the progression of neoplastic disease as well (Stowell et al. Citation2015). The glycosylation of tumor cells contributes to the evasion of an efficient immune response through a direct effect of glycosylation to create an immunosuppressive environment, often through the upregulation of self-associated molecular patterns (SAMPs) that results ultimately in immune tolerance. In addition, glycans can indirectly contribute to the immune evasion capacity by potentiating immune checkpoints such as the PD-1/PD-L1 axis. Glycosylation of PD-L1 has been shown to stabilize and modulate its binding to PD-1 (Li et al. Citation2016; Li et al. Citation2018a). Altered glycans can also impair or modulate cancer therapies (Costa et al. Citation2020).
TACAs as tumor-specific neo-antigens
TACAs, generated by the aberrant post-translational glycosylation, add a layer of neo-antigenicity to tumor-specific peptides presented by MHC class I complexes that can be recognized by a glycoform-specific TCR (Vlad et al. Citation2002; Apostolopoulos et al. Citation2003). In fact, TACAs are considered as priming agents for T-cells and for B-cells working together (Zajonc and Kronenberg Citation2009; Freire et al. Citation2010; Christiansen et al. Citation2011). TACAs are produced by structurally and functionally impaired Golgi apparatus that is converted into aberrant and distinctive cancer biosynthetic machinery (Roth et al. Citation1988; Hakomori Citation1989; Yang et al. Citation1994; Kumamoto et al. Citation2001; Brockhausen Citation2003). The differentiated features of TACAs from the normal cell glycan repertoire grants modified biophysical and protein-binding characteristics to individual tumor types (Trabbic et al. Citation2021). TACAs are absolutely indispensable for tumor development, aggressiveness, metastasis, immune evasion and chemoresistance (Tsuboi et al. Citation2011; Taniguchi and Kizuka Citation2015; Magalhães et al. Citation2017); and their presence has been traditionally used for diagnostic purposes. The clinically approved serological biomarkers for cancer diagnosis are basically glycosylated biomarkers secreted by tumors in physiological fluids. However, serum and plasma are the most commonly used human biological fluids for obtaining non-invasive cancer biomarkers ideal for disease prognosis, staging and monitoring (Hanash et al. Citation2008; Kulasingam and Diamandis Citation2008) (Table 2).
Table 2. Serum/plasma glycans and glycoproteins as cancer biomarkers.
Intriguingly, the carbohydrate moieties of glycosylated protein biomarkers are altered in cancer, and such changes are considered to be more useful markers of cancer. To illustrate, prostate-specific antigen (PSA)-specific glycosylation changes have largely supplanted measuring the concentration of PSA alone in prostate cancer patients. Due to its over-diagnosis and limitations in early detection, the US Preventative Services Task Force (USPSTF) discouraged the use of PSA for prostate cancer diagnosis since 2012 (Moyer Citation2012). Binding of Maackia amurensis agglutinin (MAA, a lectin recognizing α2–3-terminal sialic acids) to serum PSA was significantly higher (5.3-fold) for prostate cancer samples than for healthy controls. Studies have found that the combined analysis of serum PSA levels and glycoprofiling of PSA has a potential for improved detection of prostate cancer because α2–3-linked sialylation of PSA was significantly higher in prostate cancer than in benign diseases (Yoneyama et al. Citation2014; Llop et al. Citation2016; Pihikova et al. Citation2016). Therefore, increased α2–3-sialylated PSA has a great potential as a novel biomarker for biomarker for prostate cancer early detection. Similarly, the significant rates of false positive and false negative results have rendered the use of serum alpha-fetoprotein (AFP) as a diagnostic marker for hepatocellular carcinoma (HCC) obsolete. The total AFP is not HCC-specific as it can be elevated in pregnancy, hepatitis, and liver cirrhosis but its fucosylated glycoform, AFP-L3, provides better specificity for HCC (Li et al. Citation2001; Donati et al. Citation2010). The improved cancer specificity of AFP-L3 in HCC is caused by over-expression of the fucosyltransferase FUT8, an enzyme responsible for core-fucosylation (alpha-1,6 linkage) of N-glycoproteins (Meany and Chan Citation2011; Silsirivanit Citation2019). Although mass spectrometry (MS) is the gold standard method for discovering cancer glycoprotein biomarkers (An et al. Citation2009; Ruhaak et al. Citation2018; de Haan et al. Citation2020), lectin-based methods coupled with MS have proved highly useful. Lectins provide the advantage of minimizing the number of compounds to be analyzed by MS (Kaji et al. Citation2003; Drake et al. Citation2006; Zhao et al. Citation2006). So far, the discovery and quantification of the Lens culinaris agglutinin (LCA)-reactive species of AFP-L3 represent the greatest success with use of lectins for the diagnosis of malignant conditions (Kuzmanov et al. Citation2013).
Cancer-associated glycans are shared by many cancers (Hamilton et al. Citation1993), and their overexpression correlates with poor prognosis (Werther et al. Citation1996) as it is a reliable sign of tumor progression and metastasis (Bresalier et al. Citation1991). Antibodies and CAR-T cells directed against TACAs currently in ongoing clinical trials are promising cancer immunotherapeutic agents (Mereiter et al. Citation2019; Thurin Citation2021). Several mAbs targeting glycolipids have demonstrated the ability to mediate potent ADCC and CDC (Thurin Citation2021). There are three main advantageous of targeting glycans over protein targeting. Firstly, cancer-associated glycans have often-low abundance or absence on normal tissues, while very densely expressed on tumors (Kudelka et al. Citation2015; Stowell et al. Citation2015). Secondly, glycans are more accessible to administrated targeting vehicles than membrane-bound proteins that are masked by the glycan shield. Thirdly, as atypically-expressed glycans tend to amplify their expression on glycoproteins, glycosylation changes are more reflective of disease process than changes in the proteome (Fuster and Esko Citation2005; Lebrilla and An Citation2009). In a similar vein, these characteristics also make cancer-associated glycans ideal candidates for targeted glycan-based tumor imaging and for use as potential cancer glycobiomarkers (i.e. for diagnosis, follow-up, monitoring of therapeutic response, or patient stratification) (Houvast et al. Citation2020). Moreover, cancer-associated glycans were considered as promising targets for the design of anti-cancer vaccines but their clinical application was hampered by many obstacles. Luckily, several strategies were improvised to improve the immunological character of the TACA constructs that made TACA-based vaccines a new milestone for cancer immunotherapy (Feng et al. Citation2016a).
The three main changes often associated with cancer are: (1) truncation/ incomplete glycosylation of O-linked carbohydrate chains, (2) increased branching of N-glycans due to increased MGAT5 expression, and (3) augmented or changed presence of sialic acid-containing glycans (Varki et al. Citation2015b). Both upregulated sialyltransferases and fucosyltransferases lead to the overexpression of TACAs and assoacieted lack of response to chemotherapy (Table 3). Fucosyltransferases (FUTs) belong to the Golgi apparatus enzyme family that catalyzes fucosylation by the transfer of L-fucose sugars from Guanosine 5′-diphospho-β-L-fucose (GDP-fucose) donor substrates to glycoconjugates such as glycoproteins and glycolipids (Marques Citation2000). Fucosylated glycans are synthesized by fucosyltransferases. So far, 13 FUTs are known to be encoded by the human genome. FUTs are expressed in many tissues of human body and their expression levels are often altered in patients with different types of cancers (Shan et al. Citation2019). De novo or increased expression of FUTs in tumor cells is a strong risk factor for poor clinical outcomes (Feng et al. Citation2016b; Auslander et al. Citation2017; Li et al. Citation2018b; Liu et al. Citation2019a; Liu et al. Citation2019b).
Table 3. Examples of TACA overexpressed due to upregulation of sialyltransferases and fucosyltransferases.
Classification of TACAs
Cancer-associated glycans (Figure ) include:
The mucin-type O (O-GalNAc) glycans:
Figure 1. Cancer-associated glycans/tumor-associated carbohydrate antigens (TACAs). TF, the disaccharide Thomsen-Friedenreich antigen; Tn, the monosaccharide Thomsen-nouvelle antigen; sTF, sialylated TF antigen; sTn, sialylated Tn antigen; SSEA-1, stage-specific embryonic antigen-1.
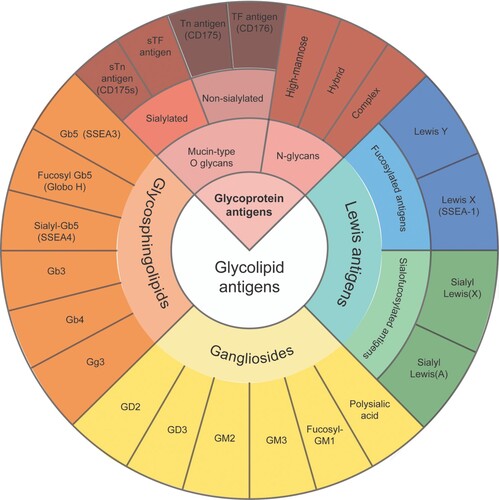
O-glycosylation is a common covalent modification of mammalian glycoproteins. O-GalNAc Glycans are initiated by N-acetylgalactosamine (GalNAc) attached to the hydroxyl of Ser or Thr residues of glycoproteins. Mucins are heavily glycosylated proteins (glycoproteins) found in mucous secretions carrying the greatest number of O-GalNAc glycans (also called mucin-type O-glycans), but this posttranslational modification is common among many glycoproteins. Mucins play an essential role in forming protective mucous barriers on epithelial surfaces. These proteins also play a role in intracellular signaling. The first mucin gene to be cloned encoded a transmembrane mucin termed Mucin1 (MUC1) that is ubiquitous in the epithelium. MUC1 (CD227) represents a prime target antigen for cancer immunotherapy because it is abundantly expressed and aberrantly glycosylated in various carcinomas. Overexpression of MUC1 is associated with increased tumor cell invasion and metastasis. Particularly, MUC1 is highly overexpressed and aberrantly O-glycosylated in adenocarcinomas. MUC-1 CART can target abnormal glycosylation of MUC-1 in intrahepatic cholangiocarcinoma, one of the most common liver malignancies. Currently, an ongoing clinical trial is evaluating the safety and efficacy of MUC-1 CART in intrahepatic cholangiocarcinoma (NCT03633773). MUC1 also appears in a variety of hematological malignancies, including T and B cell lymphomas and myelomas (Brossart et al. Citation2001), and is a potential prognostic marker and therapeutic target for several types of non-Hodgkin lymphoma (Rassidakis et al. Citation2003; Jain et al. Citation2015). Furthermore, targeting MUC16, which is primarily upregulated in ovarian cancer cells, is expected to improve the poor prognosis of ovarian cancer. MUC16, also known as carbohydrate antigen 125 (CA125), is the FDA-approved ovarian tumor marker (Bottoni and Scatena Citation2015). The first anti-MUC16 mAb investigated in clinical trials, Oregovomab (OvaRex) binds the glycosylated region of MUC16 with high affinity and induces indirect immune responses via an anti-idiotype antibody induction cascade (Schultes et al. Citation1998), and is currently being evaluated at phase 3 clinical trial in conjunction with carboplatin and paclitaxel chemotherapy for newly diagnosed ovarian cancer patients (NCT04498117). There are many anti-cancer therapeutic candidates that target MUC1 and MUC16 in clinical trials (Lee et al. Citation2021).
O-GalNAc glycan synthesis begins with the transfer of N-acetylgalactosamine (GalNAc) from UDP-GalNAc to Ser or Thr catalyzed by a polypeptide GalNAc-transferase (GALNT). A single GalNAc residue attached to Ser/Thr forms the Tn antigen. The unsubstituted core 1 O-GalNAc glycan forms the Thomsen-Friedenreich (TF or T antigen). Although Tn and T antigens are usually cryptic because they are extended by other sugars, the extension of O-GalNAc glycans beyond the first sugar is blocked in some cancer cells resulting in increased levels of Tn and TF antigens. They can also carry Sia and form sialyl-Tn or sialyl-T antigens Sialyl-Tn antigen is common in advanced tumors (Brockhausen and Stanley Citation2017). This is called truncation of O linked carbohydrate chains resulting from incomplete glycosylation and is the most common cancer-associated change in glycosylation. Truncation facilitates tumorigenesis and/or progression of solid tumors has been obtained by preventing extension of O-GalNAc in precancerous cells. Tn and TF antigens are found at increased levels in mucins from cancer cells (especially epithelial cancers including breast, prostate, ovary). The cancer-associated Tn glycoform of Mucin1 (MUC1; CD227), a neo-antigen expressed in a variety of cancers, has been used to generate specific anti-Tn-MUC1 monoclonal antibodies (Sørensen et al. Citation2006). Recently, a new study used lectin microarray glycan profiling and a 3D myoma tissue-based invasion assay to demonstrate that both increased levels of terminal GalNAc decorating several metastasis-associated glycoproteins that is recognized by Helix Pomatia lectin (HPA), and the increased binding potential of HPA itself correlated significantly with highly invasive cancer cell phenotype. The study also suggested that cancer cells differ markedly in their O-and N-glycan signatures (Khosrowabadi et al. Citation2022).
N-glycans:
N-Glycans are covalently attached to proteins at asparagine (Asn) residues by an N-glycosidic bond. Changes to the core structures of N-glycans are one of the most common aberrant glycosylations in cancer. Highly branched N-glycan structures are associated with cancer growth and metastasis (Dennis et al. Citation1987; Chen et al. Citation1998; Taniguchi et al. Citation1999; Miyoshi et al. Citation2012). Oligosaccharide modification by N-acetylglucosaminyltransferase- V (GnT-V), a glycosyltransferase encoded by the MGAT5 gene that catalyzes the formation of β1,6GlcNAc (N-acetylglucosamine) branches on N-glycans, resulting in highly branched N-glycan structures, was found to be associated with cancer growth and metastasis (Granovsky et al. Citation2000; Hou et al. Citation2016). The larger and more branched N-glycans catalyzed by the increased activity of GNT-V provides additional acceptors for terminal sialylation (Dennis et al. Citation1987; Kim and Varki Citation1997). The three processes most commonly involved in aberrant expression of N-glycan-related enzymes in hematological malignancies are sialylation, fucosylation, and bisecting β D N acetylglucosamine (GlcNAc) (Pang et al. Citation2018). More understanding of N-glycomics in cancer can be learnt from recent study of spatial and temporal analysis of glycome expression patterns in the mammalian brain using a cutting-edge experimental tool based on liquid chromatography-mass spectrometry (Lee et al. Citation2020).
The blood group Lewis antigens (fucosylated glycans linked to lipids or to proteins):
The group includes Lewis Y (LeY), Lewis X (LeX) (also known as stage-specific embryonic antigen-1, SSEA-1), and the sialofucosylated antigens Sialyl Lewis(X) (sLeX) and Sialyl Lewis(A) (sLeA). FUT1 (H enzyme) and FUT2 (secretor enzyme) are responsible for the formation of Lewis (and ABH) blood group antigens. Lewis carbohydrate antigens can be found on various glycoconjugates in most human epithelial tissues (Ravn and Dabelsteen Citation2000). They are overexpressed in colon, liver, ovarian, breast and prostate tumors. The frequent increase in fucosylated Lewis antigens expressed by cancer cells contribute to immunosuppressive environment and results ultimately in immune tolerance.
The sialylated Lewis carbohydrate antigens, sLeX and its isomer sLeA, are the E-selectin ligand glycans. sLeX is a terminal component of glycoconjugates on most circulating leukocytes and some endothelial cells, but sLeA is not found on normal leukocytes (Cheung et al. Citation2012). Both sLeX and sLeA facilitate the arrest of circulating tumor cells on endothelial cells by mediating the adhesion between tumor cells and the endothelium (Monzavi-Karbassi et al. Citation2013). They are abundantly expressed on many types of cancer cell surface, including colorectal, pancreatic, gastric, breast, prostate and lung cancer (Kannagi et al. Citation2004; Sakuma et al. Citation2012), and their expression is strongly associated with increased metastatic potential (Esposito et al. Citation2019).
The Glycosphingolipids (GSLs):
GSLs are most widely overexpressed glycolipids on tumors (Durrant et al. Citation2012). The globo-series GSLs Gb5 (known as stage-specific embryonic antigen 3, SSEA-3), SSEA-4 (sialyl-Gb5), and Globo-H (fucosyl Gb5), are specifically expressed on pluripotent stem cells and cancer cells, and are known to be associated with various biological processes such as cell recognition, cell adhesion, and signal transduction (Asano et al. Citation2019). Several GSLs are markers of hematological malignancies which result from precursor accumulation during incomplete GSL synthesis.
GSLs are ubiquitous membrane constituents, which are embedded in the cell plasma membrane (Zhang et al. Citation2004). GSLs are neutral ‘core’ structures composed of a glycan linked to a lipid ceramide (Cer). Ceramides are fatty acid derivatives of sphingoid bases Cer, a central molecule in sphingolipid metabolism, can be phosphorylated to form Cer-1-P, or glycosylated to form glucosylceramide (GlcCer). 90% of mammalian GSL biosynthesis begins with the synthesis of GlcCer (Lingwood Citation2011). When GlcCer is followed by addition of galactose, lactosylceramide (LacCer) is produced. LacCer is an important branching point, being a common precursor for the synthesis of the three major classes of GSLs: (i) the ganglio- and isoganglio-, (ii) the lacto- and neolacto-, and (iii) the globo- and isoglobo-series (Liang et al. Citation2010). The normal balance between Cer and its derivatives is disrupted in hematological malignancies, particularly those displaying chemotherapeutic resistance (Itoh et al. Citation2003). GlcCer synthase, the enzyme that attaches the Glc moiety to Cer, is co-overexpressed with Pgp in MDR leukemia cells (Turzanski et al. Citation2005; Xie et al. Citation2008), and blocks drug-induced cell cycle arrest (Rani et al. Citation1995). As GlcCer is present consistently in MDR cell lines and absent (or present at very low levels) in corresponding drug-sensitive cells, it is a marker for MDR cancers (Lucci et al. Citation1998).
The gangliosides (sialic acid-containing GSLs):
Gangliosides are mono- or multi-sialosylated GSLs mainly located in the outer layer of the plasma membrane (PM) of vertebrate cells. Gangliosides, are present in normal tissues, but are highly expressed in many human cancer cells including melanoma, lung, colon, renal, and prostate cancers (Daniotti et al. Citation2013). Complex gangliosides, such as GD3 and GD2 involved in aggressiveness and metastasis of neuroectoderm-derived tumors such as melanoma and neuroblastoma, can specifically be targeted by monoclonal antibodies and have been shown to be promising targets for immunotherapy (Ladenstein et al. Citation2018; Cavdarli et al. Citation2019). The majority of studies focused on the disialo-ganglioside GD3 (9-O-acetyl GD3) as the major carrier of terminal O-acetylated sialic acid. 9-O-acetyl GD3, an oncofetal (expressed in fetal tissue) antigen, was recognized by a crab lectin (Cancer antennarius lectin) as abiomarker in human melanoma (Ravindranaths et al. Citation1988). 9-O-acetyl GD3 is highly expressed in many cancers. Table 4 lists many cancers that overexpress cancer associated-glycans.
Table 4. Selected cancer associated-glycans expressed in several types of human cancers.
Application of lectins in cancer therapy
Lectinology
The beginnings of lectinology date back to 1888 (Tsaneva and Van Damme Citation2020). Lectins were initially discovered and described in plants (Zhang et al. Citation2000), but in subsequent years multiple lectins were identified in all organisms, from microorganisms to fungi, plants and animals. Plant and animal lectins show no primary structural homology but still they have similar preferential binding to carbohydrates highlighting the importance of lectin-glycan interactions in living systems (Ghazarian et al. Citation2011). Historically, lectins were called hemagglutinins or agglutinins because of their ability to agglutinate erythrocytes (like antibodies). The impressive antibody-like capacity of plant lectins for selecting distinct blood-group epitopes (carbohydrate determinants) on human erythrocytes (blood typing) was the cornerstone for lectins to broadly enter laboratories as tools. Serology was the first discipline of medicine that relies on the specific biological activity of lectins (Końska et al. Citation2008). Interestingly, the detection of ABO blood-group-selective activities different from immunoglobulins led to the introduction of the term ‘lectin’ by W. C. Boyd in 1954 (Gabius Citation2013).
Lectin-glycan interactions are generally achieved by hydrogen bonding and Van der Waals forces, and often depend on cations and multivalent interactions between multiple carbohydrate-recognition domains (CRDs) and multiple, clustered target glycans in order to achieve sufficient affinities for lectin activity (Varki et al. Citation2009). The unique intrinsic ability of lectins to bind specific sugar residues in glycoproteins and glycolipid complexes has made them suitable candidates for use in detection, isolation and characterization of glycoconjugates, histochemistry of cells and tissues, and delivering drugs at the site of action. Owing to their ability to recognize surface markers (modified glycan structures) predominantly expressed in tumor cells, lectins are also used to distinguish between normal and malignant cells (tumor cell recognition) in cancer research. Moreover, plant lectins exert myriad immunomodulatory effects including anti-tumor, anti-viral, anti-insect, anti-fungal, anti-parasitic and anti-bacterial activities (Mishra et al. Citation2019). These potent immunomodulatory properties were the basis for investigating plant lectins as a candidate for adjuvant/carrier formulation for use in chemotherapeutic and prophylactic vaccine production (Sander et al. Citation2019).
The search for synthetic carbohydrate receptors falls within the field of supramolecular chemistry (Kubik Citation2009). Initially, carbohydrate recognition in aqueous solution presented a difficult challenge to supramolecular chemists. After many decades of research, it has at last been possible to develop biomimetic receptors which perform well in aqueous solution (Davis Citation2009). The advent of state-of-the-art lectin engineering (protein engineering technology aiming at producing artificial lectins) where various methods by which lectin specificity and/or stability of a known lectin scaffold can be improved (Hirabayashi and Arai Citation2019) should open a new realm in the field of cancer immunology. Noteworthy, some authors prefer the term carbohydrate-binding agents (CBAs) to include both peptidic lectins and non-peptidic low-molecular-weight agents (i.e. those with a molecular weight of less than 1–2 kDa) with carbohydrate-binding capacity (François and Balzarini Citation2012) that have shown efficient suppression of viral and fungal infections. Throughout, engineered lectins will refer to both synthetic peptidic or non-peptidic carbohydrate-binding agents, unless otherwise indicated.
Lectinology of acute leukemias
The glycosylation landscape of leukemia and clinical significance of plant lectins binding to leukemic blasts were an area of particular interest in the past century. Receptors for the plant lectins peanut (Arachis hypogaea) agglutinin (PNA) and lentil (Lens culinaris) agglutinin (LCA) were found in the majority of ALL and AML blast cells (Levin et al. Citation1980; Veerman et al. Citation1985; Slesak et al. Citation1989; Slesak et al. Citation1990; Sharma et al. Citation1996). PNA has the advantage of weaker interaction with human erythrocytes and normal peripheral blood lymphocytes than other plant lectins (Reisner et al. Citation1979). The efficacy of plant lectins was recently shown by the potent and broad anti-viral activities of LCA against the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants while demonstrating very weak hemagglutination activity and no cytotoxicity (Wang et al. Citation2021). Within this context, it is likely that binding of these plant lectins to their specific glycans would activate the LP on the surface of blast cells leading to their lysis. The experimental basis of this hypothesis has been recently supported by a study showing that treatment with Urtica dioica agglutinin (UDA), a very small plant lectin, has resulted in cell death in ALL cell lines and induced apoptosis in the T-ALL cell line (Rashidbaghan et al. Citation2020). To fully exploit plant lectins clinically, it will be vital to improvise strategies to integrate them with conventional therapies.
In ALL, elevated 9-OAcGD3 was detected in ALL cells that developed resistance against vincristine or nilotinib, two drugs with different cytotoxic mechanisms. The treatment of ALL cells by a sialate acetyl esterase that cleaved the 9-O-acetyl residues from sialic acids made these cells more sensitive to both drugs (Parameswaran et al. Citation2013). Chemoresistance was due to the anti-apoptoctic effect of OAcGD3. OAcGD3 was found to protect leukemic blasts, Jurkat cells, and glioblastoma cells from apoptosis (Mukherjee et al. Citation2008; Birks et al. Citation2011). A significant amount of total membrane protein (2 mg from 107 cells) was found to be O acetylated sialoglycoprotein in ALL. Achatinin-H, a lectin, purified from the hemolymph of the African giant land snail Achatina fulica, confirmed the overexpression of glycoproteins with Neu5,9Ac2 (Neu5,9Ac2-GPs) in a large patient population of several T- and B-ALL cell lines, indicating that Neu5,9Ac2-GPsALL is a novel leukemia-specific antigen (Pal et al. Citation2004). Since Neu5,9Ac2-GPs are strongly expressed in childhood ALL at the onset of disease, then they decrease with chemotherapy and reappear with relapse, Neu5,9Ac2-GPs a potential biomarker for diagnosis and monitoring the disease status in childhood ALL (Sinha et al. Citation1999a, Citation1999b; Chowdhury et al. Citation2008; Chowdhury and Mandal Citation2009; Mandal et al. Citation2009). Therefore, Neu5,9Ac2 is an excellent biomarker for the detection of minimal residual disease (MRD), the main cause of a relapse of the leukemia (Mandal et al. Citation2015). Glycoproteins carrying Neu5,9Ac2 in B-ALLs were identified as nucleolin (NCL), suggesting it as a possible therapeutic target for B-ALL (Joo et al. Citation2018).
In AML, a recently identified myeloid lineage restricted cell surface marker was identified as the human C-type lectin-like molecule-1 (CLL-1, also known as CLEC12A, DCAL-2, and KLRL-1). CLL-1 is an attractive target for immunotherapy since it is overexpressed in over 90% of AML blasts and the majority of leukemia stem cells while absent on normal HSPCs (Zhao et al. Citation2010; Laborda et al. Citation2017). Recent results supported CLL1 as a highly promising AML target for bispecific antibodies (Lu et al. Citation2014), anti-CLL-1-ADC (Zheng et al. Citation2019a), and CAR-T-cell therapy (Tashiro et al. Citation2017). CLL-1 is also known as human myeloid inhibitory C-type lectin (hMICL) (Foster et al. Citation2015). hMICL has been independently identified by several groups and shown to be a type II transmembrane receptor which is highly N-glycosylated compared to its most closely related paralogs, lectin-like receptor for oxidized LDL, Dectin-1, 23, and CLEC-2 (Marshall et al. Citation2006). Therefore, the exclusive and high expression of CLL-1 in AML can also be an excellent therapeutic target for engineered lectins directed against CLL-1-associated N-glycans. The relevance of targeting N-glycans in AML has been confirmed by the ArtinM (also known as KM+ and Artocarpin), a D-mannose-binding lectin from Artocarpus heterophyllus (jackfruit), which efficiently inhibited AML with N-glycans containing a β1,6-GlcNAc branch linked to α1,6-mannose. This is also an illustrative example of a plant lectin targeting altered cell surface glycans being employed as a novel anti-leukemia treatment (Carvalho et al. Citation2011). Although limited efficacy was observed with CAR T cells directed at LeY, a carbohydrate antigen overexpressed by malignant myeloid cells, no overt hematopoietic toxicity was observed. The reduced efficacy of anti-LeY CAR T cells was not related to LeY expression itself but rather due to rendering the CAR T cells unresponsive by the immune suppressive BM environment in AML (Ritchie et al. Citation2013). Instead of resorting to sophisticated strategies such as enhancing CAR T cells by immune checkpoint blockade combination therapy, manufacturing engineered lectins targeting LeY antigen can be an ideal alternative for treatment of high-risk AML.
The glycosylation processes of single glycoproteins and alterations of the cancer cell glycocalyx are best characterized by applying by a systematic integrated multi-omics approach (combination of transcriptomics, proteomics and glycomics). The first multi-omics evaluation of primary BCP ALL with MLL-r patient cells uncovered that MLL-r cells undergo an extensive remodeling of their glycoprotein glycocalyx, with increased Core 2-type O-glycans and complex N-glycans as well as significant changes in sialylation and fucosylation levels. The GAG glycocalyx of MLL-r cells undergoes a major remodeling compared to control cells with a shift from CS towards HS (both are binding partners of Gal-1) (Camby et al. Citation2006). Given the strong upregulation of Gal-1 in MLL-r cells, a cell surface remodeling towards higher levels of HS might also play an important role in MLL-r cell survival within the BM environment. The multi-omics approach aids to identify novel markers that are not detectable solely by a transcriptomics approach (Oliveira et al. Citation2021). Such work can open new perspectives for potential future therapeutic targets for engineered lectins.
Lectin antagonists: interfering with immune lectins for cancer immunotherapy
Targeting galectin-ligands
Many tumors show overexpression and increased secretion of galectins. Galectins have gained increasing interest as therapeutic targets in several diseases, such as chronic inflammation and cancer (Stegmayr et al. Citation2019). Galectins bind glycans on the surface of potentially pathogenic microbes as well as function as recognition and effector factors in innate immunity (Vasta et al. Citation2017). In mammals, 16 different members of galectin family are found both in the intra- and extracellular environments (Bertuzzi et al. Citation2020). Galectins (formerly known as ‘S-type lectins’) are a family of secreted (soluble) β-galactoside- (β-Gal)-binding lectins defined by their affinity for β-d-galactopyranoside moieties and their possession of a conserved CRD which is responsible for the majority of galectin activities. Since galactosides are glycosides containing galactose, many authors define galectins as soluble carbohydrate binding proteins that can bind β-galactose-containing glycoconjugates via a CRD (Wu et al. Citation2021).
Mammalian galectins have been classified into three types: prototype galectins (Gal-1, -2, -5, -7, -10, -11, -13, -14, and -15, containing one CRD and existing as monomers or dimerizing through non-covalent interactions), tandem repeat-type galectins (Gal-4, -6, -8, -9, and -12), which exist as bivalent galectins containing two different CRDs connected by a linker peptide, and finally, Gal-3, the only chimera-type member of the galectin family. Galectins are known for their ability to cross-link glycoconjugates to form lattices that have been proposed to influence the behavior and activities of cross-linked glycoconjugates by regulating their cellular trafficking, localization, residence times, and molecular interactions (Peterson et al. Citation2018). Galectins are expressed by various cells of the TME including fibroblasts and cancer cells and regulate a multitude of processes by inducing glycoprotein clustering and membrane retention. They have been implicated in diverse events associated with cancer biology such as apoptosis, homotypic cell aggregation, angiogenesis, cell migration, and tumor-immune escape (Cagnoni et al. Citation2016).
Apart from being crucial for RTK activation, galectins have been shown to govern the immunological landscape of tumors (Cerliani et al. Citation2011). Targeting galectin-glycan interactions has evolved as a promising therapeutic approach utilizing galectins’ immune inhibitory and pro-angiogenic activities to restore anti-tumor immunity and to attenuate abnormal vascularization of established tumors (RodrÍguez et al. Citation2018). To illustrate, Gal-1, as an immunosuppressant confers immune privilege to tumor cells by promoting escape from T-cell-dependent immunity, and its blockade can allow for and potentiate effective immune responses against tumor cells, with profound implications for cancer immunotherapy (Rubinstein et al. Citation2004). Similar to Gal-1, Gal-3 signaling contributes to tilt the balance toward immunosuppressive TMEs by interacting with specific glycans, and impairing anti-tumor responses (Farhad et al. Citation2018). The major cancer cell surface carbohydrate ligand for Gal-3 is the TF antigen which is expressed in up to 90% of human carcinomas (Glinsky et al. Citation2001).
Amongst the 12 gelectin members identified in humans (Wdowiak et al. Citation2018), the role of Gal-1, -3, -9 and -12 is defined in the development and progression of different types of leukemia. To demonstrate, the serum Gal-3 levels are increased in BCP-ALL patients and are significantly high in acute promyelocytic leukemia (APL) patients. BM stromal cells also participate in drug resistance of AML and ALL by secreting Gal-3. Gal-3 expression was an independent poor prognostic factor, regardless of age, leukocyte counts or karyotype, and was proven to be reliable biomarker in AML (Cheng et al. Citation2013). Mixed-lineage leukemia gene rearranged (MLL-r) BCP-ALL selectively express the immunoregulatory carbohydrate-binding protein Gal-1 (Juszczynski et al. Citation2010; Paz et al. Citation2018). There is also significant increase in the serum levels of Gal-9 in AML patients. A large number of studies have documented the key role of galectins in development and progression of cancers including renal cell carcinoma, cervical squamous cell carcinoma, breast cancer, oral squamous cell carcinomas, esophageal and gastric cancers (Zheng et al. Citation2019b).
In view of prominent galectins’ pro-tumorigenic effects in multiple tumors, galectin inhibition through inhibition of their CRD by antagonists (or inhibitors) has become logically desirable. This provided a rationale for the design of glycomimetic inhibitory compounds as anti-galectin agents (Modenutti et al. Citation2019). For example, GB0139 (formerly TD139), a thiodigalactoside (TDG) Gal-3 CRD inhibitor, is currently being evaluated clinically for the treatment of idiopathic pulmonary fibrosis (Delaine et al. Citation2016; St-Gelais et al. Citation2020). Administration of TDG also significantly reduced tumor progression and metastasis via Gal-1 inhibition, using murine models of breast and colon adenocarcinoma (Ito and Ralph Citation2012). Galectin inhibition can also be achieved by administration of recombinant galectins. Astonishingly, treatment with recombinant Gal-9 prevents metastatic spread in various preclinical cancer models including hematological malignancies (Fujihara et al. Citation2013; Wiersma et al. Citation2013). Apart from its expression on T-cells, the Gal-9 receptor TIM-3 is also expressed on AML LSCs in most types of AML except for APL, but not on normal HSCs, myelo-erythroid or lymphoid progenitor cells which renders recombinant Gal-9 a promising AML LSC-targeted therapy (Kikushige et al. Citation2010; Gao et al. Citation2014). Moreover, the Tim-3/Gal-9 signaling pathway is described as a one of the immune checkpoints responsible for T-cell exhaustion leading to defective T cell-mediated immunity that facilitates disease progression in AML (Ustun et al. Citation2011). Thus, inhibition of Gal-9 may not only target LSCs but also restore T-cell immunity in AML (Pena et al. Citation2014). The ability of galectins to modulate different events in tumorigenesis and metastasis makes them attractive targets for cancer therapy. Initial engineering of galectins has been focused on improving their productivity and stability for pharmaceutical potential (Hu et al. Citation2015).
Targeting selectin-ligands
Selectins are vascular cell adhesion molecules (VCAMs) belonging to the C-type (calcium-dependent) transmembrane lectin family with well-characterized roles in homing and tissue recruitment of leukocytes. They mediate adhesive interactions between leukocytes and the endothelium to facilitate entry to secondary lymphoid organs and sites of inflammation during normal and abnormal inflammatory episodes. The three members of selectins are expressed on platelets (P-selectins), leukocytes (L-selectins) and endothelial cells (E- and P-selectins) and each has distinct ligand specificities (Rosen and Bertozzi Citation1994; McEver and Cummings Citation1997; Trinchera et al. Citation2017). Therapies targeting selectin-ligand interactions have been investigated in sickle cell disease (SCD), cancer cell metastasis and BM transplantation. The P-selectin inhibitor mAb crizanlizumab was associated with a significantly lower frequency of vaso-occlusive pain crises (VOCs) in patients with SCD (Ataga et al. Citation2017) which led to FDA approval of crizanlizumab as a prophylactic agent that could be administered on a dosing schedule of once every 3–4 weeks (US Food and Drug Administration Citation2019). While crizanlizumab inhibits P-selectin by blocking its interaction with its ligand, P-selectin Glycoprotein Ligand-1 (PSGL-1), sLeX-capped O-glycans in PSGL-1 are key determinant of PSGL-1/P-selectin interactions (Tanaka Citation2016). P-selectin has also been considered a risk factor for recurrent thromboembolism and cardiovascular disorders and P- selectin inhibition using a recombinant P- selectin ligand was shown to decrease thrombosis in non-human primate animal models (Myers et al. Citation2001; Myers et al. Citation2002).
Evidence indicates that selectins and their ligands are involved in cancer progression. Cancer cells express selectin ligands (mucins) that facilitate their binding to leukocytes and endothelial cells, leading to extravasation during tumor metastasis (Cell Adhesion and the Extracellular Matrix Citation2021). All members of the selectin family have been implicated, but in particular E-selectin (CD62E) was identified as a BM homing receptor and a key regulator of the switch between dormancy and proliferation of HSCs (Winkler et al. Citation2012). Leukemia cells were found to home to regions of the BM niche characterized by E-selectin expression. Overexpression of E-selectin ligands (sLeX/sLeA) is associated with poor prognosis and malignant relapse (Nakagoe et al. Citation2002). The interaction of the antigen sLeX on tumor cells and E-selectin on endothelial cells facilitates tumor cell invasion in blood microvessels, extravasation and migration into tissue by mediating adhesion of tumor cells to endothelial cells (Walz et al. Citation1990).
Recently, E-selectin-LSC interaction was characterized as a new cell-extrinsic niche-based pathway that directly mediates therapy resistance. Endothelial E-selectin is a key player of niche hijack in AML but also unique among other adhesion molecules in mediating significant intracellular pro-survival signaling for LSCs. As a hallmark of cancer, inflammation manipulates the BM niche to create tumor-associated vasculature that provides a protective niche for LSCs. Pro-inflammatory mediators generated by AML blasts increases the expression of E-selectin on the BM endothelium. On the other hand, aberrant glycosylation associated with oncogenesis increase the E-selectin binding potential of AML blasts (Hakomori Citation2002). Unlike normal conditions where only leukocytes can bind selectins, the increased expression and the coordinated actions of ST3GAL4, FUT4, FUT7 and FUT9 leads to pronounced expression of sLeX/sLeA lectin binding domains (functional selectin receptors) on malignant cells (Yang et al. Citation2012; Mondal et al. Citation2015; Pinho and Reis Citation2015). Binding of E-selectin to sLeX/sLeA on LSCs surface activates niche-mediated intracellular pro-survival signaling (AKT/NF-kB/mTOR) pathways that contribute to malignant cell regeneration, survival and chemo-resistance (Barbier et al. Citation2020). In vivo, AML blasts with highest E-selectin binding potential are 12-fold more likely to survive chemotherapy. Therefore, therapeutic blockade of this pathway could potentiate conventional AML treatment regimens to improve efficacy and OS. Interestingly, therapies designed to disrupt the interactions between malignant stem cells and their BM niches are called ‘nichotherapies’ and have evolved as a promising therapeutic strategies in order to overcome immune evasion and chemoresistance of LSCs (Levesque et al. Citation2013).
To test if blockade of E-selectin sensitizes AML LSCs to chemotherapy, led to discovery of a selective small molecule antagonist called GMI-1271 (also known as uproleselan), a novel E-selectin antagonist designed to prevent the binding of E-selectin to its ligands on leukemia blasts. Uproleselan effectively inhibits niche-mediated pro-survival signaling, dampens AML blast regeneration, strongly enhances chemotherapy response, and protects from toxicity such as mucositis with improved survival in vivo (DeAngelo et al. Citation2019). A Phase I/II study of uproleselan added to salvage chemotherapy in adults with R/R AML showed promising remission rates and survival outcomes, and reduced rates of mucositis. Study data were consistent with previous findings demonstrating that higher E-selectin ligand expression contributes to clinical chemotherapy resistance, particularly increasing the risk of relapse (DeAngelo Citation2019). The Phase I/II study was succeeded by the currently ongoing pivotal phase 3 study (NCT03616470) aiming to assess the efficacy and safety of uproleselan with standard salvage chemotherapy in adults with R/R AML with results expected in late 2023. E-selectin ligand expression is also associated with poor survival in a small cohort of pediatric AML patients treated on COG AAML1031 (Leonti et al. Citation2019). Plans are being made for an early phase trial of chemotherapy ± uproleselan in children with R/R AML through the Pediatric Acute Leukemia (PedAL) Initiative (Chen and Glasser Citation2020).
Positive outcomes from selectin inhibition, as showcased by mAbs (e.g. crizanlizumab for SCD) and glycomimetics (e.g. uproleselan for AML) have reinvigorated the field of glycobiology-targeted therapeutics and encouraged promising endeavors to add more selectin blockers to the pipeline (Smith and Bertozzi Citation2021). The cessation E-selectin binding potential when both FUT4 and FUT7 were absent and the drastically reduced LSC survival in uproleselan-treated mice with AML confirm the absolute importance of TACA in AML and provide a rational basis for targeting E-selectin ligands by engineered lectins. Despite the E-selecton critical role in AML chemo-resistance, E-selectin blockade is only associated with a small (<1%) relocation of AML blasts from the BM to the blood stream. As a new idea, since P-selectin is more efficient mediator of leukocyte homing, its blockade would predictably lead to more efficient relocation of AML blasts to the blood stream that render them more susceptible to chemotherapy (Figure ). Patients with AML may, therefore, represent a target population for deploying a combination of E-selectin and P-selectin blockers in order to boost the anti-tumoral immunity. In this context, by targeting selectin-ligands, engineered lectins can be employed as novel targeted therapies where inhibiting selectins is crucial in the management. This includes diseases like SCD, inflammatory skin disorders, acute myocardial infarction, atherosclerosis, glomerulonephritis, systemic lupus erythematosus, stroke and malignancies (Ley Citation2003).
Figure 2. The effect of P-selectin antagonism. The upregulation of P-selectin on tumor associated vasculature (in contrast to E-selectin) may be less about induction of survival and regeneration of malignant cells but more as a factor directly contributing to metastatic cell homing. The simultaneous increased expression of P-selectin on endothelial cells induced by inflammatory cytokines in conjunction with increased expression of P-selectin ligands induced by aberrant glycosylation renders them hyperadhesive. By blocking P-selectin ligands, engineered lectins or glycomimetics can relocate AML leukemic stem cells (LSCs) to the peripheral blood followed by their elimination by chemotherapy. Thereferofer, P-selectin blockade can predictably prevent therapy resistance.
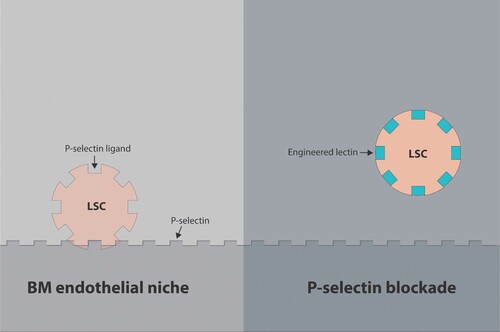
Targeting Siglec-ligands
In a manner similar to the selectins, the discovery of sialoadhesin (Siglec-1/ CD169) as a prominent mediator of macrophage adhesion to BM led to discovery of siglecs. The Siglecs are a family of I-type (immunoglobulin superfamily) transmembrane immunomodulatory receptors. Siglecs bind to sialoglycans that are expressed by most mammalian cells as markers of ‘self’ and so Siglecs can function as receptors for ‘self.’ In humans, the family of Siglecs comprises 15 members expressed almost exclusively on immune cells. Based on their amino acid sequence and chemical phylogeny, Siglecs divide into two groups – the rapidly evolving CD33-related (CD33 or Siglec-3, Siglec-5, -6, -7, -8, -9, -10, -11, -12, -14, and -16) and the conserved non-CD33-related (Siglec-1, -2, -4, and -15) (Bornhöfft et al. Citation2018).
Distinct Siglec- sialoglycan interactions initiate molecular and cellular responses important to the function of the cells on which they are expressed (Gonzalez-Gil and Schnaar Citation2021). Most individual Siglecs show restricted patterns of expression on specific cell types (Bednar et al. Citation2017), which makes them useful cell surface markers, and reflects cell-type specific functions mediated by these Siglecs (Bochner and Zimmermann Citation2015). For example, Siglec-2 (CD22) is exclusively expressed on mature B-cells and is perhaps a better target than CD19 which suffers from acquired resistance due to surface CD19 antigen loss; and in this scenrio, CD22 is an ideal target as it usually retains expression in CD19neg tumors. Siglec-3 (CD33) is expressed on the surface of myeloid progenitor cells, monocytes, macrophages, and microglia. Unfortunately, pursuing anti-Siglec based immunotherapies for clinical use in hematological malignancies has been limited to the ADC format. These therapeutic applications constitute the only FDA- approved Siglec- targeted therapies: the anti- CD22 Ino and the anti-CD33 GO. Details on Siglec family members are beyond the scope of this review and the reader is referred to an excellent review on roles all 15 human Siglecs (including Siglec XII) in cancer biology (Lim et al. Citation2021).
Through inhibitory signaling pathways and shared ability to recognize a wide range of configurations of sialic acids, Siglecs are important part of mammalian innate immune ‘self’ recognition, a mechanism for the host to discriminate between infectious nonself and noninfectious self. The amount of sialoglycans on the surface of normal cells far exceeds that of pathogens, and because of this, sialoglycans are self (SAMPs) rather than non-self glycans that bind Siglecs (Hitchcock et al. Citation2021). Unfortunately, pathogens have developed the ability, through cell-surface glycocalyx mimicry, to display sialic acids that are mistakenly seen as self. The transmembrane CD33-related Siglecs receptors play a crucial role in this process. Normally, sialoglycans have been shown to serve as SAMPs by binding to CD33-related Siglecs, a family of pattern-recognition immune receptors on immune cells containing intracellular immunoreceptor tyrosine-based inhibitory motifs (ITIM) that regulate the cells of the innate and adaptive immune response. Binding between CD33-related Siglecs appear to suppress autoimmune responses. On the other hand, the evolutionary pressure exerted by pathogens can mimic sialoglycan-SAMPs (Sia-SAMPs) and bind to inhibitory CD33-related Siglecs to escape innate immune control. To illustrate, the interections between Siglecs (especially Siglec-7 and Siglec-9) on leukocytes and specific sialoglycans on bacteria (e.g. group B Streptococcus, Pseudomonas, Campylobacter, and Neisseria) initiate inhibitory responses in those leukocytes that dampens their anti-pathogen responses leading to innate immune evasion (Carlin et al. Citation2009; Chang and Nizet Citation2014).
In a similar way, hypersialylation is also associated with tumorigenesis. Upregulation of sialoglycan expression is commonly found on various tumor types. Numerous studies have shown that tumor cells can exploit sialoglycan–Siglec interactions to inhibit effector immune cell activity and modulate myeloid cell functions to create an immunosuppressive TME, thus contributing to tumor immune evasion and sustained tumor growth (Sanford Citation1967; Macauley et al. Citation2014). The CD33-related Siglecs Siglec-7 (p75/AIRM1, CD328) and Siglec-9 (CD329) are closely related and are of vital importance for tumor immunotherapy. Similar to pathogens, hypersialylation on cancer cell surfaces is recognized by Siglec receptors found on leukocytes as ‘Sia-SAMPs.’ Recent evidence suggests that increased Sia-SAMP inhibit cell-mediated tumor cell killing via inhibitory CD33-related Siglecs, especially Siglec-7 and Siglec-9 receptors (Stanczak et al. Citation2018). The resultant down-regulation of immune responsiveness leads to tumor growth. Engagement of Siglec-7 and Siglec-9 inhibits NK cell-mediated tumor cell killing in vitro. In that vein, the role of NK cells as anti-cancer effectors cannot be overstated. NK cells normally act as early responders to pathogens and cancerous cells, leading to cell death and clearance (Kumar and McNerney Citation2005). For example, MUC16, which binds to Siglec-9-positive leukocytes via α2,3-linked sialic acids, facilitate survival of ovarian cancer cells by decreasing the expression of NK cell stimulating protein CD16, leading to reduced NK cell activity (Patankar et al. Citation2005; Belisle et al. Citation2007). NK cells activity is positively correlated with reducing metastasis and their reduced activation contribute to increased metastasis in ovarian cancer. Siglec-7/9 are abundantly present on NK cells with Siglec-7 being a pan-marker for human NK cells and Siglec-9 being present on a population of CD56dim CD16+ NK cells (Crocker et al. Citation2007). Ligands for Siglec-7/9 were found on various human cancers because hypersialylation offers a selective advantage to tumor cells under pressure from NK immunosurveillance by increasing Siglec ligands (Hudak et al. Citation2014). Consequently, tumors expressing high levels of Siglec ligands (especially ligands for Siglec-7 and Siglec-9) can evade NK cell killing (Bochner and Zimmermann Citation2015) and, vice versa, removing sialic acids from tumor cells increases their susceptibility to cytotoxicity from NK cells (Jandus et al. Citation2014). Because NK cells have a well-documented role in the early stages of tumorigenesis, NK cell response to increasing sialoglycans on tumor cells is an important determinant of tumor engraftment. Changes in sialylation may also modulate tumor cell invasion or metastasis (Miyazaki et al. Citation2012).
Furthermore, signaling through the Siglec-sialoglycan axis in the TME favors tumor immune evasion similar to the PD-1/PD-L1 signaling pathway. This is attributed to that majority of Siglecs contains immune inhibitory motifs (ITIM domains) similar to the IC receptor PD-1 (van Houtum et al. Citation2021). In agreement with previously published works, immune inhibitory Siglecs are now considered as pivotal glyco-IC receptors (van de Wall et al. Citation2020; Wielgat et al. Citation2020), while Siglec7/9 are considered specifically as myeloid IC as their blockade prevent the formation of immunosuppressive TME by influencing the myeloid arm of innate immune responses (Avril et al. Citation2004; Kim et al. Citation2021; Poggi and Zocchi Citation2021; Rosenstock and Kaufmann Citation2021). In particular, Siglec-9, the typical pattern-recognition immune receptor for Sia-SAMPs, has also been implicated in myeloid cell-mediated cancer progression via promoting an immunosuppressive TME through engagement with tumor-associated mucins. Cancer cells release MUC1-associated sTn antigens which bind Siglec-9 expressed on immature DCs promoting an immature DC phenotype that sequentially reduces T-cell effector functions (Bhatia et al. Citation2019). Moreover, binding of the cancer-associated MUC1-ST, a glycoform of the mucin MUC1 carrying the sialyl-T antigen (ST antigen), to Siglec-9 expressed by primary macrophages in breast cancer and pancreatic ductal adenocarcinoma induces an immunosuppressive TAM phenotype characterized by increased expression of PD-L1 (Burchell et al. Citation1999; Beatson et al. Citation2016). Tumor sialoglycan–Siglec interactions contribute to the immunosuppressive TME by modulating the function of DCs. DCs are the dominant APCs of the immune system and central regulators of the adaptive immune response that govern the transition from innate to the adaptive immune repertoires (Crespo et al. Citation2013; Gardner and Ruffell Citation2016).
In conclusion, aberrant sialoglycan expression has strong immunosuppressive characteristics that contribute to the formation of an immunosuppressive TME. Targeting tumor sialoglycans and/or sialoglycan–Siglec interactions represents a promising therapeutic strategy to augment anti-tumor immunity in the TME. One group has demonstrated that blocking sialoglycan expression in mouse B16-F10 melanoma and 9464D neuroblastoma tumors by intratumoral injections with Ac53FaxNeu5Ac suppressed tumor growth and significantly altered the immune cell composition of the TME. The selective sialoglycan biosynthesis inhibitor Ac53FaxNeu5Ac is a synthetic fluorinated sialic acid mimetic that blocks sialic acid expression by inhibiting sialyltransferase activity (glycomimetic) (Büll et al. Citation2018). Since myeloid checkpoints that can be effectively used as immunotherapy targets are still lacking, targeting the myeloid receptors Siglec-7 and Siglec-9 can be exploited as ICBs to further release the brakes of the immune system beyond targeting the PD-1/PD-L1 and CTLA4 pathways (Ibarlucea-Benitez et al. Citation2021). Therefore, sialoglycans are potentially attractive targets that should inspire the development of engineered lectins targeting tumor sialoglycans as novel Siglec-based therapeutics and a new pillar in cancer immunotherapy.
Employing lectins in cancer immunotherapy
The tumor-virus mimicry
Like tumors, viruses have evolved ways to achieve molecular mimicry of host glycans (Varki Citation2017). Molecular mimicry serves to (1) down-regulate innate immune responses by engaging the inhibitory Siglec recognition of cell surface sialoglycans that imitate endogenous SAMPs, (2) recruit the soluble complement regulator sialic acid-binding protein Factor H (the main regulator of the alternative complement pathway activity) for complement immune evasion (Agrawal et al. Citation2017), and (3) mask antibody recognition through the glycan shield. Most viruses simply hijack the host glycosylation machinery to heavily glycosylate their own surface proteins during replication. In addition to immune escape strategy of enveloped viruses, glycans on the viral envelope often have a crucial role in enabling an efficient transmission of the pathogen and/or entry into its susceptible target cells.
Viral glycosylation has been investigated in the context of a number of envelope glycoproteins, including the envelope glycoprotein (Env) of human immunodeficiency virus-1 (HIV-1), HA (the ‘H’ in ‘H1N1’) of influenza virus, coronavirus glycoprotein spike (S), glycoprotein (GP) of Ebola virus, glycoprotein complex (GPC) of Lassa virus, and envelope (E) glycoprotein of dengue, Zika, and other flaviviruses (Watanabe et al. Citation2019). The evolutionary development of viral glycosylation as a mechanism to both enhance viral uptake and evade host organism defenses has led to co-evolution of anti-viral lectins interacting predominantly with high-mannose glycan structures added as post-translational modifications to the envelope proteins of viruses. For example, both human immunodeficiency virus-1 (HIV-1) gp120 and influenza HA protein bear high-mannose-type N-glycans that are susceptible to host lectins. Intriguingly, many natural lectins identified in bacteria, plants and marine algae have been also reported to have anti-viral activity by inhibiting viral replication through interacting with viral envelope glycoproteins (Mitchell et al. Citation2017). These anti-viral lectins have been successfully used against hepatitis C, influenza A/B, herpes, Japanese encephalitis, HIV and Ebola (Ghafoor et al. Citation2021). The therapeutic potential of host and natural lectins as anti-viral agents led to the development of molecularly engineered anti-viral lectins that target virus-associated glycans. Engineered lectins were broadly effective anti-viral agents either directly by binding to glycans of the viral envelope leading to blocking virus entry, or indirectly by forcing pathogens to mutate (or delete) their protective envelope glycan shields (Balzarini Citation2007b). The latter mechanism is termed lectin pressure which adds an exciting new dimension to lectins as a novel conceptual class of anti-microbial agents.
The importance of lectins as first-line host defense against infections is demonstrated by MBL deficiency, one of the most common primary immunodeficiencies with increased predisposition to infections in immunocompromised patients (Neth et al. Citation2001; Neth et al. Citation2005; Jacobson et al. Citation2020). Remarkably, MBL deficiency is deleterious in HIV infection (Eisen et al. Citation2008). Plasma-derived MBL has been used in reconstitution therapy but concerns over viral contamination and production capacity led to the production of recombinant MBL (rMBL) (Vorup-Jensen et al. Citation2001; Jensenius et al. Citation2003). rMBL has therapeutic potential as anti-viral agent through LP activation, opsonophagocytosis and direct spatial blocking of virus-receptor interactions and entry. Unfortunately, due to its complex quaternary structure, rMBL is complicated and expensive to produce. However, recombinant chimeric lectin (RCL) consisting of MBL and L-ficolin exhibits superior protective potency, cost-effectiveness and safety profile than rMBL (Mason and Tarr Citation2015). RCLs have been generated by replacing various portions of the collagenous domain of MBL with that of L-FCN, thus utilizing MBL-CRD for target recognition. In the case of IAV, RCLs have surpassed rMBL for anti-viral activities and showed significantly higher dose-dependent activation of the LP. Although the RCL-mediated LP activation is augmented by MASP supplementation, all RCLs activate the LP without MASP (in comparison to rMBLs which require 1000-fold more protein concentration to activate the LP without MASP). Unlike rMBL, RCLs also have the therapeutic advantage of diminished interference on the host coagulation. The anti-IAV effects of RCLs were efficacious both in vitro and in vivo (Chang et al. Citation2011; Takahashi et al. Citation2013).
The potential mitogenic properties, cost of production, and isolation and purification in mass quantities are the main pharmacological hurdles for developing clinically useful anti-viral lectins. Luckily, anti-viral proteins have the potential to be improved by changes to their structure that will render them more suitable for clinical development. Research was conducted to mutate lectins were engineered to eliminate their potentially harmful mitogenicity (ability to cause immune cell proliferation) while still retaining their very potent anti-viral activity (Swanson et al. Citation2015). Moreover, genetic engineering strategies to increase the population of lectins adopting a suitable conformation for glycan binding are effective for creating more powerful lectins with enhanced anti-viral activity (Matoba et al. Citation2021). Recently, the H84T BanLec, a recombinant BanLec engineered to lose its mitogenicity while preserving the virucidal activity of the lectin by mutating a single amino acid, was found highly efficacious against influenza infection in vitro and in vivo via intranasal administration. H84T is highly effective even against oseltamivir- and adamantane-resistant strains (Covés-Datson et al. Citation2020). The H84T BanLec activity against viruses containing high-mannose N-glycans also included HIV-1 and -2, hepatitis C virus and Ebola virus (Covés-Datson et al. Citation2019). Another approach was incorporating a benzoboroxole (BzB) moiety into a variety of biocompatible polymer backbones and polymeric constructs which can be synthetically modified to enhance their activity and specificity. The resultant BzB-based synthetic lectins present an affordable and scalable microbicide (Mahalingam et al. Citation2011).
It is worth mentioning that recombinant DNA technology has been traditionally used for cloning and characterizing newly discovered lectins that benefit from the tailor-made design associated with recombinant production. Recombinant DNA technology produces lectins with several advantages in terms of the production and purification processes. The use of recombinant expression systems has allowed the large-scale production of many lectins with interesting activities, which would be very difficult otherwise to obtain. Genetically modified lectins with specific properties e.g. recombinant lectins with anti-HIV activity may be obtained by using recombinant techniques (Oliveira et al. Citation2013). In order the overcome various challenges of plant lectins (Nascimento et al. Citation2012), recombinant prokaryotic lectins (RPLs) have emerged as an alternative. Prokaryotic lectins offer significant advantages over eukaryotic lectins for the development of enhanced glycoselective tools (Keogh et al. Citation2014). The directed evolution strategy can tailor novel lectins for novel specificity or biological functions (Hu et al. Citation2014). Recently, novel recombinant prokaryotic lectins (RPLs) were developed through employing directed evolution particularly iterative focussed site directed mutagenesis, expressed, and produced in Escherichia coli, specific to different sugar moieties: α-mannose, β-galactose, fucose and sialic acid. Besides exhibiting similar specificity to different glycoproteins and BSA glycan conjugates with different glycosylation patterns, the presented RPLs displayed substantially higher binding activities to the selected targets than their analogous plant lectins with similar binding targets. The majority of RPLs also showed exceptional analytical performance that could be of great benefit in the characterization of glycoproteins, evaluations of monoclonal antibodies and development of biosensors (Fernandez-Poza et al. Citation2021). The tailor-made design associated with recombinant DNA technology should aid in cloning and characterizing recombinant lectins targeting tumors and microbes including SARS-CoV-2 incriminated in the deadly coronavirus disease 2019 (COVID-19) (Saad Citation2021).
Lectin-protease conjugates
In case administering natural or engineered lectins alone does not result in sufficient activation of the LP on the surface of leukemic blast cells then administering lectin-protease conjugates (e.g. a PNA – associated serine protease conjugate) reminiscent of the lectin–MASP complexes of the natural LP is a another potential option. The MASP component of the natural LP is basically an enzyme that belongs to family of modular serine proteases (peptidases) that catalyze proteolysis by hydrolyzing peptide bonds in proteins. Synthetic proteases can be coupled with lectins by linkers. This is feasible since proteases represent the largest segment of the industrial enzyme market. In fact, substrate specificity experiments have showed that recombinant protease (MASP-2) displayed a rather wide, C1s-like esterolytic activity, and specifically cleaved complement proteins C2 and C4, with relative efficiencies 3- and 23-fold higher, respectively, than human C1s (Rossi et al. Citation2001). This model (Figure ) can be utilized in the treatment of tumors by specifically targeting their specific glycans. Therefore, the utilization of the LP in innovative therapeutics could lead to a quantum leap forward for the medical science.
Figure 3. The lectin-protease conjugate. Binding of a lectin-protease conjugate to membrane glycans activates the lectin pathway in a manner similar to the classic pathway where cleaving both C4 and C2 generates the C3 convertase (C4b2a). The C3 convertase cleaves C3 to give rise to a smaller C3a fragment and a larger C3b fragment. The C3b molecules associate with C4bC2a or C3bBb complexes to form the C5 convertase (not shown). The C5 convertase cleaves C5 and initiates activation and assembly of the terminal components, C5, C6, C7, C8, and C9, into the C5b-9 membrane attack complex (MAC) on the surface of target cells.
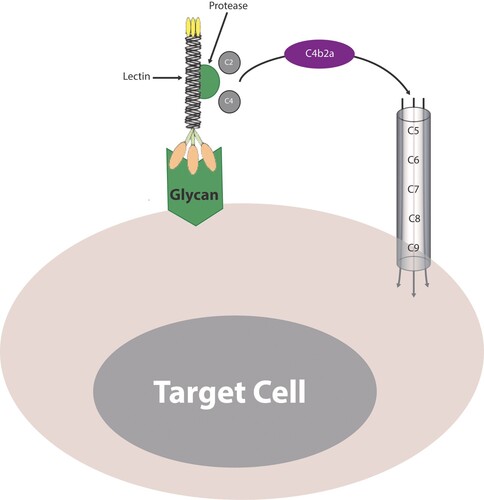
Possible resistance to engineered lectins
Complement depletion can be replenished with complement proteins-replete FFP in conjunction with engineered lectins. Just like mAbs, mCRPs can theoretically also pose a threat to engineered lectins by inhibiting complement activation. However, the mCRPs CD55 and CD59 are basically glycoconjugates (GPI-APs). The covalent attachment of GPI moiety to the C-terminus of at least 150 human proteins anchors these proteins in lipid rafts on the cell surface to form GPI-APs. GPI-APs are integral membrane proteins that play critical roles in embryogenesis, neurogenesis, immunity, and fertilization (Liu and Fujita Citation2020).
The GPI moiety of GPI-APs consists of the conserved core glycan, phosphatidylinositol (PI) and glycan side chains. The structure of the conserved core glycan is EtNP-6Manα2-Manα6-(EtNP)2Manα4-GlNα6-myoinositol-phospholipid (where EtNP, Man, and GlcN are ethanolamine phosphate, mannose, and glucosamine, respectively). Despite the highly conserved core structure of GPI-APs among all eukaryotes, some diversity in GPI-anchor biosynthesis exists owing to the great variability in the glycan side chains (Ferguson et al. Citation2017; Sarnataro et al. Citation2017; Kinoshita Citation2020). Engineered lectins can be designed to target those specific glycan side chains possessed by CD55 and CD59. Therefore, combining anti- GPI-AP engineered lectins with therapeutic engineered lectins directed against cancer-associated glycans would predictably neutralize tumor resistance. As mentioned earlier, GPI-APs are highly specific cancer biomarkers that play critical role in cancer progression. Several GPI-AP-targeted immunotherapies have been designed for targeting GPI-AP blockade on tumor cells (Hussein et al. Citation2020). Hence, anti-GPI-APs engineered lectins may have dual effects manifested by counteracting tumor resistance concurrently with tumor-killing effect through the GPI-AP blockade itself.
Conclusion
While there are an ever-increasing number of targeted therapies to treat leukemia, durable benefits remain mostly elusive as reflected by the unchanging poor patients’ outcome (Haltalli and Lo Celso Citation2020). Adaptive immune resistance by which cancer cells evade tumor-specific T-cell responses and grow indefinitely imposes enormous challenges for the future clinical application of the T-cell mediated immunotherapies. Alternatively, the innate immune lectins can distinguish self from non-self with considerable specificity and are crucial defenders for vertebrates and invertebrates as they do not rely on adaptive immunity for pathogen clearance (Fujita et al. Citation2004). As altered expression of cell-surface glycans catalyzed by the malignant transformation is an intrinsic target for lectin recognition, the burgeoning need for truly tumor-specific antigens and novel targeted therapies for hematological malignancies should be sought in the potential avenues of glycan-based cancer neo-antigens. The higher treatment specificity and reduced off-target effects entailed with targeting TACA would also be encouraging to be exploited in soild tumors where the quest for extremely delicate targets is rather more demanding (Townsend et al. Citation2018). Moreover, as innate LP activation is contingent on glycan recognition, the antibody-like specific reactivity of lectins for cancer-associated glycans make lectins ideal non-antibody constructs for effective target cell killing with favorable off-target toxicity profile. The opsonization/CDC-powered lectins clearly seems superior to the perforin-powered T-cell-mediated immunotherapies for target cell death. The deeper we delve into the complex glycobiological landscapes of pathogens and tumors, it becomes clear that both play the game of molecular mimicry by masquerading as SAMPS in order to evade host immune responses. Congruent with the recent success of anti-viral engineered lectins, it is apparent that LP can readily be skewed towards cancer-associated glycans and, therefore, engineered lectins that target cancer-associated glycans should be pursued for clinical development and trialed for incorporation into either upfront or salvage therapy. Researchers should also seek to create bispecific engineered lectins (i.e. similar to bispecific antibodies) that are capable of binding two distinct targets (e.g. one that binds glycans on target cells and another that binds glycans on phagocytes for opsonization (Tang et al. Citation2017; Park et al. Citation2021)) in order to boost the killing mechanism of lectins. Scientific research on therapeutic approaches targeting malignancies that integrates both cancer glycomics and cancer genomics has become imperative in fighting humans’ most dreaded diseases.
Graphic designer
Eng. Yasmin Adel.
Acknowledgements
The author apologizes to the numerous excellent scientists whose work is not adequately discussed or cited in this review, and especially thanks Ingrid G. Winkler, Johanna Erbani, Valerie Barbier, Jean-Pierre Levesque, Stefan Mereiter, Meritxell Balmaña, Diana Campos, Joana Gomes, Celso A. Reis, Ana Filipa Costa, Catarina Gomes, Carolyn R. Bertozzi, Benjamin A. H. Smith, Ruben D. Houvast, Mireille Vankemmelbeke, Lindy G. Durrant, Manfred Wuhrer, Victor M. Baart, Peter J. K. Kuppen, Lioe-Fee de Geus-Oei, Alexander L. Vahrmeijer, Cornelis F. M. Sier, Yi-Chen Chan, Hsien-Ya Lin, Zhijay Tu, Yen-Hsi Kuo, Shang-Te Danny Hsu, Chun-Hung Lin, Danyang Feng, Abdul Sami Shaikh, Fengshan Wang, Jan Balzarini, Kazue Takahashi, Jun Hirabayashi, Ryoichi Arai, Bruce S. Bochner, Nives Zimmermann, Carter A. Mitchell, Koreen Ramessar, Barry R O’Keefe, Maurizio Chiriva-Internati, Alejandro J. Cagnoni, Juan M. Pérez Sáez, Gabriel A. Rabinovich, Karina V. Mariño, Valerie R. Wiersma, Chitra Mandal Reinhard Schwartz-Albiez, Reinhard Vlasak, Jose L. Daniotti, Ernesto RodrÍguez, Sjoerd T. T. Schetters and Yvette van Kooyk for their invaluable contribution for ushering in a new era of cancer immunotherapy based on lectin–glycan interactions as a fundamental axis in cancer and an excellent source of novel targeted cancer therapies.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- Agrawal P, Nawadkar R, Ojha H, Kumar J, Sahu A. 2017. Complement evasion strategies of viruses: an overview. Front Microbiol. 8:1117. doi:10.3389/fmicb.2017.01117.
- Ailles LE, Weissman IL. 2007. Cancer stem cells in solid tumors. Curr Opin Biotechnol. 18(5):460–466. doi:10.1016/j.copbio.2007.10.007.
- Akella NM, Ciraku L, Reginato MJ. 2019. Fueling the fire: emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biol. 17(1):52. doi:10.1186/s12915-019-0671-3.
- Aldoss I, Song J, Stiller T. 2017. Correlates of resistance and relapse during blinatumomab therapy for relapsed/refractory acute lymphoblastic leukemia. Am J Hematol. 92(9):858–865. doi:10.1002/ajh.24783.
- Alfonso S, Valdés-Zayas A, Santiesteban ER. 2014. A randomized, multicenter, placebo-controlled clinical trial of racotumomab-alum vaccine as switch maintenance therapy in advanced non-small cell lung cancer patients. Clin Cancer Res. 20(14):3660–3671. doi:10.1158/1078-0432.CCR-13-1674.
- Ali YM, Lynch NJ, Haleem KS. 2012. The lectin pathway of complement activation is a critical component of the innate immune response to pneumococcal infection. PLoS Pathog. 8(7):e1002793. doi:10.1371/journal.ppat.1002793.
- Aller CT, Kucuk O, Springer GF, Gilman-Sachs A. 1996. Flow cytometric analysis of T and Tn epitopes on chronic lymphocytic leukemia cells. Am J Hematol. 52(1):29–38. doi:10.1002/(SICI)1096-8652(199605)52:1<29::AID-AJH5>3.0.CO;2-8.
- Alvarez-Rueda N, Desselle A, Cochonneau D. 2011. A monoclonal antibody to O-acetyl-GD2 ganglioside and not to GD2 shows potent anti-tumor activity without peripheral nervous system cross-reactivity. PLoS One. 6(9):e25220. doi:10.1371/journal.pone.0025220.
- Amado M, Carneiro F, Seixas M, Clausen H, Sobrinho-Simões M. 1998. Dimeric sialyl-Le(x) expression in gastric carcinoma correlates with venous invasion and poor outcome. Gastroenterology. 114(3):462–470. doi:10.1016/s0016-5085(98)70529-3.
- An HJ, Kronewitter SR, de Leoz ML, Lebrilla CB. 2009. Glycomics and disease markers. Curr Opin Chem Biol. 13(5-6):601–607. doi:10.1016/j.cbpa.2009.08.015.
- Anguille S, Van Tendeloo VF, Berneman ZN. 2012. Leukemia-associated antigens and their relevance to the immunotherapy of acute myeloid leukemia. Leukemia. 26(10):2186–2196. doi:10.1038/leu.2012.145.
- Apostolopoulos V, Yuriev E, Ramsland PA. 2003. A glycopeptide in complex with MHC class I uses the GalNAc residue as an anchor. Proc Natl Acad Sci USA. 100(25):15029–15034. doi:10.1073/pnas.2432220100.
- Apweiler R, Hermjakob H, Sharon N. 1999. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta. 1473(1):4–8. doi:10.1016/s0304-4165(99)00165-8.
- Arnaud J, Audfray A, Imberty A. 2013. Binding sugars: from natural lectins to synthetic receptors and engineered neolectins. Chem Soc Rev. 42(11):4798–4813. doi:10.1039/c2cs35435g.
- Asano S, Pal R, Tanaka HN. 2019. Development of fluorescently labeled SSEA-3, SSEA-4, and Globo-H glycosphingolipids for elucidating molecular interactions in the cell membrane. Int J Mol Sci. 20(24):6187. doi:10.3390/ijms20246187.
- Asiry S, Kim G, Filippou PS. 2021. The cancer cell dissemination machinery as an immunosuppressive niche: a new obstacle towards the era of cancer immunotherapy. Front Immunol. 12:654877. doi:10.3389/fimmu.2021.654877.
- Ataga KI, Kutlar A, Kanter J. 2017. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 376(5):429–439. doi:10.1056/NEJMoa1611770.
- Auslander N, Cunningham CE, Toosi BM. 2017. An integrated computational and experimental study uncovers FUT9 as a metabolic driver of colorectal cancer. Mol Syst Biol. 13(12):956. doi:10.15252/msb.20177739.
- Avril T, Floyd H, Lopez F, Vivier E, Crocker PR. 2004. The membrane-proximal immunoreceptor tyrosine-based inhibitory motif is critical for the inhibitory signaling mediated by Siglecs-7 and -9, CD33-related Siglecs expressed on human monocytes and NK cells. J Immunol. 173:6841–6849. doi:10.4049/jimmunol.173.11.6841.
- Azizidoost S, Vijay V, Cogle CR, Khodadi E, Saki N. 2017. The role and clinical implications of the endosteal niche and osteoblasts in regulating leukemia. Clin Transl Oncol. 19(9):1059–1066. doi:10.1007/s12094-017-1642-1.
- Badar T, Szabo A, Litzow M. 2021. Multi-institutional study evaluating clinical outcome with allogeneic hematopoietic stem cell transplantation after blinatumomab in patients with B-cell acute lymphoblastic leukemia: real-world data. Bone Marrow Transplant. 56(8):1998–2004. doi:10.1038/s41409-021-01279-w.
- Bajwa R, Schechter T, Soni S. 2013. Outcome of children who experience disease relapse following allogeneic hematopoietic SCT for hematologic malignancies. Bone Marrow Transplant. 48(5):661–665. doi:10.1038/bmt.2012.209.
- Baldus SE, Zirbes TK, Mönig SP. 1998. Histopathological subtypes and prognosis of gastric cancer are correlated with the expression of mucin-associated sialylated antigens: sialosyl-Lewis(a), Sialosyl-Lewis(x) and sialosyl-Tn. Tumour Biol. 19(6):445–453. doi:10.1159/000030036.
- Ballehaninna UK, Chamberlain RS. 2012. The clinical utility of serum CA 19-9 in the diagnosis, prognosis and management of pancreatic adenocarcinoma: An evidence based appraisal. J Gastrointest Oncol. 3(2):105–119. doi:10.3978/j.issn.2078-6891.2011.021.
- Balzarini J. 2007a. Carbohydrate-binding agents: a potential future cornerstone for the chemotherapy of enveloped viruses? Antivir Chem Chemother. 18(1):1–11. doi:10.1177/095632020701800101.
- Balzarini J. 2007b. Targeting the glycans of glycoproteins: a novel paradigm for antiviral therapy. Nat Rev Microbiol. 5(8):583–597. doi:10.1038/nrmicro1707.
- Barbier V, Erbani J, Fiveash C. 2020. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat Commun. 11(1):2042. doi:10.1038/s41467-020-15817-5.
- Beatson R, Tajadura-Ortega V, Achkova D. 2016. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin Siglec-9. Nat Immunol. 17(11):1273–1281. doi:10.1038/ni.3552.
- Bednar KJ, Shanina E, Ballet R. 2017. Human CD22 inhibits murine B cell receptor activation in a human CD22 transgenic mouse model. J Immunol. 199(9):3116–3128. doi:10.4049/jimmunol.1700898.
- Bejanyan N, Weisdorf DJ, Logan BR. 2015. Survival of patients with acute myeloid leukemia relapsing after allogeneic hematopoietic cell transplantation: a center for international blood and marrow transplant research study. Biol Blood Marrow Transplant. 21(3):454–459. doi:10.1016/j.bbmt.2014.11.007.
- Belardi B, Bertozzi CR. 2015. Chemical lectinology: tools for probing the ligands and dynamics of mammalian lectins in vivo. Chem Biol. 22(8):983–993. doi:10.1016/j.chembiol.2015.07.009.
- Belisle JA, Gubbels JA, Raphael CA. 2007. Peritoneal natural killer cells from epithelial ovarian cancer patients show an altered phenotype and bind to the tumour marker MUC16 (CA125). Immunology. 122(3):418–429. doi:10.1111/j.1365-2567.2007.02660.x.
- Bellone S, Roque D, Cocco E. 2012. Downregulation of membrane complement inhibitors CD55 and CD59 by siRNA sensitises uterine serous carcinoma overexpressing Her2/neu to complement and antibody-dependent cell cytotoxicity in vitro: implications for trastuzumab-based immunotherapy. Br J Cancer. 106(9):1543–1550. doi:10.1038/bjc.2012.132.
- Bergfeld AK, Pearce OM, Diaz SL, Pham T, Varki A. 2012. Metabolism of vertebrate amino sugars with N-glycolyl groups: elucidating the intracellular fate of the non-human sialic acid N-glycolylneuraminic acid. J Biol Chem. 287(34):28865–28881. doi:10.1074/jbc.M112.363549.
- Bertuzzi S, Quintana JI, Ardá A, Gimeno A, Jiménez-Barbero J. 2020. Targeting galectins with glycomimetics. Front Chem. 8:593. doi:10.3389/fchem.2020.00593.
- Beyer-Sehlmeyer G, Hiddemann W, Wörmann B, Bertram J. 1999. Suppressive subtractive hybridisation reveals differential expression of serglycin, sorcin, bone marrow proteoglycan and prostate-tumour-inducing gene I (PTI-1) in drug-resistant and sensitive tumour cell lines of haematopoetic origin. Eur J Cancer. 35(12):1735–1742. doi:10.1016/s0959-8049(99)00202-6.
- Bhatia R, Gautam SK, Cannon A. 2019. Cancer-associated mucins: role in immune modulation and metastasis. Cancer Metastasis Rev. 38(1-2):223–236. doi:10.1007/s10555-018-09775-0.
- Birks SM, Danquah JO, King L, Vlasak R, Gorecki DC, Pilkington GJ. 2011. Targeting the GD3 acetylation pathway selectively induces apoptosis in glioblastoma. Neuro Oncol. 13(9):950–960. doi:10.1093/neuonc/nor108.
- Blaeschke F, Willier S, Stenger D. 2020. Leukemia-induced dysfunctional TIM-3+CD4+ bone marrow T cells increase risk of relapse in pediatric B-precursor ALL patients. Leukemia. 34(10):2607–2620. doi:10.1038/s41375-020-0793-1.
- Blsakova A, Kveton F, Kasak P, Tkac J. 2019. Antibodies against aberrant glycans as cancer biomarkers. Expert Rev Mol Diagn. 19(12):1057–1068. doi:10.1080/14737159.2020.1687295.
- Bochner BS, Zimmermann N. 2015. Role of siglecs and related glycan-binding proteins in immune responses and immunoregulation. J Allergy Clin Immunol. 135(3):598–608. doi:10.1016/j.jaci.2014.11.031.
- Bornhöfft KF, Goldammer T, Rebl A, Galuska SP. 2018. Siglecs: a journey through the evolution of sialic acid-binding immunoglobulin-type lectins. Dev Comp Immunol. 86:219–231. doi:10.1016/j.dci.2018.05.008.
- Boroughs LK, DeBerardinis RJ. 2015. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 17(4):351–359. doi:10.1038/ncb3124.
- Bottoni P, Scatena R. 2015. The role of CA 125 as tumor marker: biochemical and clinical aspects. Adv Exp Med Biol. 867:229–244. doi:10.1007/978-94-017-7215-0_14.
- Bowles WHD, Gloster TM. 2021. Sialidase and sialyltransferase inhibitors: targeting pathogenicity and disease. Front Mol Biosci. 8:705133. doi:10.3389/fmolb.2021.705133.
- Brändlein S, Pohle T, Ruoff N, Wozniak E, Müller-Hermelink HK, Vollmers HP. 2003. Natural IgM antibodies and immunosurveillance mechanisms against epithelial cancer cells in humans. Cancer Res. 63(22):7995–8005.
- Bremer EG, Levery SB, Sonnino S. 1984. Characterization of a glycosphingolipid antigen defined by the monoclonal antibody MBr1 expressed in normal and neoplastic epithelial cells of human mammary gland. J Biol Chem. 259(23):14773–14777.
- Bresalier RS, Niv Y, Byrd JC. 1991. Mucin production by human colonic carcinoma cells correlates with their metastatic potential in animal models of colon cancer metastasis. J Clin Invest. 87(3):1037–1045. doi:10.1172/JCI115063.
- Bride KL, Vincent TL, Im SY. 2018. Preclinical efficacy of daratumumab in T-cell acute lymphoblastic leukemia. Blood. 131(9):995–999. doi:10.1182/blood-2017-07-794214.
- Brockhausen I. 2003. Glycodynamics of mucin biosynthesis in gastrointestinal tumor cells. Adv Exp Med Biol. 535:163–188. doi:10.1007/978-1-4615-0065-0_11.
- Brockhausen I, Stanley P. 2017. O-GalNAc glycans. In: Varki A, Cummings RD, Esko JD, editor. Essentials of glycobiology. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; p. 2015–2017. https://www.ncbi.nlm.nih.gov/books/NBK453030/ doi: 10.1101/glycobiology.3e.010.
- Brodsky RA. 2014. Paroxysmal nocturnal hemoglobinuria. Blood. 124(18):2804–2811. doi:10.1182/blood-2014-02-522128.
- Brossart P, Schneider A, Dill P. 2001. The epithelial tumor antigen MUC1 is expressed in hematological malignancies and is recognized by MUC1-specific cytotoxic T-lymphocytes. Cancer Res. 61(18):6846–6850.
- Brown PA, Ji L, Xu X. 2021. Effect of postreinduction therapy consolidation with blinatumomab vs chemotherapy on disease-free survival in children, adolescents, and young adults with first relapse of B-cell acute lymphoblastic leukemia: a randomized clinical trial. JAMA. 325(9):833–842. doi:10.1001/jama.2021.0669.
- Buckley SA, Walter RB. 2015. Antigen-specific immunotherapies for acute myeloid leukemia. Hematol Am Soc Hematol Educ Program. 2015:584–595. doi:10.1182/asheducation-2015.1.584.
- Büll C, Boltje TJ, Balneger N. 2018. Sialic acid blockade suppresses tumor growth by enhancing T-cell-mediated tumor immunity. Cancer Res. 78(13):3574–3588. doi:10.1158/0008-5472.CAN-17-3376.
- Büll C, Stoel MA, den Brok MH, Adema GJ. 2014. Sialic acids sweeten a tumor’s life. Cancer Res. 74(12):3199–3204. doi:10.1158/0008-5472.CAN-14-0728.
- Burchell J, Poulsom R, Hanby A. 1999. An alpha2,3 sialyltransferase (ST3Gal I) is elevated in primary breast carcinomas. Glycobiology. 9(12):1307–1311. doi:10.1093/glycob/9.12.1307.
- Cagnoni AJ, Pérez Sáez JM, Rabinovich GA, Mariño KV. 2016. Turning-off signaling by siglecs, selectins, and galectins: chemical inhibition of glycan-dependent interactions in cancer. Front Oncol. 6:109. doi:10.3389/fonc.2016.00109.
- Camby I, Le Mercier M, Lefranc F, Kiss R. 2006. Galectin-1: a small protein with major functions. Glycobiology. 16(11):137R–157R. doi:10.1093/glycob/cwl025.
- Carlin AF, Uchiyama S, Chang YC, Lewis AL, Nizet V, Varki A. 2009. Molecular mimicry of host sialylated glycans allows a bacterial pathogen to engage neutrophil Siglec-9 and dampen the innate immune response. Blood. 113(14):3333–3336. doi:10.1182/blood-2008-11-187302.
- Carr A, Rodríguez E, Arango Mdel C. 2003. Immunotherapy of advanced breast cancer with a heterophilic ganglioside (NeuGcGM3) cancer vaccine. J Clin Oncol. 21(6):1015–1021. doi:10.1200/JCO.2003.02.124.
- Carroll MV, Sim RB. 2011. Complement in health and disease. Adv Drug Deliv Rev. 63(12):965–975. doi:10.1016/j.addr.2011.06.005.
- Carter JL, Hege K, Yang J. 2020. Targeting multiple signaling pathways: the new approach to acute myeloid leukemia therapy. Signal Transduct Target Ther. 5(1):288. doi:10.1038/s41392-020-00361-x.
- Caruana I, Diaconu I, Dotti G. 2014. From monoclonal antibodies to chimeric antigen receptors for the treatment of human malignancies. Semin Oncol. 41(5):661–666. doi:10.1053/j.seminoncol.2014.08.005.
- Carvalho FC, Soares SG, Tamarozzi MB, Rego EM, Roque-Barreira MC. 2011. The recognition of N-glycans by the lectin ArtinM mediates cell death of a human myeloid leukemia cell line. PLoS One. 6(11):e27892. doi:10.1371/journal.pone.0027892.
- Cavdarli S, Dewald JH, Yamakawa N. 2019. Identification of 9-O-acetyl-N-acetylneuraminic acid (Neu5,9Ac2) as main O-acetylated sialic acid species of GD2 in breast cancer cells. Glycoconj J. 36(1):79–90. doi:10.1007/s10719-018-09856-w.
- Cavdarli S, Schröter L, Albers M. 2021. Role of sialyl-O-acetyltransferase CASD1 on GD2 ganglioside O-acetylation in breast cancer cells. Cells. 10(6):1468. doi:10.3390/cells10061468.
- Cedzyński M, Świerzko AS. 2020. Components of the lectin pathway of complement in haematologic malignancies. Cancers. 12(7):1792. doi:10.3390/cancers12071792.
- Cell Adhesion and the Extracellular Matrix. 2021. Goodman’s medical cell biology. p. 203–247. doi:10.1016/b978-0-12-817927-7.00007-7.
- Cerliani JP, Stowell SR, Mascanfroni ID, Arthur CM, Cummings RD, Rabinovich GA. 2011. Expanding the universe of cytokines and pattern recognition receptors: galectins and glycans in innate immunity. J Clin Immunol. 31(1):10–21. doi:10.1007/s10875-010-9494-2.
- Chakraborty A, Dorsett KA, Trummell HQ. 2018. ST6Gal-I sialyltransferase promotes chemoresistance in pancreatic ductal adenocarcinoma by abrogating gemcitabine-mediated DNA damage. J Biol Chem. 293(3):984–994. doi:10.1074/jbc.M117.808584.
- Chang WC, Hartshorn KL, White MR. 2011. Recombinant chimeric lectins consisting of mannose-binding lectin and L-ficolin are potent inhibitors of influenza A virus compared with mannose-binding lectin. Biochem Pharmacol. 81(3):388–395. doi:10.1016/j.bcp.2010.10.012.
- Chang YC, Nizet V. 2014. The interplay between Siglecs and sialylated pathogens. Glycobiology. 24(9):818–825. doi:10.1093/glycob/cwu067.
- Charkhchi P, Cybulski C, Gronwald J, Wong FO, Narod SA, Akbari MR. 2020. CA125 and ovarian cancer: a comprehensive review. Cancers. 12(12):3730. doi:10.3390/cancers12123730.
- Cheever MA, Allison JP, Ferris AS. 2009. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 15(17):5323–5337. doi:10.1158/1078-0432.CCR-09-0737.
- Chen CY, Jan YH, Juan YH. 2013. Fucosyltransferase 8 as a functional regulator of nonsmall cell lung cancer. Proc Natl Acad Sci USA. 110(2):630–635. doi:10.1073/pnas.1220425110.
- Chen J, Glasser CL. 2020. New and emerging targeted therapies for pediatric acute myeloid leukemia (AML). Children. 7(2):12. doi:10.3390/children7020012.
- Chen L, Zhang W, Fregien N, Pierce M. 1998. The her-2/neu oncogene stimulates the transcription of N-acetylglucosaminyltransferase V and expression of its cell surface oligosaccharide products. Oncogene. 17(16):2087–2093. doi:10.1038/sj.onc.1202124.
- Cheng CL, Hou HA, Lee MC. 2013. Higher bone marrow LGALS3 expression is an independent unfavorable prognostic factor for overall survival in patients with acute myeloid leukemia. Blood. 121(16):3172–3180. doi:10.1182/blood-2012-07-443762.
- Cheung LS-L, Raman PS, Wirtz D, Konstantopoulos K. 2012. Biophysics of selectin-mediated cell adhesion. In: Edward H. Egelman, editor. Comprehensive biophysics. Elsevier; p. 10–32. doi:10.1016/B978-0-12-374920-8.00703-7
- Cho RW, Clarke MF. 2008. Recent advances in cancer stem cells. Curr Opin Genet Dev. 18(1):48–53. doi:10.1016/j.gde.2008.01.017.
- Chowdhury S, Bandyopadhyay S, Mandal C, Chandra S, Mandal C. 2008. Flow-cytometric monitoring of disease-associated expression of 9-O-acetylated sialoglycoproteins in combination with known CD antigens, as an index for MRD in children with acute lymphoblastic leukaemia: a two-year longitudinal follow-up study. BMC Cancer. 8:40. doi:10.1186/1471-2407-8-40.
- Chowdhury S, Mandal C. 2009. O-acetylated sialic acids: multifaceted role in childhood acute lymphoblastic leukaemia. Biotechnol J. 4(3):361–374. doi:10.1002/biot.200800253.
- Christiansen D, Vaughan HA, Milland J. 2011. Antibody responses to glycolipid-borne carbohydrates require CD4+ T cells but not CD1 or NKT cells [published correction appears in Immunol Cell Biol. 2011 May;89(4):574. Miland, Julie [corrected to Milland, Julie]. Immunol Cell Biol. 89(4):502–510. doi:10.1038/icb.2010.166.
- Chua JX, Durrant L. 2017. Monoclonal antibodies against tumour-associated carbohydrate antigens, carbohydrate, Mahmut Caliskan, I. Halil Kavakli and Gul Cevahir Oz, IntechOpen, doi:10.5772/66996. https://www.intechopen.com/chapters/53645.
- Cogle CR, Bosse RC, Brewer T. 2016. Acute myeloid leukemia in the vascular niche. Cancer Lett. 380(2):552–560. doi:10.1016/j.canlet.2015.05.007.
- Cohen D. 2009. Complications: tracking down the data on oseltamivir [published correction appears in BMJ. 2010; 340:c406]. Br Med J. 339:b5387. doi:10.1136/bmj.b5387.
- Coley WB. 1893. The treatment of malignant tumors by repeated inoculations of erysipelas. Am J Med Sci. 105:487–510.
- Contessa JN, Bhojani MS, Freeze HH, Rehemtulla A, Lawrence TS. 2008. Inhibition of N-linked glycosylation disrupts receptor tyrosine kinase signaling in tumor cells. Cancer Res. 68(10):3803–3809. doi:10.1158/0008-5472.CAN-07-6389.
- Cordo V, van der Zwet J, Canté-Barrett K, Pieters R, Meijerink J. 2021. T-cell acute lymphoblastic leukemia: A roadmap to targeted therapies. Blood Cancer Discov. 2:19–31.
- Costa AF, Campos D, Reis CA, Gomes C. 2020. Targeting glycosylation: a new road for cancer drug discovery. Trends Cancer. 6(9):757–766. doi:10.1016/j.trecan.2020.04.002.
- Covés-Datson EM, Dyall J, DeWald LE. 2019. Inhibition of Ebola virus by a molecularly engineered banana lectin. PLoS Negl Trop Dis. 13(7):e0007595. doi:10.1371/journal.pntd.0007595.
- Covés-Datson EM, King SR, Legendre M. 2020. A molecularly engineered antiviral banana lectin inhibits fusion and is efficacious against influenza virus infection in vivo. Proc Natl Acad Sci USA. 117(4):2122–2132. doi:10.1073/pnas.1915152117.
- Crespo HJ, Lau JT, Videira PA. 2013. Dendritic cells: a spot on sialic acid. Front Immunol. 4:491. doi:10.3389/fimmu.2013.00491.
- Crocker PR, Paulson JC, Varki A. 2007. Siglecs and their roles in the immune system. Nat Rev Immunol. 7(4):255–266. doi:10.1038/nri2056.
- Cummins KD, Gill S. 2019. Will CAR T cell therapy have a role in AML? Promises and pitfalls. Semin Hematol. 56(2):155–163. doi:10.1053/j.seminhematol.2018.08.008.
- Curran E, Stock W. 2019. Taking a “BiTE out of ALL”: blinatumomab approval for MRD-positive ALL. Blood. 133(16):1715–1719. doi:10.1182/blood-2018-12-852376.
- D’Addio M, Frey J, Otto VI. 2020. The manifold roles of sialic acid for the biological functions of endothelial glycoproteins. Glycobiology. 30(8):490–499. doi:10.1093/glycob/cwaa008.
- Daniotti JL, Vilcaes AA, Torres Demichelis V, Ruggiero FM, Rodriguez-Walker M. 2013. Glycosylation of glycolipids in cancer: basis for development of novel therapeutic approaches. Front Oncol. 3:306. doi:10.3389/fonc.2013.00306.
- Daver N, Alotaibi AS, Bücklein V, Subklewe M. 2021. T-cell-based immunotherapy of acute myeloid leukemia: current concepts and future developments. Leukemia. 35(7):1843–1863. doi:10.1038/s41375-021-01253-x.
- Davis AP. 2009. Synthetic lectins. Org Biomol Chem. 7(18):3629–3638. doi:10.1039/b909856a.
- DeAngelo DJ. 2019. High E- selectin ligand expression contributes to chemotherapy-resistance in poor risk relapsed and refractory (R/R) acute myeloid leukemia (AML) patients and can be overcome with the addition of uproleselan. Blood. 134:2690.
- DeAngelo DJ, Erba HP, Jonas BA. 2019. A phase III trial to evaluate the efficacy of uproleselan (GMI-1271) with chemotherapy in patients with relapsed/refractory acute myeloid leukemia. J Clin Oncol. 37(Suppl. 15). doi:10.1200/JCO.2019.37.15_suppl.TPS7066
- Decker WK, da Silva RF, Sanabria MH. 2017. Cancer immunotherapy: historical perspective of a clinical revolution and emerging preclinical animal models. Front Immunol. 8:829. doi:10.3389/fimmu.2017.00829.
- Decker WK, Safdar A. 2009. Bioimmunoadjuvants for the treatment of neoplastic and infectious disease: Coley’s legacy revisited. Cytokine Growth Factor Rev. 20(4):271–281. doi:10.1016/j.cytogfr.2009.07.004.
- de Haan N, Wuhrer M, Ruhaak LR. 2020. Mass spectrometry in clinical glycomics: the path from biomarker identification to clinical implementation. Clin Mass Spectrom. 18:1–12. doi:10.1016/j.clinms.2020.08.001.
- Delaine T, Collins P, MacKinnon A. 2016. Galectin-3-binding glycomimetics that strongly reduce bleomycin-induced lung fibrosis and modulate intracellular glycan recognition. Chembiochem. 17(18):1759–1770. doi:10.1002/cbic.201600285.
- Delcommenne M, Klingemann HG. 2012. Detection and characterization of syndecan-1-associated heparan sulfate 6-O-sulfated motifs overexpressed in multiple myeloma cells using single chain antibody variable fragments. Hum Antibodies. 21(1-2):29–40. doi:10.3233/HAB-2012-0259.
- Dennis JW, Laferté S, Waghorne C, Breitman ML, Kerbel RS. 1987. Beta 1-6 branching of Asn-linked oligosaccharides is directly associated with metastasis. Science. 236(4801):582–585. doi:10.1126/science.2953071.
- de Vries JF, Zwaan CM, De Bie M. 2012. The novel calicheamicin-conjugated CD22 antibody inotuzumab ozogamicin (CMC-544) effectively kills primary pediatric acute lymphoblastic leukemia cells. Leukemia. 26(2):255–264. doi:10.1038/leu.2011.206.
- Dhar C, Sasmal A, Varki A. 2019. From “serum sickness” to “xenosialitis”: past, present, and future significance of the non-human sialic acid Neu5Gc. Front Immunol. 10:807. doi:10.3389/fimmu.2019.00807.
- Diaz SL, Padler-Karavani V, Ghaderi D. 2009. Sensitive and specific detection of the non-human sialic acid N-glycolylneuraminic acid in human tissues and biotherapeutic products. PLoS One. 4(1):e4241. doi:10.1371/journal.pone.0004241.
- Dick JE. 2008. Stem cell concepts renew cancer research. Blood. 112(13):4793–4807. doi:10.1182/blood-2008-08-077941.
- Dijoseph JF, Dougher MM, Armellino DC, Evans DY, Damle NK. 2007. Therapeutic potential of CD22-specific antibody-targeted chemotherapy using inotuzumab ozogamicin (CMC-544) for the treatment of acute lymphoblastic leukemia. Leukemia. 21(11):2240–2245. doi:10.1038/sj.leu.2404866.
- DiJoseph JF, Dougher MM, Evans DY, Zhou BB, Damle NK. 2011. Preclinical anti-tumor activity of antibody-targeted chemotherapy with CMC-544 (inotuzumab ozogamicin), a CD22-specific immunoconjugate of calicheamicin, compared with non-targeted combination chemotherapy with CVP or CHOP. Cancer Chemother Pharmacol. 67(4):741–749. doi:10.1007/s00280-010-1342-9.
- Dinner S, Lee D, Liedtke M. 2014. Current therapy and novel agents for relapsed or refractory acute lymphoblastic leukemia. Leuk Lymphoma. 55(8):1715–1724. doi:10.3109/10428194.2013.856428.
- Diorio C, Teachey DT. 2020. Harnessing immunotherapy for pediatric T-cell malignancies. Expert Rev Clin Immunol. 16(4):361–371. doi:10.1080/1744666X.2020.1732819.
- Dobie C, Skropeta D. 2021. Insights into the role of sialylation in cancer progression and metastasis. Br J Cancer. 124(1):76–90. doi:10.1038/s41416-020-01126-7.
- Dolgin E. 2020. Immunotherapy takes aim at exhausted T cells. Nat Biotechnol. 38(1):3–5. doi:10.1038/s41587-019-0381-y.
- Donati M, Brancato G, Donati A. 2010. Clinical biomarkers in hepatocellular carcinoma (HCC). Front Biosci. 2(2):571–577. doi:10.2741/s86.
- Drake RR, Schwegler EE, Malik G. 2006. Lectin capture strategies combined with mass spectrometry for the discovery of serum glycoprotein biomarkers. Mol Cell Proteom. 5(10):1957–1967. doi:10.1074/mcp.M600176-MCP200.
- Duffy MJ, Shering S, Sherry F, McDermott E, O’Higgins N. 2000. CA 15-3: a prognostic marker in breast cancer. Int J Biol Markers. 15(4):330–333.
- Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. 2002. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 3(11):991–998. doi:10.1038/ni1102-991.
- Dunn GP, Old LJ, Schreiber RD. 2004a. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 21(2):137–148. doi:10.1016/j.immuni.2004.07.017.
- Dunn GP, Old LJ, Schreiber RD. 2004b. The three Es of cancer immunoediting. Annu Rev Immunol. 22:329–360. doi:10.1146/annurev.immunol.22.012703.104803.
- Durrant LG, Noble P, Spendlove I. 2012. Immunology in the clinic review series; focus on cancer: glycolipids as targets for tumour immunotherapy. Clin Exp Immunol. 167(2):206–215. doi:10.1111/j.1365-2249.2011.04516.x.
- Ehninger A, Kramer M, Röllig C. 2014. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 4(6):e218. doi:10.1038/bcj.2014.39.
- Eisen S, Dzwonek A, Klein NJ. 2008. Mannose-binding lectin in HIV infection. Future Virol. 3(3):225–233. doi:10.2217/17460794.3.3.225.
- Ernst B, Magnani JL. 2009. From carbohydrate leads to glycomimetic drugs. Nat Rev Drug Discov. 8(8):661–677. doi:10.1038/nrd2852.
- Esposito M, Mondal N, Greco TM. 2019. Bone vascular niche E-selectin induces mesenchymal-epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat Cell Biol. 21(5):627–639. doi:10.1038/s41556-019-0309-2.
- Essers MA, Trumpp A. 2010. Targeting leukemic stem cells by breaking their dormancy. Mol Oncol. 4(5):443–450. doi:10.1016/j.molonc.2010.06.001.
- Faderl S, O’Brien S, Pui CH. 2010. Adult acute lymphoblastic leukemia: concepts and strategies. Cancer. 116(5):1165–1176. doi:10.1002/cncr.24862.
- Fahr C, Schauer R. 2001. Detection of sialic acids and gangliosides with special reference to 9-O-acetylated species in basaliomas and normal human skin. J Invest Dermatol. 116(2):254–260. doi:10.1046/j.1523-1747.2001.01237.x.
- Farhad M, Rolig AS, Redmond WL. 2018. The role of Galectin-3 in modulating tumor growth and immunosuppression within the tumor microenvironment. Oncoimmunology. 7(6):e1434467. doi:10.1080/2162402X.2018.1434467.
- Feng D, Shaikh AS, Wang F. 2016a. Recent advance in tumor-associated carbohydrate antigens (TACAs)-based antitumor vaccines. ACS Chem Biol. 11(4):850–863. doi:10.1021/acschembio.6b00084.
- Feng X, Zhao L, Gao S. 2016b. Increased fucosylation has a pivotal role in multidrug resistance of breast cancer cells through miR-224-3p targeting FUT4. Gene. 578(2):232–241. doi:10.1016/j.gene.2015.12.028.
- Ferens-Sieczkowska M, Kratz EM, Kossowska B, Passowicz-Muszyńska E, Jankowska R. 2013. Comparison of haptoglobin and alpha 1-acid glycoprotein glycosylation in the sera of small cell and non-small cell lung cancer patients. Postepy Hig Med Dosw. 67:828–836. doi:10.5604/17322693.1061788.
- Ferguson MAJ, Hart GW, Kinoshita T. 2017. Glycosylphosphatidylinositol anchors. In: Varki A, Cummings RD, Esko JD, editor. Essentials of glycobiology. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; p. 137–150.
- Fernandez-Poza S, Padros A, Thompson R. 2021. Tailor-made recombinant prokaryotic lectins for characterisation of glycoproteins. Anal Chim Acta. 1155:338352. doi:10.1016/j.aca.2021.338352.
- Ferris RL, Blumenschein G, Fayette J. 2016. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 375(19):1856–1867. doi:10.1056/NEJMoa1602252.
- Feucht J, Kayser S, Gorodezki D. 2016. T-cell responses against CD19+ pediatric acute lymphoblastic leukemia mediated by bispecific T-cell engager (BiTE) are regulated contrarily by PD-L1 and CD80/CD86 on leukemic blasts. Oncotarget. 7(47):76902–76919. doi:10.18632/oncotarget.12357.
- Finn OJ. 2018. A believer’s overview of cancer immunosurveillance and immunotherapy. J Immunol. 200(2):385–391. doi:10.4049/jimmunol.1701302.
- Fleischer LC, Spencer HT, Raikar SS. 2019. Targeting T cell malignancies using CAR-based immunotherapy: challenges and potential solutions. J Hematol Oncol. 12(1):141. doi:10.1186/s13045-019-0801-y.
- Flitsch SL. 2012. Enzymatic carbohydrate synthesis. In: Erick M. Carreira, Hisashi Yamamoto, editor. Comprehensive chirality. Elsevier; p. 454–464.
- Foster AJ, Bird JH, Timmer MSM, Stocker BL. 2015. The ligands of C-type lectins. C-type lectin receptors in immunity. 191–215. doi:10.1007/978-4-431-56015-9_13.
- François KO, Balzarini J. 2012. Potential of carbohydrate-binding agents as therapeutics against enveloped viruses. Med Res Rev. 32(2):349–387. doi:10.1002/med.20216.
- Freire T, Zhang X, Dériaud E. 2010. Glycosidic Tn-based vaccines targeting dermal dendritic cells favor germinal center B-cell development and potent antibody response in the absence of adjuvant. Blood. 116(18):3526–3536. doi:10.1182/blood-2010-04-279133.
- Fujihara S, Mori H, Kobara H. 2013. Galectin-9 in cancer therapy. Recent Pat Endocr Metab Immune Drug Discov. 7(2):130–137. doi:10.2174/1872214811307020006.
- Fujita T, Matsushita M, Endo Y. 2004. The lectin-complement pathway – its role in innate immunity and evolution. Immunol Rev. 198:185–202. doi:10.1111/j.0105-2896.2004.0123.x.
- Fuster MM, Esko JD. 2005. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer. 5(7):526–542. doi:10.1038/nrc1649.
- Gabius HJ. 2013. The history of lectinology. In: Fang E., Ng T, editor. Antitumor potential and other emerging medicinal properties of natural compounds. Dordrecht: Springer. doi:10.1007/978-94-007-6214-5_2
- Gao L, Yu S, Zhang X. 2014. Hypothesis: Tim-3/galectin-9, a new pathway for leukemia stem cells survival by promoting expansion of myeloid-derived suppressor cells and differentiating into tumor-associated macrophages. Cell Biochem Biophys. 70(1):273–277. doi:10.1007/s12013-014-9900-0.
- Gardner A, Ruffell B. 2016. Dendritic cells and cancer immunity. Trends Immunol. 37(12):855–865. doi:10.1016/j.it.2016.09.006.
- Geis N, Zell S, Rutz R. 2010. Inhibition of membrane complement inhibitor expression (CD46, CD55, CD59) by siRNA sensitizes tumor cells to complement attack in vitro. Curr Cancer Drug Targets. 10(8):922–931. doi:10.2174/156800910793357952.
- Geller A, Yan J. 2019. The role of membrane bound complement regulatory proteins in tumor development and cancer immunotherapy. Front Immunol. 10:1074. doi:10.3389/fimmu.2019.01074.
- Gessner P, Riedl S, Quentmaier A, Kemmner W. 1993. Enhanced activity of CMP-neuAc:Gal beta 1-4GlcNAc:alpha 2,6-sialyltransferase in metastasizing human colorectal tumor tissue and serum of tumor patients. Cancer Lett. 75(3):143–149. doi:10.1016/0304-3835(93)90056-f.
- Ghafoor D, Ahmed S, Zaman N. 2021. Lectins A hope of treatment for COVID-19. Am J Biomed Sci Res. doi:10.34297/AJBSR.2021.12.001757.
- Ghazarian H, Idoni B, Oppenheimer SB. 2011. A glycobiology review: carbohydrates, lectins and implications in cancer therapeutics. Acta Histochem. 113(3):236–247. doi:10.1016/j.acthis.2010.02.004.
- Ghosh S. 2020. Sialic acid and biology of life: an introduction. Sialic acids and sialoglycoconjugates in the biology of life, health and disease. p. 1–61. doi:10.1016/B978-0-12-816126-5.00001-9.
- Gianchecchi E, Delfino DV, Fierabracci A. 2013. Recent insights into the role of the PD-1/PD-L1 pathway in immunological tolerance and autoimmunity. Autoimmun Rev. 12(11):1091–1100. doi:10.1016/j.autrev.2013.05.003.
- Gibson HM, Mishra A, Chan DV, Hake TS, Porcu P, Wong HK. 2013. Impaired proteasome function activates GATA3 in T cells and upregulates CTLA-4: relevance for Sézary syndrome. J Invest Dermatol. 133(1):249–257. doi:10.1038/jid.2012.265.
- Gill S, Tasian SK, Ruella M. 2014. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells [published correction appears in blood. 2016 Nov 24;128(21):2585]. Blood. 123(15):2343–2354. doi:10.1182/blood-2013-09-529537.
- Gion M, Mione R, Leon AE. 2001. CA27.29: a valuable marker for breast cancer management. A confirmatory multicentric study on 603 cases. Eur J Cancer. 37(3):355–363. doi:10.1016/s0959-8049(00)00396-8.
- Glinsky VV, Glinsky GV, Rittenhouse-Olson K. 2001. The role of Thomsen-Friedenreich antigen in adhesion of human breast and prostate cancer cells to the endothelium. Cancer Res. 61(12):4851–4857.
- Golay J, Zaffaroni L, Vaccari T. 2000. Biologic response of B lymphoma cells to anti-CD20 monoclonal antibody rituximab in vitro: CD55 and CD59 regulate complement-mediated cell lysis. Blood. 95(12):3900–3908.
- Gomes C, Osório H, Pinto MT, Campos D, Oliveira MJ, Reis CA. 2013. Expression of ST3GAL4 leads to SLe(x) expression and induces c-Met activation and an invasive phenotype in gastric carcinoma cells. PLoS One. 8(6):e66737. doi:10.1371/journal.pone.0066737.
- Gonzalez-Gil A, Schnaar RL. 2021. Siglec ligands. Cells. 10(5):1260. doi:10.3390/cells10051260.
- Granovsky M, Fata J, Pawling J, Muller WJ, Khokha R, Dennis JW. 2000. Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat Med. 6(3):306–312. doi:10.1038/73163.
- Griffin ME, Hsieh-Wilson LC. 2016. Glycan engineering for cell and developmental biology. Cell Chem Biol. 23(1):108–121. doi:10.1016/j.chembiol.2015.12.007.
- Gupta YK, Meenu M, Mohan P. 2015. The Tamiflu fiasco and lessons learnt. Indian J Pharmacol. 47(1):11–16. doi:10.4103/0253-7613.150308.
- Hakomori S. 1989. Aberrant glycosylation in tumors and tumor-associated carbohydrate antigens. Adv Cancer Res. 52:257–331. doi:10.1016/s0065-230x(08)60215-8.
- Hakomori S. 2001. Tumor-associated carbohydrate antigens defining tumor malignancy: basis for development of anti-cancer vaccines. Adv Exp Med Biol. 491:369–402. doi:10.1007/978-1-4615-1267-7_24.
- Hakomori S. 2002. Glycosylation defining cancer malignancy: new wine in an old bottle. Proc Natl Acad Sci USA. 99(16):10231–10233. doi:10.1073/pnas.172380699.
- Haltalli MLR, Lo Celso C. 2020. Targeting adhesion to the vascular niche to improve therapy for acute myeloid leukemia. Nat Commun. 11(1):3691. doi:10.1038/s41467-020-17594-7.
- Hamilton WB, Helling F, Lloyd KO, Livingston PO. 1993. Ganglioside expression on human malignant melanoma assessed by quantitative immune thin-layer chromatography. Int J Cancer. 53(4):566–573. doi:10.1002/ijc.2910530407.
- Hanash SM, Pitteri SJ, Faca VM. 2008. Mining the plasma proteome for cancer biomarkers. Nature. 452(7187):571–579. doi:10.1038/nature06916.
- Hao C, Tian J, Liu H, Li F, Niu H, Zhu B. 2017. Efficacy and safety of anti-PD-1 and anti-PD-1 combined with anti-CTLA-4 immunotherapy to advanced melanoma: a systematic review and meta-analysis of randomized controlled trials. Medicine. 96(26):e7325. doi:10.1097/MD.0000000000007325.
- Haubner S, Perna F, Köhnke T. 2019. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia. 33(1):64–74. doi:10.1038/s41375-018-0180-3.
- He J, Liu Y, Xie X. 2010. Identification of cell surface glycoprotein markers for glioblastoma-derived stem-like cells using a lectin microarray and LC-MS/MS approach. J Proteome Res. 9(5):2565–2572. doi:10.1021/pr100012p.
- Heijs B, Holst-Bernal S, de Graaff MA. 2020. Molecular signatures of tumor progression in myxoid liposarcoma identified by N-glycan mass spectrometry imaging. Lab Invest. 100(9):1252–1261. doi:10.1038/s41374-020-0435-2.
- Heise T, Langereis JD, Rossing E. 2018. Selective inhibition of sialic acid-based molecular mimicry in haemophilus influenzae abrogates serum resistance. Cell Chem Biol. 25(10):1279–1285. doi:10.1016/j.chembiol.2018.05.018.
- Hellicar J, Stevenson NL, Stephens DJ, Lowe M. 2022. Supply chain logistics – the role of the Golgi complex in extracellular matrix production and maintenance. J Cell Sci. 135(1):jcs258879. doi:10.1242/jcs.258879.
- Hendriks D, Choi G, de Bruyn M, Wiersma VR, Bremer E. 2017. Antibody-based cancer therapy: successful agents and novel approaches. Int Rev Cell Mol Biol. 331:289–383. doi:10.1016/bs.ircmb.2016.10.002.
- Hevey R. 2019. Strategies for the development of glycomimetic drug candidates. Pharmaceuticals. 12(2):55. doi:10.3390/ph12020055.
- Higashi H, Hirabayashi Y, Fukui Y. 1985. Characterization of N-glycolylneuraminic acid-containing gangliosides as tumor-associated Hanganutziu-Deicher antigen in human colon cancer. Cancer Res. 45(8):3796–3802.
- Hira VVV, Van Noorden CJF, Carraway HE, Maciejewski JP, Molenaar RJ. 2017. Novel therapeutic strategies to target leukemic cells that hijack compartmentalized continuous hematopoietic stem cell niches. Biochim Biophys Acta Rev Cancer. 1868(1):183–198. doi:10.1016/j.bbcan.2017.03.010.
- Hirabayashi J, Arai R. 2019. Lectin engineering: the possible and the actual. Interface Focus. 9(2):20180068. doi:10.1098/rsfs.2018.0068.
- Hiraiwa N, Yabuta T, Yoritomi K. 2003. Transactivation of the fucosyltransferase VII gene by human T-cell leukemia virus type 1 tax through a variant cAMP-responsive element. Blood. 101(9):3615–3621. doi:10.1182/blood-2002-07-2301.
- Hitchcock CL, Povoski SP, Mojzisik CM, Martin EW. 2021. Survival advantage following TAG-72 antigen-directed cancer surgery in patients with colorectal carcinoma: proposed mechanisms of action. Front Oncol. 11:731350. doi:10.3389/fonc.2021.731350.
- Ho TC, LaMere M, Stevens BM. 2016. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood. 128(13):1671–1678. doi:10.1182/blood-2016-02-695312.
- Hoffman AE, Schoonmade LJ, Kaspers GJ. 2021. Pediatric relapsed acute myeloid leukemia: a systematic review. Expert Rev Anticancer Ther. 21(1):45–52. doi:10.1080/14737140.2021.1841640.
- Hou S, Hang Q, Isaji T, Lu J, Fukuda T, Gu J. 2016. Importance of membrane-proximal N-glycosylation on integrin β1 in its activation and complex formation. FASEB J. 30(12):4120–4131. doi:10.1096/fj.201600665R.
- Houvast RD, Vankemmelbeke M, Durrant LG. 2020. Targeting glycans and heavily glycosylated proteins for tumor imaging. Cancers. 12(12):3870. doi:10.3390/cancers12123870.
- Hu D, Tateno H, Hirabayashi J. 2014. Directed evolution of lectins by an improved error-prone PCR and ribosome display method. Methods Mol Biol. 1200:527–538. doi:10.1007/978-1-4939-1292-6_43.
- Hu D, Tateno H, Hirabayashi J. 2015. Lectin engineering, a molecular evolutionary approach to expanding the lectin utilities. Molecules. 20(5):7637–7656. doi:10.3390/molecules20057637.
- Huang S, Day TW, Choi MR, Safa AR. 2009. Human beta-galactoside alpha-2,3-sialyltransferase (ST3Gal III) attenuated Taxol-induced apoptosis in ovarian cancer cells by downregulating caspase-8 activity. Mol Cell Biochem. 331(1-2):81–88. doi:10.1007/s11010-009-0147-9.
- Hudak JE, Canham SM, Bertozzi CR. 2014. Glycocalyx engineering reveals a Siglec-based mechanism for NK cell immunoevasion. Nat Chem Biol. 10(1):69–75. doi:10.1038/nchembio.1388.
- Hussein NH, Amin NS, El Tayebi HM. 2020. GPI-AP: unraveling a new class of malignancy mediators and potential immunotherapy targets. Front Oncol. 10:537311. doi:10.3389/fonc.2020.537311.
- Ibarlucea-Benitez I, Weitzenfeld P, Smith P, Ravetch JV. 2021. Siglecs-7/9 function as inhibitory immune checkpoints in vivo and can be targeted to enhance therapeutic antitumor immunity. Proc Natl Acad Sci USA. 118(26):e2107424118. doi:10.1073/pnas.2107424118.
- Ishii K, Pouzolles M, Chien CD. 2020. Perforin-deficient CAR T cells recapitulate late-onset inflammatory toxicities observed in patients. J Clin Invest. 130(10):5425–5443. doi:10.1172/JCI130059.
- Ishikawa F, Yoshida S, Saito Y. 2007. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 25(11):1315–1321. doi:10.1038/nbt1350.
- Ito K, Ralph SJ. 2012. Inhibiting galectin-1 reduces murine lung metastasis with increased CD4(+) and CD8 (+) T cells and reduced cancer cell adherence. Clin Exp Metastasis. 29(6):561–572. doi:10.1007/s10585-012-9471-7.
- Itoh M, Kitano T, Watanabe M. 2003. Possible role of ceramide as an indicator of chemoresistance: decrease of the ceramide content via activation of glucosylceramide synthase and sphingomyelin synthase in chemoresistant leukemia. Clin Cancer Res. 9(1):415–423.
- Jacob F, Goldstein DR, Bovin NV. 2012. Serum antiglycan antibody detection of nonmucinous ovarian cancers by using a printed glycan array. Int J Cancer. 130(1):138–146. doi:10.1002/ijc.26002.
- Jacobson S, Larsson P, Åberg AM, Johansson G, Winsö O, Söderberg S. 2020. Levels of mannose-binding lectin (MBL) associates with sepsis-related in-hospital mortality in women. J Inflamm. 17:28. doi:10.1186/s12950-020-00257-1.
- Jacoby E. 2019. Relapse and resistance to CAR-T cells and blinatumomab in hematologic malignancies. Clin Hematol Int. 1:79–84.
- Jacoby E, Nguyen SM, Fountaine TJ. 2016. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat Commun. 7:12320. doi:10.1038/ncomms12320.
- Jain S, Stroopinsky D, Yin L. 2015. Mucin 1 is a potential therapeutic target in cutaneous T-cell lymphoma. Blood. 126(3):354–362. doi:10.1182/blood-2015-02-628149.
- Jain T, Litzow MR. 2018. No free rides: management of toxicities of novel immunotherapies in ALL, including financial. Hematol Am Soc Hematol Educ Program. 2018(1):25–34. doi:10.1182/asheducation-2018.1.25.
- Jain T, Litzow MR. 2020. Management of toxicities associated with novel immunotherapy agents in acute lymphoblastic leukemia. Ther Adv Hematol. 11. doi:10.1177/2040620719899897
- Jandus C, Boligan KF, Chijioke O. 2014. Interactions between Siglec-7/9 receptors and ligands influence NK cell-dependent tumor immunosurveillance. J Clin Invest. 124(4):1810–1820. doi:10.1172/JCI65899.
- Jefferson T, Jones M, Doshi P, Spencer EA, Onakpoya I, Heneghan CJ. 2014b. Oseltamivir for influenza in adults and children: systematic review of clinical study reports and summary of regulatory comments. Br Med J. 348:g2545. doi:10.1136/bmj.g2545.
- Jefferson T, Jones MA, Doshi P. 2014a. Neuraminidase inhibitors for preventing and treating influenza in healthy adults and children. Cochrane Database Syst Rev. 2014(4):CD008965. doi:10.1002/14651858.CD008965.pub4.
- Jemal A, Siegel R, Ward E. 2006. Cancer statistics, 2006. CA Cancer J Clin. 56(2):106–130. doi:10.3322/canjclin.56.2.106.
- Jensenius JC, Jensen PH, McGuire K, Larsen JL, Thiel S. 2003. Recombinant mannan-binding lectin (MBL) for therapy. Biochem Soc Trans. 31(4):763–767. doi:10.1042/bst0310763.
- Jiang W, He Y, He W. 2021. Exhausted CD8+ T cells in the tumor immune microenvironment: new pathways to therapy. Front Immunol. 11:622509. doi:10.3389/fimmu.2020.622509.
- Joo EJ, Wasik BR, Parrish C. 2018. Pre-B acute lymphoblastic leukemia expresses cell surface nucleolin as a 9-O-acetylated sialoglycoprotein. Sci Rep. 8(1):17174. doi:10.1038/s41598-018-33873-2.
- Julien S, Bobowski M, Steenackers A, Le Bourhis X, Delannoy P. 2013. How do gangliosides regulate RTKs signaling? Cells. 2(4):751–767. doi:10.3390/cells2040751.
- Julien S, Videira PA, Delannoy P. 2012. Sialyl-tn in cancer: (how) did we miss the target? Biomolecules. 2(4):435–466. doi:10.3390/biom2040435.
- Jurianz K, Maslak S, Garcia-Schüler H, Fishelson Z, Kirschfink M. 1999. Neutralization of complement regulatory proteins augments lysis of breast carcinoma cells targeted with rhumAb anti-HER2. Immunopharmacology. 42(1-3):209–218. doi:10.1016/s0162-3109(99)00006-5.
- Juszczynski P, Rodig SJ, Ouyang J. 2010. MLL-rearranged B lymphoblastic leukemias selectively express the immunoregulatory carbohydrate-binding protein galectin-1. Clin Cancer Res. 16(7):2122–2130. doi:10.1158/1078-0432.CCR-09-2765.
- Kailemia MJ, Park D, Lebrilla CB. 2017. Glycans and glycoproteins as specific biomarkers for cancer. Anal Bioanal Chem. 409(2):395–410. doi:10.1007/s00216-016-9880-6.
- Kaji H, Saito H, Yamauchi Y. 2003. Lectin affinity capture, isotope-coded tagging and mass spectrometry to identify N-linked glycoproteins. Nat Biotechnol. 21(6):667–672. doi:10.1038/nbt829.
- Kandeel EZ, El Sharkawy N, Hanafi M, Samra M, Kamel A. 2020. Tracing leukemia stem cells and their influence on clinical course of adult acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 20(6):383–393. doi:10.1016/j.clml.2019.11.018.
- Kannagi R, Izawa M, Koike T, Miyazaki K, Kimura N. 2004. Carbohydrate-mediated cell adhesion in cancer metastasis and angiogenesis. Cancer Sci. 95(5):377–384. doi:10.1111/j.1349-7006.2004.tb03219.x.
- Kantarjian HM, Stock W, Cassaday RD. 2021. Inotuzumab ozogamicin for relapsed/refractory acute lymphoblastic leukemia in the INO-VATE trial: CD22 pharmacodynamics, efficacy, and safety by baseline CD22. Clin Cancer Res. 27(10):2742–2754. doi:10.1158/1078-0432.CCR-20-2399.
- Kaszuba K, Grzybek M, Orłowski A. 2015. N-Glycosylation as determinant of epidermal growth factor receptor conformation in membranes. Proc Natl Acad Sci USA. 112(14):4334–4339. doi:10.1073/pnas.1503262112.
- Kawai T, Kato A, Higashi H, Kato S, Naiki M. 1991. Quantitative determination of N-glycolylneuraminic acid expression in human cancerous tissues and avian lymphoma cell lines as a tumor-associated sialic acid by gas chromatography-mass spectrometry. Cancer Res. 51(4):1242–1246.
- Kennedy LB, Salama AKS. 2020. A review of cancer immunotherapy toxicity. CA Cancer J Clin. 70(2):86–104. doi:10.3322/caac.21596.
- Keogh D, Thompson R, Larragy R. 2014. Generating novel recombinant prokaryotic lectins with altered carbohydrate binding properties through mutagenesis of the PA-IL protein from Pseudomonas aeruginosa. Biochim Biophys Acta. 1840(6):2091–2104. doi:10.1016/j.bbagen.2014.01.020.
- Keyel ME, Reynolds CP. 2018. Spotlight on dinutuximab in the treatment of high-risk neuroblastoma: development and place in therapy. Biologics. 13:1–12. doi:10.2147/BTT.S114530.
- Khan N, Hills RK, Virgo P. 2017. Expression of CD33 is a predictive factor for effect of gemtuzumab ozogamicin at different doses in adult acute myeloid leukaemia. Leukemia. 31(5):1059–1068. doi:10.1038/leu.2016.309.
- Khosrowabadi E, Wenta T, Keskitalo S, Manninen A, Kellokumpu S. 2022. Altered glycosylation of several metastasis-associated glycoproteins with terminal GalNAc defines the highly invasive cancer cell phenotype. Oncotarget. 13:73–89. doi:10.18632/oncotarget.28167.
- Kieber-Emmons T, Monzavi-Karbassi B, Hutchins LF, Pennisi A, Makhoul I. 2017. Harnessing benefit from targeting tumor associated carbohydrate antigens. Hum Vaccin Immunother. 13(2):323–331. doi:10.1080/21645515.2017.1264789.
- Kikushige Y, Shima T, Takayanagi S. 2010. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 7(6):708–717. doi:10.1016/j.stem.2010.11.014.
- Kim MY, Yu KR, Kenderian SS. 2018. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell. 173(6):1439–1453. doi:10.1016/j.cell.2018.05.013.
- Kim N, Lee DH, Choi WS, Yi E, Kim H, Kim JM, Jin HS, Kim HS. 2021. Harnessing NK cells for cancer immunotherapy: immune checkpoint receptors and chimeric antigen receptors. BMB Rep. 54:44–58. doi:10.5483/BMBRep.2021.54.1.214.
- Kim YJ, Varki A. 1997. Perspectives on the significance of altered glycosylation of glycoproteins in cancer. Glycoconj J. 14(5):569–576. doi:10.1023/a:1018580324971.
- Kinoshita T. 2020. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biol. 10(3):190290. doi:10.1098/rsob.190290.
- Knaus HA, Berglund S, Hackl H. 2018. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI Insight. 3(21):e120974. doi:10.1172/jci.insight.120974.
- Kniep B, Monner DA, Burrichter H, Diehl V, Mühlradt PF. 1983. Gangliotriaosylceramide (asialo GM2), a glycosphingolipid marker for cell lines derived from patients with Hodgkin’s disease. J Immunol. 131(3):1591–1594.
- Koda T, Aosasa M, Asaoka H, Nakaba H, Matsuda H. 2003. Application of tyramide signal amplification for detection of N-glycolylneuraminic acid in human hepatocellular carcinoma. Int J Clin Oncol. 8(5):317–321. doi:10.1007/s10147-003-0346-4.
- Kohla G, Stockfleth E, Schauer R. 2002. Gangliosides with O-acetylated sialic acids in tumors of neuroectodermal origin. Neurochem Res. 27(7-8):583–592. doi:10.1023/a:1020211714104.
- Köhnke T, Krupka C, Tischer J, Knösel T, Subklewe M. 2015. Increase of PD-L1 expressing B-precursor ALL cells in a patient resistant to the CD19/CD3-bispecific T cell engager antibody blinatumomab. J Hematol Oncol. 8:111. doi:10.1186/s13045-015-0213-6.
- Końska G, Wójtowicz U, Pituch-Noworolska A. 2008. Mozliwości zastosowania lektyn w diagnostyce i terapii. Cz.I. Zastosowanie diagnostyczne [Possible application of lectins in diagnostics and therapy. Part I. Diagnostic application]. Przegl Lek. 65(4):189–194.
- Kooner AS, Yu H, Chen X. 2019. Synthesis of N-glycolylneuraminic acid (Neu5Gc) and its glycosides. Front Immunol. 10:2004. doi:10.3389/fimmu.2019.02004.
- Koppenol WH, Bounds PL, Dang CV. 2011. Otto Warburg’s contributions to current concepts of cancer metabolism [published correction appears in Nat Rev Cancer. 2011 Aug;11(8):618]. Nat Rev Cancer. 11(5):325–337. doi:10.1038/nrc3038.
- Kovbasnjuk O, Mourtazina R, Baibakov B. 2005. The glycosphingolipid globotriaosylceramide in the metastatic transformation of colon cancer. Proc Natl Acad Sci USA. 102(52):19087–19092. doi:10.1073/pnas.0506474102.
- Kubik S. 2009. Synthetic lectins. Angew Chem Int Ed Engl. 48(10):1722–1725. doi:10.1002/anie.200805497.
- Kudelka MR, Ju T, Heimburg-Molinaro J, Cummings RD. 2015. Simple sugars to complex disease–mucin-type O-glycans in cancer. Adv Cancer Res. 126:53–135. doi:10.1016/bs.acr.2014.11.002.
- Kudo D, Rayman P, Horton C. 2003. Gangliosides expressed by the renal cell carcinoma cell line SK-RC-45 are involved in tumor-induced apoptosis of T cells. Cancer Res. 63(7):1676–1683.
- Kudo T, Ikehara Y, Togayachi A. 1998. Up-regulation of a set of glycosyltransferase genes in human colorectal cancer. Lab Invest. 78(7):797–811.
- Kulasingam V, Diamandis EP. 2008. Strategies for discovering novel cancer biomarkers through utilization of emerging technologies. Nat Clin Pract Oncol. 5(10):588–599. doi:10.1038/ncponc1187.
- Kumamoto K, Goto Y, Sekikawa K. 2001. Increased expression of UDP-galactose transporter messenger RNA in human colon cancer tissues and its implication in synthesis of Thomsen-Friedenreich antigen and sialyl Lewis A/X determinants. Cancer Res. 61(11):4620–4627.
- Kumar V, McNerney ME. 2005. A new self: MHC-class-I-independent natural-killer-cell self-tolerance. Nat Rev Immunol. 5(5):363–374. doi:10.1038/nri1603.
- Kuzmanov U, Kosanam H, Diamandis EP. 2013. The sweet and sour of serological glycoprotein tumor biomarker quantification. BMC Med. 11:31. doi:10.1186/1741-7015-11-31.
- Laborda E, Mazagova M, Shao S. 2017. Development of a chimeric antigen receptor targeting C-type lectin-like molecule-1 for human acute myeloid leukemia. Int J Mol Sci. 18(11):2259. doi:10.3390/ijms18112259.
- LaCasse EC, Bray MR, Patterson B. 1999. Shiga-like toxin-1 receptor on human breast cancer, lymphoma, and myeloma and absence from CD34(+) hematopoietic stem cells: implications for ex vivo tumor purging and autologous stem cell transplantation. Blood. 94(8):2901–2910.
- Ladenstein R, Pötschger U, Valteau-Couanet D. 2018. Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): a multicentre, randomised, phase 3 trial. Lancet Oncol. 19(12):1617–1629. doi:10.1016/S1470-2045(18)30578-3.
- Lamble AJ, Tasian SK. 2019. Opportunities for immunotherapy in childhood acute myeloid leukemia. Blood Adv. 3(22):3750–3758. doi:10.1182/bloodadvances.2019000357.
- Lan Y, Hao C, Zeng X. 2016. Serum glycoprotein-derived N- and O-linked glycans as cancer biomarkers. Am J Cancer Res. 6(11):2390–2415.
- Lanza F, Maffini E, Rondoni M, Massari E, Faini AC, Malavasi F. 2020. CD22 expression in B-cell acute lymphoblastic leukemia: biological significance and implications for inotuzumab therapy in adults. Cancers. 12(2):303. doi:10.3390/cancers12020303.
- Lau KS, Partridge EA, Grigorian A. 2007. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell. 129(1):123–134. doi:10.1016/j.cell.2007.01.049.
- Läubli H, Borsig L. 2019. Altered cell adhesion and glycosylation promote cancer immune suppression and metastasis. Front Immunol. 10:2120. doi:10.3389/fimmu.2019.02120.
- Lebrilla CB, An HJ. 2009. The prospects of glycan biomarkers for the diagnosis of diseases. Mol Biosyst. 5(1):17–20. doi:10.1039/b811781k.
- Lee DH, Choi S, Park Y, Jin HS. 2021. Mucin1 and Mucin16: therapeutic targets for cancer therapy. Pharmaceuticals. 14(10):1053. doi:10.3390/ph14101053.
- Lee J, Ha S, Kim M. 2020. Spatial and temporal diversity of glycome expression in mammalian brain. Proc Natl Acad Sci USA. 117(46):28743–28753. doi:10.1073/pnas.2014207117.
- Leonard J, Stock W. 2017. Progress in adult ALL: incorporation of new agents to frontline treatment. Hematol Am Soc Hematol Educ Program. 2017(1):28–36. doi:10.1182/asheducation-2017.1.28.
- Leonti AR, Pardo L, Alonzo TA, Gerbing RB, Eidenschink Brodersen L, Ries RE, Smith JL, Le Q, Aplenc R, Kolb EA. 2019. Transcriptome profiling of glycosylation genes defines correlation with e-selectin ligand expression and clinical outcome in AML. Blood. 134:3772.
- Levesque JP, Winkler IG, Rasko JE. 2013. Nichotherapy for stem cells: there goes the neighborhood. Bioessays. 35(3):183–190. doi:10.1002/bies.201200111.
- Leviatan Ben-Arye S, Yu H, Chen X, Padler-Karavani V. 2017. Profiling anti-Neu5Gc IgG in human sera with a sialoglycan microarray assay. J Vis Exp. 125:56094. doi:10.3791/56094.
- Levin S, Russell EC, Blanchard D, McWilliams NB, Maurer HM, Mohanakumar T. 1980. Receptors for peanut agglutinin (Arachus hypogea) in childhood acute lymphoblastic leukemia: possible clinical significance. Blood. 55(1):37–39.
- Ley K. 2003. The role of selectins in inflammation and disease. Trends Mol Med. 9(6):263–268. doi:10.1016/s1471-4914(03)00071-6.
- Li CW, Lim SO, Chung EM. 2018a. Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell. 33(2):187–201. doi:10.1016/j.ccell.2018.01.009.
- Li CW, Lim SO, Xia W. 2016. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 7:12632. doi:10.1038/ncomms12632.
- Li D, Mallory T, Satomura S. 2001. AFP-L3: a new generation of tumor marker for hepatocellular carcinoma. Clin Chim Acta. 313(1-2):15–19. doi:10.1016/s0009-8981(01)00644-1.
- Li D, Sun H, Bai G. 2018b. α-1,3-Fucosyltransferase-VII siRNA inhibits the expression of SLex and hepatocarcinoma cell proliferation. Int J Mol Med. 42(5):2700–2708. doi:10.3892/ijmm.2018.3850.
- Liang YJ, Kuo HH, Lin CH. 2010. Switching of the core structures of glycosphingolipids from globo- and lacto- to ganglio-series upon human embryonic stem cell differentiation. Proc Natl Acad Sci USA. 107(52):22564–22569. doi:10.1073/pnas.1007290108.
- Liberti MV, Locasale JW. 2016. The Warburg effect: how does it benefit cancer cells? [published correction appears in Trends Biochem Sci. 2016 Mar;41(3):287] [published correction appears in Trends Biochem Sci. 2016 Mar;41(3):287]. Trends Biochem Sci. 41(3):211–218. doi:10.1016/j.tibs.2015.12.001.
- Lichtenstein DA, Schischlik F, Shao L. 2021. Characterization of HLH-like manifestations as a CRS variant in patients receiving CD22 CAR T cells. Blood. 138(24):2469–2484. doi:10.1182/blood.2021011898.
- Lih Y-H, Wu C-Y. 2017. Chemical synthesis of sialosides. In: C.S. Bennett, editor. Selective glycosylations: synthetic methods and catalysts. doi:10.1002/9783527696239.ch16
- Lim ABM, Curley C, Fong CY. 2018. Acute myeloid leukaemia relapsing after allogeneic haemopoietic stem cell transplantation: prognostic factors and impact of initial therapy of relapse. Intern Med J. 48(3):276–285. doi:10.1111/imj.13522.
- Lim J, Sari-Ak D, Bagga T. 2021. Siglecs as therapeutic targets in cancer. Biology. 10(11):1178. doi:10.3390/biology10111178.
- Linenberger ML, Hong T, Flowers D. 2001. Multidrug-resistance phenotype and clinical responses to gemtuzumab ozogamicin. Blood. 98(4):988–994. doi:10.1182/blood.v98.4.988.
- Lingwood CA. 2011. Glycosphingolipid functions [published correction appears in Cold Spring Harb Perspect Biol. 2011 Sep;3(9):a011874]. Cold Spring Harb Perspect Biol. 3(7):a004788. doi:10.1101/cshperspect.a004788.
- Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. 1994. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors [published correction appears in immunity 1995 Feb;2(2):following 203]. Immunity. 1(9):793–801. doi:10.1016/s1074-7613(94)80021-9.
- Liu B, Ma H, Liu Q. 2019b. MiR-29b/Sp1/FUT4 axis modulates the malignancy of leukemia stem cells by regulating fucosylation via Wnt/β-catenin pathway in acute myeloid leukemia. J Exp Clin Cancer Res. 38(1):200. doi:10.1186/s13046-019-1179-y.
- Liu C, Li Z, Wang S. 2019a. FUT4 is involved in PD-1-related immunosuppression and leads to worse survival in patients with operable lung adenocarcinoma. J Cancer Res Clin Oncol. 145(1):65–76. doi:10.1007/s00432-018-2761-y.
- Liu YS, Fujita M. 2020. Mammalian GPI-anchor modifications and the enzymes involved. Biochem Soc Trans. 48(3):1129–1138. doi:10.1042/BST20191142.
- Llop E, Ferrer-Batallé M, Barrabés S. 2016. Improvement of prostate cancer diagnosis by detecting PSA glycosylation-specific changes [published correction appears in theranostics. 2018 Jan 1;8(3):746-748]. Theranostics. 6(8):1190–1204. doi:10.7150/thno.15226.
- Locasale JW, Cantley LC. 2011. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 14(4):443–451. doi:10.1016/j.cmet.2011.07.014.
- Locatelli F, Zugmaier G, Rizzari C. 2021. Effect of blinatumomab vs chemotherapy on event-free survival among children with high-risk first-relapse B-cell acute lymphoblastic leukemia: a randomized clinical trial. JAMA. 325(9):843–854. doi:10.1001/jama.2021.0987.
- Löffler A, Kufer P, Lutterbüse R. 2000. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood. 95(6):2098–2103.
- Long AH, Haso WM, Shern JF. 2015. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 21(6):581–590. doi:10.1038/nm.3838.
- Lou YW, Wang PY, Yeh SC. 2014. Stage-specific embryonic antigen-4 as a potential therapeutic target in glioblastoma multiforme and other cancers. Proc Natl Acad Sci USA. 111(7):2482–2487. doi:10.1073/pnas.1400283111.
- Lu H, Zhou Q, Deshmukh V. 2014. Targeting human C-type lectin-like molecule-1 (CLL1) with a bispecific antibody for immunotherapy of acute myeloid leukemia. Angew Chem Int Ed Engl. 53(37):9841–9845. doi:10.1002/anie.201405353.
- Lucci A, Cho WI, Han TY, Giuliano AE, Morton DL, Cabot MC. 1998. Glucosylceramide: a marker for multiple-drug resistant cancers. Anticancer Res. 18(1B):475–480.
- Ma H, Zhou H, Song X, Shi S, Zhang J, Jia L. 2015. Modification of sialylation is associated with multidrug resistance in human acute myeloid leukemia. Oncogene. 34(6):726–740. doi:10.1038/onc.2014.7.
- Ma Z, Vosseller K. 2014. Cancer metabolism and elevated O-GlcNAc in oncogenic signaling. J Biol Chem. 289(50):34457–34465. doi:10.1074/jbc.R114.577718.
- Maak M, Nitsche U, Keller L. 2011. Tumor-specific targeting of pancreatic cancer with Shiga toxin B-subunit. Mol Cancer Ther. 10(10):1918–1928. doi:10.1158/1535-7163.MCT-11-0006.
- Macauley MS, Crocker PR, Paulson JC. 2014. Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol. 14(10):653–666. doi:10.1038/nri3737.
- Magalhães A, Duarte HO, Reis CA. 2017. Aberrant glycosylation in cancer: a novel molecular mechanism controlling metastasis. Cancer Cell. 31(6):733–735. doi:10.1016/j.ccell.2017.05.012.
- Mahalingam A, Geonnotti AR, Balzarini J, Kiser PF. 2011. Activity and safety of synthetic lectins based on benzoboroxole-functionalized polymers for inhibition of HIV entry. Mol Pharm. 8(6):2465–2475. doi:10.1021/mp2002957.
- Makhoul I, Hutchins L, Emanuel PD. 2015. Moving a carbohydrate mimetic peptide into the clinic. Hum Vaccine Immunother. 11(1):37–44. doi:10.4161/hv.34300.
- Mamidi S, Höne S, Teufel C, Sellner L, Zenz T, Kirschfink M. 2015. Neutralization of membrane complement regulators improves complement-dependent effector functions of therapeutic anticancer antibodies targeting leukemic cells. Oncoimmunology. 4(3):e979688. doi:10.4161/2162402X.2014.979688.
- Mandal C, Schwartz-Albiez R, Vlasak R. 2015. Functions and biosynthesis of O-acetylated sialic acids. Top Curr Chem. 366:1–30. doi:10.1007/128_2011_310.
- Mandal C, Srinivasan GV, Chowdhury S. 2009. High level of sialate-O-acetyltransferase activity in lymphoblasts of childhood acute lymphoblastic leukaemia (ALL): enzyme characterization and correlation with disease status. Glycoconj J. 26(1):57–73. doi:10.1007/s10719-008-9163-3.
- Mangeney M, Lingwood CA, Taga S, Caillou B, Tursz T, Wiels J. 1993. Apoptosis induced in Burkitt’s lymphoma cells via Gb3/CD77, a glycolipid antigen. Cancer Res. 53(21):5314–5319.
- Mangeney M, Richard Y, Coulaud D, Tursz T, Wiels J. 1991. CD77: an antigen of germinal center B cells entering apoptosis. Eur J Immunol. 21(5):1131–1140. doi:10.1002/eji.1830210507.
- Mardiana S, Gill S. 2020. CAR T cells for acute myeloid leukemia: state of the art and future directions. Front Oncol. 10:697. doi:10.3389/fonc.2020.00697.
- Mardiros A, Forman SJ, Budde LE. 2015. T cells expressing CD123 chimeric antigen receptors for treatment of acute myeloid leukemia. Curr Opin Hematol. 22(6):484–488. doi:10.1097/MOH.0000000000000190.
- Marques ETA Jr. 2000. Fucosyltransferases. In: Carbohydrate in chemistry and biology. Weinheim: Wiley-VCH; p. 197–211. doi:10.1002/9783527618255.ch50.
- Marquina G, Waki H, Fernandez LE. 1996. Gangliosides expressed in human breast cancer. Cancer Res. 56(22):5165–5171.
- Marshall AS, Willment JA, Pyz E. 2006. Human MICL (CLEC12A) is differentially glycosylated and is down-regulated following cellular activation. Eur J Immunol. 36(8):2159–2169. doi:10.1002/eji.200535628.
- Martinez-Palomo A, Braislovsky C, Bernhard W. 1969. Ultrastructural modifications of the cell surface and intercellular contacts of some transformed cell strains. Cancer Res. 29(4):925–937.
- Masih KE, Ligon JA, Yates B. 2021. Consequences of hemophagocytic lymphohistiocytosis-like cytokine release syndrome toxicities and concurrent bacteremia. Pediatr Blood Cancer. 68(10):e29247. doi:10.1002/pbc.29247.
- Mason CP, Tarr AW. 2015. Human lectins and their roles in viral infections. Molecules. 20(2):2229–2271. doi:10.3390/molecules20022229.
- Matoba Y, Sato Y, Oda K, Hatori Y, Morimoto K. 2021. Lectins engineered to favor a glycan-binding conformation have enhanced antiviral activity. J Biol Chem. 296:100698. doi:10.1016/j.jbc.2021.100698.
- Matsuura N, Narita T, Hiraiwa N. 1998. Gene expression of fucosyl- and sialyl-transferases which synthesize sialyl Lewisx, the carbohydrate ligands for E-selectin, in human breast cancer. Int J Oncol. 12(5):1157–1164. doi:10.3892/ijo.12.5.1157.
- Maude SL. 2018. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 378:439–448.
- Mazalovska M, Kouokam JC. 2018. Lectins as promising therapeutics for the prevention and treatment of HIV and other potential coinfections. Biomed Res Int. 2018:3750646. doi:10.1155/2018/3750646.
- McEver RP, Cummings RD. 1997. Role of PSGL-1 binding to selectins in leukocyte recruitment. J Clin Invest. 100(Suppl. 11):S97–S103.
- Meads MB, Hazlehurst LA, Dalton WS. 2008. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res. 14(9):2519–2526. doi:10.1158/1078-0432.CCR-07-2223.
- Meany DL, Chan DW. 2011. Aberrant glycosylation associated with enzymes as cancer biomarkers. Clin Proteom. 8(1):7. doi:10.1186/1559-0275-8-7.
- Mereiter S, Balmaña M, Campos D, Gomes J, Reis CA. 2019. Glycosylation in the era of cancer-targeted therapy: where are we heading? Cancer Cell. 36(1):6–16. doi:10.1016/j.ccell.2019.06.006.
- Michiels B, Van Puyenbroeck K, Verhoeven V, Vermeire E, Coenen S. 2013. The value of neuraminidase inhibitors for the prevention and treatment of seasonal influenza: a systematic review of systematic reviews. PLoS One. 8(4):e60348. doi:10.1371/journal.pone.0060348.
- Misaghian N, Ligresti G, Steelman LS. 2009. Targeting the leukemic stem cell: the Holy Grail of leukemia therapy. Leukemia. 23(1):25–42. doi:10.1038/leu.2008.246.
- Mishra A, Behura A, Mawatwal S. 2019. Structure-function and application of plant lectins in disease biology and immunity. Food Chem Toxicol. 134:110827. doi:10.1016/j.fct.2019.110827.
- Mitchell CA, Ramessar K, O’Keefe BR. 2017. Antiviral lectins: selective inhibitors of viral entry. Antiviral Res. 142:37–54. doi:10.1016/j.antiviral.2017.03.007.
- Miyagi T. 2008. Aberrant expression of sialidase and cancer progression. Proc Jpn Acad Ser B Phys Biol Sci. 84(10):407–418. doi:10.2183/pjab.84.407.
- Miyagi T, Yamaguchi K. 2007. Comprehensive glycoscience. In: Kamerling J. P., Boons G., Lee Y. C., Suzuki A., Taniguchi N., Voragen A. G. J., editor. Biochemistry of glycans: sialic acids. Amsterdam: Elsevier; p. 297–322.
- Miyazaki K, Sakuma K, Kawamura YI. 2012. Colonic epithelial cells express specific ligands for mucosal macrophage immunosuppressive receptors siglec-7 and -9. J Immunol. 188(9):4690–4700. doi:10.4049/jimmunol.1100605.
- Miyoshi E, Terao M, Kamada Y. 2012. Physiological roles of N-acetylglucosaminyltransferase V(GnT-V) in mice. BMB Rep. 45(10):554–559. doi:10.5483/bmbrep.2012.45.10.190.
- Mnich ME, van Dalen R, van Sorge NM. 2020. C-type lectin receptors in host defense against bacterial pathogens. Front Cell Infect Microbiol. 10:309. doi:10.3389/fcimb.2020.00309.
- Modenutti CP, Capurro JIB, Di Lella S, Martí MA. 2019. The structural biology of galectin-ligand recognition: current advances in modeling tools, protein engineering, and inhibitor design. Front Chem. 7:823. doi:10.3389/fchem.2019.00823.
- Mohseni M, Uludag H, Brandwein JM. 2018. Advances in biology of acute lymphoblastic leukemia (ALL) and therapeutic implications. Am J Blood Res. 8(4):29–56.
- Mondal N, Buffone A, Stolfa G. 2015. ST3Gal-4 is the primary sialyltransferase regulating the synthesis of E-, P-, and L-selectin ligands on human myeloid leukocytes. Blood. 125(4):687–696. doi:10.1182/blood-2014-07-588590.
- Mondal S, Chandra S, Mandal C. 2010. Elevated mRNA level of hST6Gal I and hST3Gal V positively correlates with the high risk of pediatric acute leukemia. Leuk Res. 34(4):463–470. doi:10.1016/j.leukres.2009.07.042.
- Monzavi-Karbassi B, Pashov A, Kieber-Emmons T. 2013. Tumor-associated glycans and immune surveillance. Vaccines. 1(2):174–203. doi:10.3390/vaccines1020174.
- Moremen KW, Tiemeyer M, Nairn AV. 2012. Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol. 13(7):448–462. doi:10.1038/nrm3383.
- Morita K, Wang F, Jahn K. 2020. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics [published correction appears in Nat Commun. 2020 Nov 19;11(1):5996] [published correction appears in Nat Commun. 2021 May 10;12(1):2823]. Nat Commun. 11(1):5327. doi:10.1038/s41467-020-19119-8.
- Morton DL, Barth A. 1996. Vaccine therapy for malignant melanoma. CA Cancer J Clin. 46(4):225–244. doi:10.3322/canjclin.46.4.225.
- Moscona A. 2005. Neuraminidase inhibitors for influenza. N Engl J Med. 353(13):1363–1373. doi:10.1056/NEJMra050740.
- Moyer VA. 2012. Screening for prostate cancer: U.S. preventive services task force recommendation statement. Ann Intern Med. 157(2):120–134. doi:10.7326/0003-4819-157-2-201207170-00459.
- Moynihan KD, Irvine DJ. 2017. Roles for innate immunity in combination immunotherapies. Cancer Res. 77(19):5215–5221. doi:10.1158/0008-5472.CAN-17-1340.
- Mujoo K, Cheresh DA, Yang HM, Reisfeld RA. 1987. Disialoganglioside GD2 on human neuroblastoma cells: target antigen for monoclonal antibody-mediated cytolysis and suppression of tumor growth. Cancer Res. 47(4):1098–1104.
- Mukherjee K, Chava AK, Mandal C. 2008. O-acetylation of GD3 prevents its apoptotic effect and promotes survival of lymphoblasts in childhood acute lymphoblastic leukaemia. J Cell Biochem. 105(3):724–734. doi:10.1002/jcb.21867.
- Munkley J, Elliott DJ. 2016. Hallmarks of glycosylation in cancer. Oncotarget. 7(23):35478–35489. doi:10.18632/oncotarget.8155.
- Myers D, Wrobleski S, Londy F. 2002. New and effective treatment of experimentally induced venous thrombosis with anti-inflammatory rPSGL-Ig. Thromb Haemost. 87(3):374–382.
- Myers DD, Schaub R, Wrobleski SK. 2001. P-selectin antagonism causes dose-dependent venous thrombosis inhibition. Thromb Haemost. 85(3):423–429.
- Nakagoe T, Fukushima K, Tanaka K. 2002. Evaluation of sialyl Lewis(a), sialyl Lewis(x), and sialyl Tn antigens expression levels as predictors of recurrence after curative surgery in node-negative colorectal cancer patients. J Exp Clin Cancer Res. 21(1):107–113.
- Nakagoe T, Sawai T, Tsuji T. 2001. Circulating sialyl Lewis(x), sialyl Lewis(a), and sialyl Tn antigens in colorectal cancer patients: multivariate analysis of predictive factors for serum antigen levels. J Gastroenterol. 36(3):166–172. doi:10.1007/s005350170124.
- Nakamura M, Ogino H, Nojiri H, Kitagawa S, Saito M. 1989. Characteristic incorporation of ganglioside GM3, which induces monocytic differentiation in human myelogenous leukemia HL-60 cells. Biochem Biophys Res Commun. 161(2):782–789. doi:10.1016/0006-291x(89)92668-5.
- Nascimento KS, Cunha AI, Nascimento KS, Cavada BS, Azevedo AM, Aires-Barros MR. 2012. An overview of lectins purification strategies. J Mol Recognit. 25(11):527–541. doi:10.1002/jmr.2200.
- Neth O, Hann I, Turner MW, Klein NJ. 2001. Deficiency of mannose-binding lectin and burden of infection in children with malignancy: a prospective study. Lancet. 358(9282):614–618. doi:10.1016/S0140-6736(01)05776-2.
- Neth OW, Bajaj-Elliott M, Turner MW, Klein NJ. 2005. Susceptibility to infection in patients with neutropenia: the role of the innate immune system. Br J Haematol. 129(6):713–722. doi:10.1111/j.1365-2141.2005.05462.x.
- Nghiem PT, Bhatia S, Lipson EJ. 2016. PD-1 blockade with pembrolizumab in advanced merkel-cell carcinoma. N Engl J Med. 374(26):2542–2552. doi:10.1056/NEJMoa1603702.
- Niemann CU, Kjeldsen L, Ralfkiaer E, Jensen MK, Borregaard N. 2007. Serglycin proteoglycan in hematologic malignancies: a marker of acute myeloid leukemia. Leukemia. 21(12):2406–2410. doi:10.1038/sj.leu.2404975.
- Notova S, Bonnardel F, Rosato F. 2022. A pore-forming β-trefoil lectin with specificity for the tumor-related glycosphingolipid Gb3. bioRxiv. doi:10.1101/2022.02.10.479907.
- Oberley MJ, Gaynon PS, Bhojwani D. 2018. Myeloid lineage switch following chimeric antigen receptor T-cell therapy in a patient with TCF3-ZNF384 fusion-positive B-lymphoblastic leukemia. Pediatr Blood Cancer. 65(9):e27265. doi:10.1002/pbc.27265.
- O’Brien RM, Cannon A, Reynolds JV, Lysaght J, Lynam-Lennon N. 2021. Complement in tumourigenesis and the response to cancer therapy. Cancers. 13(6):1209. doi:10.3390/cancers13061209.
- Oiseth SJ, Aziz MSA-E. 2017. Cancer immunotherapy: a brief review of the history, possibilities, and challenges ahead. J Cancer Metastasis Treat. 3:250–261.
- Ok CY, Young KH. 2017. Checkpoint inhibitors in hematological malignancies. J Hematol Oncol. 10(1):103. doi:10.1186/s13045-017-0474-3.
- Okada M, Furukawa K, Yamashiro S. 1996. High expression of ganglioside alpha-2,8-sialyltransferase (GD3 synthase) gene in adult T-cell leukemia cells unrelated to the gene expression of human T-lymphotropic virus type I. Cancer Res. 56(12):2844–2848.
- Oliveira C, Teixeira JA, Domingues L. 2013. Recombinant lectins: an array of tailor-made glycan-interaction biosynthetic tools. Crit Rev Biotechnol. 33(1):66–80. doi:10.3109/07388551.2012.670614.
- Oliveira T, Zhang M, Joo EJ. 2021. Glycoproteome remodeling in MLL-rearranged B-cell precursor acute lymphoblastic leukemia. Theranostics. 11(19):9519–9537. doi:10.7150/thno.65398.
- Ono M, Hakomori S. 2004. Glycosylation defining cancer cell motility and invasiveness. Glycoconj J. 20(1):71–78. doi:10.1023/B:GLYC.0000018019.22070.7d.
- Opdenakker G, Rudd PM, Ponting CP, Dwek RA. 1993. Concepts and principles of glycobiology. FASEB J. 7(14):1330–1337. doi:10.1096/fasebj.7.14.8224606.
- Orleans-Lindsay JK, Barber LD, Prentice HG, Lowdell MW. 2001. Acute myeloid leukaemia cells secrete a soluble factor that inhibits T and NK cell proliferation but not cytolytic function–implications for the adoptive immunotherapy of leukaemia. Clin Exp Immunol. 126(3):403–411. doi:10.1046/j.1365-2249.2001.01692.x.
- Pal S, Ghosh S, Mandal C. 2004. Purification and characterization of 9-O-acetylated sialoglycoproteins from leukemic cells and their potential as immunological tool for monitoring childhood acute lymphoblastic leukemia. Glycobiology. 14(10):859–870. doi:10.1093/glycob/cwh111.
- Pang X, Li H, Guan F, Li X. 2018. Multiple roles of glycans in hematological malignancies. Front Oncol. 8:364. doi:10.3389/fonc.2018.00364.
- Parameswaran R, Lim M, Arutyunyan A. 2013. O-acetylated N-acetylneuraminic acid as a novel target for therapy in human pre-B acute lymphoblastic leukemia. J Exp Med. 210(4):805–819. doi:10.1084/jem.20121482.
- Park DD, Chen J, Kudelka MR. 2021. Resident and elicited murine macrophages differ in expression of their glycomes and glycan-binding proteins. Cell Chem Biol. 28(4):567–582. doi:10.1016/j.chembiol.2020.12.005.
- Park JJ, Lee M. 2013. Increasing the α 2, 6 sialylation of glycoproteins may contribute to metastatic spread and therapeutic resistance in colorectal cancer. Gut Liver. 7(6):629–641. doi:10.5009/gnl.2013.7.6.629.
- Paszek MJ, DuFort CC, Rossier O. 2014. The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature. 511(7509):319–325. doi:10.1038/nature13535.
- Patankar MS, Jing Y, Morrison JC. 2005. Potent suppression of natural killer cell response mediated by the ovarian tumor marker CA125. Gynecol Oncol. 99(3):704–713. doi:10.1016/j.ygyno.2005.07.030.
- Patil S, Rao RS, Majumdar B. 2015. T-cell exhaustion and cancer immunotherapy. J Int Oral Health. 7(8):i–ii.
- Paul S, Rausch CR, Nasnas PE, Kantarjian H, Jabbour EJ. 2019. Treatment of relapsed/refractory acute lymphoblastic leukemia. Clin Adv Hematol Oncol. 17(3):166–175.
- Paz H, Joo EJ, Chou CH. 2018. Treatment of B-cell precursor acute lymphoblastic leukemia with the galectin-1 inhibitor PTX008. J Exp Clin Cancer Res. 37(1):67. doi:10.1186/s13046-018-0721-7.
- Pearce OM, Varki A. 2010. Chemo-enzymatic synthesis of the carbohydrate antigen N-glycolylneuraminic acid from glucose. Carbohydr Res. 345(9):1225–1229. doi:10.1016/j.carres.2010.04.003.
- Peixoto A, Relvas-Santos M, Azevedo R, Santos LL, Ferreira JA. 2019. Protein glycosylation and tumor microenvironment alterations driving cancer hallmarks. Front Oncol. 9:380. doi:10.3389/fonc.2019.00380.
- Pena C, Mirandola L, Figueroa JA. 2014. Galectins as therapeutic targets for hematological malignancies: a hopeful sweetness. Ann Transl Med. 2(9):87. doi:10.3978/j.issn.2305-5839.2014.09.14.
- Peterson K, Kumar R, Stenström O. 2018. Systematic tuning of fluoro-galectin-3 interactions provides thiodigalactoside derivatives with single-digit nM affinity and high selectivity. J Med Chem. 61(3):1164–1175. doi:10.1021/acs.jmedchem.7b01626.
- Petretti T, Schulze B, Schlag PM, Kemmner W. 1999. Altered mRNA expression of glycosyltransferases in human gastric carcinomas. Biochim Biophys Acta. 1428(2-3):209–218. doi:10.1016/s0304-4165(99)00080-x.
- Petrov JC, Wada M, Pinz KG. 2018. Compound CAR T-cells as a double-pronged approach for treating acute myeloid leukemia. Leukemia. 32(6):1317–1326. doi:10.1038/s41375-018-0075-3.
- Pieters R, Kaspers GJ, van Wering ER. 1993. Cellular drug resistance profiles that might explain the prognostic value of immunophenotype and age in childhood acute lymphoblastic leukemia. Leukemia. 7(3):392–397.
- Pihikova D, Kasak P, Kubanikova P, Sokol R, Tkac J. 2016. Aberrant sialylation of a prostate-specific antigen: electrochemical label-free glycoprofiling in prostate cancer serum samples. Anal Chim Acta. 934:72–79. doi:10.1016/j.aca.2016.06.043.
- Pillai V, Muralidharan K, Meng W. 2019. CAR T-cell therapy is effective for CD19-dim B-lymphoblastic leukemia but is impacted by prior blinatumomab therapy. Blood Adv. 3(22):3539–3549. doi:10.1182/bloodadvances.2019000692.
- Pinho SS, Reis CA. 2015. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 15(9):540–555. doi:10.1038/nrc3982.
- Ploessl C, Pan A, Maples KT, Lowe DK. 2016. Dinutuximab: an anti-GD2 monoclonal antibody for high-risk neuroblastoma. Ann Pharmacother. 50(5):416–422. doi:10.1177/1060028016632013.
- Poggi A, Zocchi MR. 2021. Natural killer cells and immune-checkpoint inhibitor therapy: current knowledge and new challenges. Mol Ther Oncolyt. 24:26–42. doi:10.1016/j.omto.2021.11.016.
- Pollard JA, Alonzo TA, Loken M. 2012. Correlation of CD33 expression level with disease characteristics and response to gemtuzumab ozogamicin containing chemotherapy in childhood AML. Blood. 119(16):3705–3711. doi:10.1182/blood-2011-12-398370.
- Ponath P, Menezes D, Pan C. 2018. A novel, fully human anti-fucosyl-GM1 antibody demonstrates potent in vitro and in vivo antitumor activity in preclinical models of small cell lung cancer. Clin Cancer Res. 24(20):5178–5189. doi:10.1158/1078-0432.CCR-18-0018.
- Portell CA, Wenzell CM, Advani AS. 2013. Clinical and pharmacologic aspects of blinatumomab in the treatment of B-cell acute lymphoblastic leukemia. Clin Pharmacol. 5(Suppl. 1):5–11. doi:10.2147/CPAA.S42689.
- Posey AD, Schwab RD, Boesteanu AC. 2016. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity. 44(6):1444–1454. doi:10.1016/j.immuni.2016.05.014.
- Prendergast GC. 2008. Immune escape as a fundamental trait of cancer: focus on IDO. Oncogene. 27(28):3889–3900. doi:10.1038/onc.2008.35.
- Pukel CS, Lloyd KO, Travassos LR, Dippold WG, Oettgen HF, Old LJ. 1982. GD3, a prominent ganglioside of human melanoma. Detection and characterisation by mouse monoclonal antibody. J Exp Med. 155(4):1133–1147. doi:10.1084/jem.155.4.1133.
- Purushothaman A, Toole BP. 2014. Serglycin proteoglycan is required for multiple myeloma cell adhesion, in vivo growth, and vascularization. J Biol Chem. 289(9):5499–5509. doi:10.1074/jbc.M113.532143.
- Qin H, Ishii K, Nguyen S. 2018. Murine pre-B-cell ALL induces T-cell dysfunction not fully reversed by introduction of a chimeric antigen receptor. Blood. 132(18):1899–1910. doi:10.1182/blood-2017-12-815548.
- Qin W, Hu L, Zhang X. 2019. The diverse function of PD-1/PD-L pathway beyond cancer. Front Immunol. 10:2298. doi:10.3389/fimmu.2019.02298.
- Rack B, Jückstock J, Trapp E. 2016. CA27.29 as a tumour marker for risk evaluation and therapy monitoring in primary breast cancer patients. Tumour Biol. 37(10):13769–13775. doi:10.1007/s13277-016-5171-2.
- Raetz EA, Teachey DT. 2016. T-cell acute lymphoblastic leukemia. Hematol Am Soc Hematol Educ Program. 2016(1):580–588. doi:10.1182/asheducation-2016.1.580.
- Ragoonanan D, Khazal SJ, Abdel-Azim H. 2021. Diagnosis, grading and management of toxicities from immunotherapies in children, adolescents and young adults with cancer [published correction appears in Nat Rev Clin Oncol. 2021 Mar 17]. Nat Rev Clin Oncol. 18(7):435–453. doi:10.1038/s41571-021-00474-4.
- Rani CS, Abe A, Chang Y. 1995. Cell cycle arrest induced by an inhibitor of glucosylceramide synthase. Correlation with cyclin-dependent kinases. J Biol Chem. 270(6):2859–2867. doi:10.1074/jbc.270.6.2859.
- Rasche L, Vago L, Mutis T. 2022. Tumour escape from CAR-T cells. In: Kröger N., Gribben J., Chabannon C., Yakoub-Agha I., Einsele H, editor. The EBMT/EHA CAR-T cell handbook. Cham: Springer. doi:10.1007/978-3-030-94353-0_4
- Rashidbaghan A, Mostafaie A, Yazdani Y, Mansouri K. 2020. Urtica dioica agglutinin (a plant lectin) has a caspase-dependent apoptosis induction effect on the acute lymphoblastic leukemia cell line. Cell Mol Biol. 66(6):121–126.
- Rassidakis GZ, Goy A, Medeiros LJ. 2003. Prognostic significance of MUC-1 expression in systemic anaplastic large cell lymphoma. Clin Cancer Res. 9(6):2213–2220.
- Ravindranaths MH, Paulson JC, Irie RF. 1988. Human melanoma antigen O-acetylated ganglioside GD3 is recognized by cancer antennarius lectin. J Biol Chem. 263(4):2079–2086.
- Ravn V, Dabelsteen E. 2000. Tissue distribution of histo-blood group antigens. APMIS. 108(1):1–28. doi:10.1034/j.1600-0463.2000.d01-1.x.
- Recchi MA, Hebbar M, Hornez L, Harduin-Lepers A, Peyrat JP, Delannoy P. 1998. Multiplex reverse transcription polymerase chain reaction assessment of sialyltransferase expression in human breast cancer. Cancer Res. 58(18):4066–4070.
- Reily C, Stewart TJ, Renfrow MB, Novak J. 2019. Glycosylation in health and disease. Nat Rev Nephrol. 15(6):346–366. doi:10.1038/s41581-019-0129-4.
- Reismüller B, Attarbaschi A, Peters C. 2009. Long-term outcome of initially homogenously treated and relapsed childhood acute lymphoblastic leukaemia in Austria – a population-based report of the Austrian Berlin-Frankfurt-münster (BFM) study group. Br J Haematol. 144(4):559–570. doi:10.1111/j.1365-2141.2008.07499.x.
- Reisner Y, Biniaminov M, Rosenthal E, Sharon N, Ramot B. 1979. Interaction of peanut agglutinin with normal human lymphocytes and with leukemic cells. Proc Natl Acad Sci USA. 76(1):447–451. doi:10.1073/pnas.76.1.447.
- Rettig M, Godwin J, Vey N. 2017. Preliminary translational results from an ongoing phase 1 study of flotetuzumab, a CD123 x CD3 Dart®, in AML/MDS: rationale for combining flotetuzumab and anti-PD-1/PD-L1 immunotherapies [abstract]. Blood. 130(Suppl. 1).
- Reya T, Morrison SJ, Clarke MF, Weissman IL. 2001. Stem cells, cancer, and cancer stem cells. Nature. 414(6859):105–111. doi:10.1038/35102167.
- Rezvani AR, Maloney DG. 2011. Rituximab resistance. Best Pract Res Clin Haematol. 24(2):203–216. doi:10.1016/j.beha.2011.02.009.
- Richardson PG, Corbacioglu S. 2020. Veno-occlusive disease/sinusoidal obstruction syndrome in patients with prior gemtuzumab ozogamicin: literature analysis of survival after defibrotide treatment. Blood Cancer J. 10(3):29. doi:10.1038/s41408-020-0286-5.
- Richter M, Yumul R, Saydaminova K. 2016. Preclinical safety, pharmacokinetics, pharmacodynamics, and biodistribution studies with Ad35K++ protein: a novel rituximab cotherapeutic. Mol Ther Methods Clin Dev. 5:16013. doi:10.1038/mtm.2016.13.
- Riley JL, Mao M, Kobayashi S. 2002. Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc Natl Acad Sci USA. 99(18):11790–11795. doi:10.1073/pnas.162359999.
- Ritchie DS, Neeson PJ, Khot A. 2013. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. 21(11):2122–2129. doi:10.1038/mt.2013.154.
- RodrÍguez E, Schetters STT, van Kooyk Y. 2018. The tumour glyco-code as a novel immune checkpoint for immunotherapy. Nat Rev Immunol. 18(3):204–211. doi:10.1038/nri.2018.3.
- Rodrigues Mantuano N, Natoli M, Zippelius A, Läubli H. 2020. Tumor-associated carbohydrates and immunomodulatory lectins as targets for cancer immunotherapy. J Immunother Cancer. 8(2):e001222. doi:10.1136/jitc-2020-001222.
- Rodriguez E, Boelaars K, Brown K. 2021. Sialic acids in pancreatic cancer cells drive tumour-associated macrophage differentiation via the Siglec receptors Siglec-7 and Siglec-9. Nat Commun. 12(1):1270. doi:10.1038/s41467-021-21550-4.
- Rosen SD, Bertozzi CR. 1994. The selectins and their ligands. Curr Opin Cell Biol. 6(5):663–673. doi:10.1016/0955-0674(94)90092-2.
- Rosenstock P, Kaufmann T. 2021. Sialic acids and their influence on human NK cell function. Cells. 10:263. doi:10.3390/cells10020263.
- Rossi V, Cseh S, Bally I, Thielens NM, Jensenius JC, Arlaud GJ. 2001. Substrate specificities of recombinant mannan-binding lectin-associated serine proteases-1 and -2. J Biol Chem. 276(44):40880–40887. doi:10.1074/jbc.M105934200.
- Roth J, Zuber C, Wagner P. 1988. Reexpression of poly(sialic acid) units of the neural cell adhesion molecule in Wilms tumor. Proc Natl Acad Sci USA. 85(9):2999–3003. doi:10.1073/pnas.85.9.2999.
- Roth P, Winklhofer S, Müller AMS. 2021. Neurological complications of cancer immunotherapy. Cancer Treat Rev. 97:102189. doi:10.1016/j.ctrv.2021.102189.
- Rubinstein N, Alvarez M, Zwirner NW. 2004. Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection; A potential mechanism of tumor-immune privilege. Cancer Cell. 5(3):241–251. doi:10.1016/s1535-6108(04)00024-8.
- Rubnitz JE, Kaspers GJ. 2021. How I treat pediatric acute myeloid leukemia. Blood. 138(12):1009–1018. doi:10.1182/blood.2021011694.
- Ruhaak LR, Xu G, Li Q, Goonatilleke E, Lebrilla CB. 2018. Mass spectrometry approaches to glycomic and glycoproteomic analyses. Chem Rev. 118(17):7886–7930. doi:10.1021/acs.chemrev.7b00732.
- Rupp LJ, Schumann K, Roybal KT. 2017. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 7(1):737. doi:10.1038/s41598-017-00462-8.
- Saad AAD. 2021. Recombinant lectins as pioneering anti-viral agents against COVID-19. Hematol Transfus Int J. 9(4):77–79. doi:10.15406/htij.2021.09.00259.
- Sakuma K, Aoki M, Kannagi R. 2012. Transcription factors c-Myc and CDX2 mediate E-selectin ligand expression in colon cancer cells undergoing EGF/bFGF-induced epithelial-mesenchymal transition. Proc Natl Acad Sci USA. 109(20):7776–7781. doi:10.1073/pnas.1111135109.
- Samraj AN, Bertrand KA, Luben R. 2018. Polyclonal human antibodies against glycans bearing red meat-derived non-human sialic acid N-glycolylneuraminic acid are stable, reproducible, complex and vary between individuals: total antibody levels are associated with colorectal cancer risk. PLoS One. 13(6):e0197464. doi:10.1371/journal.pone.0197464.
- Samraj AN, Läubli H, Varki N, Varki A. 2014. Involvement of a non-human sialic acid in human cancer. Front Oncol. 4:33. doi:10.3389/fonc.2014.00033.
- Sander VA, Corigliano MG, Clemente M. 2019. Promising plant-derived adjuvants in the development of coccidial vaccines. Front Vet Sci. 6:20. doi:10.3389/fvets.2019.00020.
- Sanford BH. 1967. An alteration in tumor histocompatibility induced by neuraminidase. Transplantation. 5(5):1273–1279. doi:10.1097/00007890-196709000-00005.
- Santamaria S, Delgado M, Kremer L, Garcia-Sanz JA. 2017. Will a mAb-based immunotherapy directed against cancer stem cells Be feasible? Front Immunol. 8:1509. doi:10.3389/fimmu.2017.01509.
- Sarnataro D, Pepe A, Zurzolo C. 2017. Cell biology of prion protein. Prog Mol Biol Transl Sci. 150:57–82. doi:10.1016/bs.pmbts.2017.06.018.
- Schauer R. 2000. Achievements and challenges of sialic acid research. Glycoconj J. 17(7–9):485–499. doi:10.1023/a:1011062223612.
- Schauer R, Srinivasan GV, Wipfler D, Kniep B, Schwartz-Albiez R. 2011. O-acetylated sialic acids and their role in immune defense. Adv Exp Med Biol. 705:525–548. doi:10.1007/978-1-4419-7877-6_28.
- Schmid C, Labopin M, Nagler A. 2012. Treatment, risk factors, and outcome of adults with relapsed AML after reduced intensity conditioning for allogeneic stem cell transplantation. Blood. 119(6):1599–1606. doi:10.1182/blood-2011-08-375840.
- Schultes BC, Baum RP, Niesen A, Noujaim AA, Madiyalakan R. 1998. Anti-idiotype induction therapy: anti-CA125 antibodies (Ab3) mediated tumor killing in patients treated with Ovarex mAb B43.13 (Ab1). Cancer Immunol Immunother. 46(4):201–212. doi:10.1007/s002620050479.
- Schwartz-Albiez R, Dörken B, Möller P, Brodin NT, Monner DA, Kniep B. 1990. Neutral glycosphingolipids of the globo-series characterize activation stages corresponding to germinal center B cells. Int Immunol. 2(10):929–936. doi:10.1093/intimm/2.10.929.
- Scott AM, Lee FT, Hopkins W. 2001. Specific targeting, biodistribution, and lack of immunogenicity of chimeric anti-GD3 monoclonal antibody KM871 in patients with metastatic melanoma: results of a phase I trial. J Clin Oncol. 19(19):3976–3987. doi:10.1200/JCO.2001.19.19.3976.
- Sewell R, Bäckström M, Dalziel M. 2006. The ST6GalNAc-I sialyltransferase localizes throughout the Golgi and is responsible for the synthesis of the tumor-associated sialyl-Tn O-glycan in human breast cancer. J Biol Chem. 281(6):3586–3594. doi:10.1074/jbc.M511826200.
- Shah NN, Fry TJ. 2019. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 16(6):372–385. doi:10.1038/s41571-019-0184-6.
- Shah NN, Maatman T, Hari P, Johnson B. 2019. Multi targeted CAR-T cell therapies for B-cell malignancies. Front Oncol. 9:146. doi:10.3389/fonc.2019.00146.
- Shah NN, O’Brien MM, Yuan C, Ji L, Xu X, Rheingold SR. 2020. Evaluation of CD22 modulation as a mechanism of resistance to inotuzumab ozogamicin (InO): results from central CD22 testing on the Children’s Oncology Group (COG) phase II trial of INO in children and young adults with CD22+ B-acute lymphoblastic leukemia (B-ALL). J Clin Oncol. 38(Suppl. 15):10519.
- Shan M, Yang D, Dou H, Zhang L. 2019. Fucosylation in cancer biology and its clinical applications. Prog Mol Biol Transl Sci. 162:93–119. doi:10.1016/bs.pmbts.2019.01.002.
- Shang Y, Zhou F. 2019. Current advances in immunotherapy for acute leukemia: an overview of antibody, chimeric antigen receptor, immune checkpoint, and natural killer. Front Oncol. 9:917. doi:10.3389/fonc.2019.00917.
- Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. 2017. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 168(4):707–723. doi:10.1016/j.cell.2017.01.017.
- Sharma V, Vijayan M, Surolia A. 1996. Imparting exquisite specificity to peanut agglutinin for the tumor-associated Thomsen-Friedenreich antigen by redesign of its combining site. J Biol Chem. 271(35):21209–21213. doi:10.1074/jbc.271.35.21209.
- Shewell LK, Day CJ, Kutasovic JR, Abrahams JL, Wang J, Poole J, Niland C, Ferguson K, Saunus JM, Lakhani SR, et al. 2021. N-glycolylneuraminic acid serum biomarker levels are elevated in breast cancer patients at all stages of disease. bioRxiv: n. pag.
- Shintani Y, Takashima S, Asano Y. 2006. Glycosaminoglycan modification of neuropilin-1 modulates VEGFR2 signaling. EMBO J. 25(13):3045–3055. doi:10.1038/sj.emboj.7601188.
- Short NJ, Konopleva M, Kadia TM. 2020. Advances in the Treatment of acute myeloid leukemia: new drugs and new challenges. Cancer Discov. 10(4):506–525. doi:10.1158/2159-8290.CD-19-1011.
- Shukla N, Sulis ML. 2021. Blinatumomab for treatment of children with high-risk relapsed B-cell acute lymphoblastic leukemia. JAMA. 325(9):830–832. doi:10.1001/jama.2021.1395.
- Silsirivanit A. 2019. Chapter five – glycosylation markers in cancer. In: Gregory S. Makowski, editor. Advances in clinical chemistry. Elsevier; p. 189–213. doi:10.1016/bs.acc.2018.12.005
- Singh JP, Zhang K, Wu J, Yang X. 2015. O-GlcNAc signaling in cancer metabolism and epigenetics. Cancer Lett. 356(2 Pt A):244–250. doi:10.1016/j.canlet.2014.04.014.
- Singrang N, Laophetsakunchai S, Tran BN, Matsudaira PT, Tassanakajon A, Wangkanont K. 2021. Biochemical and structural characterization of a recombinant fibrinogen-related lectin from Penaeus monodon. Sci Rep. 11(1):2934. doi:10.1038/s41598-021-82301-5.
- Sinha D, Bhattacharya DK, Mandal C. 1999a. A colorimetric assay to evaluate the chemotherapeutic response of children with acute lymphoblastic leukemia (ALL) employing achatininH: a 9-O-acetyl sialic acid binding lectin. Leuk Res. 23(9):803–809. doi:10.1016/s0145-2126(99)00093-4.
- Sinha D, Mandal C, Bhattacharya DK. 1999b. Development of a simple, blood based lymphoproliferation assay to assess the clinical status of patients with acute lymphoblastic leukemia. Leuk Res. 23(5):433–439. doi:10.1016/s0145-2126(98)00184-2.
- Siukstaite L, Imberty A, Römer W. 2021. Structural diversities of lectins binding to the glycosphingolipid Gb3. Front Mol Biosci. 8:704685. doi:10.3389/fmolb.2021.704685.
- Skliris A, Labropoulou VT, Papachristou DJ, Aletras A, Karamanos NK, Theocharis AD. 2013. Cell-surface serglycin promotes adhesion of myeloma cells to collagen type I and affects the expression of matrix metalloproteinases. FEBS J. 280(10):2342–2352. doi:10.1111/febs.12179.
- Slesak B, Bogusławska-Jaworska J, Pejcz J, Harłozińska A. 1989. Binding of lectins to human leukaemic cells. Folia Haematol Int Mag Klin Morphol Blutforsch. 116(2):251–259.
- Slesak B, Harłozińska-Szmyrka A, Frydecka I. 1990. Lectin binding ability of B-chronic lymphocytic leukaemia cells. Folia Haematol Int Mag Klin Morphol Blutforsch. 117(1):31–35.
- Smith BAH, Bertozzi CR. 2021. The clinical impact of glycobiology: targeting selectins, Siglecs and mammalian glycans [published correction appears in Nat Rev Drug Discov. 2021 Feb 8;:]. Nat Rev Drug Discov. 20(3):217–243. doi:10.1038/s41573-020-00093-1.
- Smithson M, Irwin R, Williams G. 2022. Sialyltransferase ST6GAL-1 mediates resistance to chemoradiation in rectal cancer [published online ahead of print, 2022 Jan 15]. J Biol Chem. 101594. doi:10.1016/j.jbc.2022.101594.
- Sørensen AL, Reis CA, Tarp MA. 2006. Chemoenzymatically synthesized multimeric Tn/STn MUC1 glycopeptides elicit cancer-specific anti-MUC1 antibody responses and override tolerance. Glycobiology. 16(2):96–107. doi:10.1093/glycob/cwj044.
- Stanczak MA, Siddiqui SS, Trefny MP. 2018. Self-associated molecular patterns mediate cancer immune evasion by engaging Siglecs on T cells. J Clin Invest. 128(11):4912–4923. doi:10.1172/JCI120612.
- Stanley P. 2011. Golgi glycosylation. Cold Spring Harb Perspect Biol. 3(4):a005199. doi:10.1101/cshperspect.a005199.
- Stegmayr J, Zetterberg F, Carlsson MC. 2019. Extracellular and intracellular small-molecule galectin-3 inhibitors. Sci Rep. 9(1):2186. doi:10.1038/s41598-019-38497-8.
- Stencel-Baerenwald JE, Reiss K, Reiter DM, Stehle T, Dermody TS. 2014. The sweet spot: defining virus-sialic acid interactions. Nat Rev Microbiol. 12(11):739–749. doi:10.1038/nrmicro3346.
- St-Gelais J, Denavit V, Giguère D. 2020. Efficient synthesis of a galectin inhibitor clinical candidate (TD139) using a Payne rearrangement/azidation reaction cascade. Org Biomol Chem. 18(20):3903–3907. doi:10.1039/d0ob00910e.
- Stimmer L, Dehay S, Nemati F. 2014. Human breast cancer and lymph node metastases express Gb3 and can be targeted by STxB-vectorized chemotherapeutic compounds. BMC Cancer. 14:916. doi:10.1186/1471-2407-14-916.
- Stokke JL, Bhojwani D. 2021. Antibody-drug conjugates for the treatment of acute pediatric leukemia. J Clin Med. 10(16):3556. doi:10.3390/jcm10163556.
- Stowell SR, Ju T, Cummings RD. 2015. Protein glycosylation in cancer. Annu Rev Pathol. 10:473–510. doi:10.1146/annurev-pathol-012414-040438.
- Strohl WR, Naso M. 2019. Bispecific T-cell redirection versus chimeric antigen receptor (CAR)-T cells as approaches to kill cancer cells. Antibodies. 8(3):41. doi:10.3390/antib8030041.
- Sun S, Hao H, Yang G, Zhang Y, Fu Y. 2018. Immunotherapy with CAR-modified T cells: toxicities and overcoming strategies. J Immunol Res. 2018:2386187. doi:10.1155/2018/2386187.
- Swanson MD, Boudreaux DM, Salmon L. 2015. Engineering a therapeutic lectin by uncoupling mitogenicity from antiviral activity. Cell. 163(3):746–758. doi:10.1016/j.cell.2015.09.056.
- Świerzko AS, Cedzyński M. 2020. The influence of the Lectin pathway of complement activation on infections of the respiratory system. Front Immunol. 11:585243. doi:10.3389/fimmu.2020.585243.
- Takahashi K, Moyo P, Chigweshe L, Chang WC, White MR, Hartshorn KL. 2013. Efficacy of recombinant chimeric lectins, consisting of mannose binding lectin and L-ficolin, against influenza A viral infection in mouse model study. Virus Res. 178(2):495–501. doi:10.1016/j.virusres.2013.10.001.
- Tanaka T. 2016. Leukocyte adhesion molecules. In: Michael J.H. Ratcliffe, editor. Encyclopedia of immunobiology. Academic Press; p. 505–511. doi:10.1016/B978-0-12-374279-7.07015-6
- Tang J, Lin G, Langdon WY, Tao L, Zhang J. 2018. Regulation of C-type lectin receptor-mediated antifungal immunity. Front Immunol. 9:123. doi:10.3389/fimmu.2018.00123.
- Tang L, Zhang Y, Hu Y, Mei H. 2021. T cell exhaustion and CAR-T immunotherapy in hematological malignancies. Biomed Res Int. 2021:6616391. doi:10.1155/2021/6616391.
- Tang XL, Yuan CH, Ding Q, Zhou Y, Pan Q, Zhang XL. 2017. Selection and identification of specific glycoproteins and glycan biomarkers of macrophages involved in Mycobacterium tuberculosis infection. Tuberculosis. 104:95–106. doi:10.1016/j.tube.2017.03.010.
- Taniguchi N, Kizuka Y. 2015. Glycans and cancer: role of N-glycans in cancer biomarker, progression and metastasis, and therapeutics. Adv Cancer Res. 126:11–51. doi:10.1016/bs.acr.2014.11.001.
- Taniguchi N, Miyoshi E, Ko JH, Ikeda Y, Ihara Y. 1999. Implication of N-acetylglucosaminyltransferases III and V in cancer: gene regulation and signaling mechanism. Biochim Biophys Acta. 1455(2–3):287–300. doi:10.1016/s0925-4439(99)00066-6.
- Tashiro H, Sauer T, Shum T. 2017. Treatment of acute myeloid leukemia with T cells expressing chimeric antigen receptors directed to C-type lectin-like molecule 1. Mol Ther. 25(9):2202–2213. doi:10.1016/j.ymthe.2017.05.024.
- Tasian SK, Hunger SP. 2017. Targeted therapy and precision medicine. Childhood acute lymphoblastic leukemia.
- Teachey DT, Pui CH. 2019. Comparative features and outcomes between paediatric T-cell and B-cell acute lymphoblastic leukaemia. Lancet Oncol. 20(3):e142–e154. doi:10.1016/S1470-2045(19)30031-2.
- Teachey DT, Rheingold SR, Maude SL. 2013. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 121(26):5154–5157. doi:10.1182/blood-2013-02-485623.
- Testa U, Riccioni R, Militi S. 2002. Elevated expression of IL-3Ralpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood. 100(8):2980–2988. doi:10.1182/blood-2002-03-0852.
- Thielens NM, Cseh S, Thiel S. 2001. Interaction properties of human mannan-binding lectin (MBL)-associated serine proteases-1 and -2, MBL-associated protein 19, and MBL. J Immunol. 166(8):5068–5077. doi:10.4049/jimmunol.166.8.5068.
- Thomas X. 2014. Profile of inotuzumab ozogamicin and its potential in the treatment of acute lymphoblastic leukemia. Blood Lymphat Cancer. 4:1–8. doi:10.2147/BLCTT.S49048.
- Thurin M. 2021. Tumor-associated glycans as targets for immunotherapy: the Wistar institute experience/legacy. Monoclon Antib Immunodiagn Immunother. 40(3):89–100. doi:10.1089/mab.2021.0024.
- Topp MS, Gökbuget N, Stein AS. 2015. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study [published correction appears in Lancet Oncol. 2015 Apr;16(4):e158]. Lancet Oncol. 16(1):57–66. doi:10.1016/S1470-2045(14)71170-2.
- Townsend MH, Shrestha G, Robison RA, O’Neill KL. 2018. The expansion of targetable biomarkers for CAR T cell therapy. J Exp Clin Cancer Res. 37(1):163. doi:10.1186/s13046-018-0817-0.
- Trabbic KR, Kleski KA, Barchi JJ. 2021. A stable gold nanoparticle-based vaccine for the targeted delivery of tumor-associated glycopeptide antigens. ACS Bio Med Chem Au. 1(1):31–43. doi:10.1021/acsbiomedchemau.1c00021.
- Trinchera M, Aronica A, Dall’Olio F. 2017. Selectin ligands Sialyl-Lewis a and Sialyl-Lewis x in gastrointestinal cancers. Biology. 6(1):16. doi:10.3390/biology6010016.
- Tsaneva M, Van Damme EJM. 2020. 130 years of plant lectin research. Glycoconj J. 37(5):533–551. doi:10.1007/s10719-020-09942-y.
- Tsuboi S, Sutoh M, Hatakeyama S. 2011. A novel strategy for evasion of NK cell immunity by tumours expressing core2 O-glycans. EMBO J. 30(15):3173–3185. doi:10.1038/emboj.2011.215.
- Turzanski J, Grundy M, Shang S, Russell N, Pallis M. 2005. P-glycoprotein is implicated in the inhibition of ceramide-induced apoptosis in TF-1 acute myeloid leukemia cells by modulation of the glucosylceramide synthase pathway. Exp Hematol. 33(1):62–72. doi:10.1016/j.exphem.2004.10.005.
- US Food and Drug Administration. 2019. FDA approves crizanlizumab-tmca for sickle cell disease. FDA.
- Ustun C, Miller JS, Munn DH, Weisdorf DJ, Blazar BR. 2011. Regulatory T cells in acute myelogenous leukemia: is it time for immunomodulation? Blood. 118(19):5084–5095. doi:10.1182/blood-2011-07-365817.
- Uy GL, Aldoss I, Foster MC, Sayre PH, Wieduwilt MJ, Advani AS, Godwin JE, Arellano ML, Sweet KL, Emadi A, et al. 2021. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood. 137(6):751–762. doi:10.1182/blood.2020007732.
- Vajaria BN, Patel PS. 2017. Glycosylation: a hallmark of cancer? Glycoconj J. 34(2):147–156. doi:10.1007/s10719-016-9755-2.
- van Cruijsen H, Ruiz MG, van der Valk P, de Gruijl TD, Giaccone G. 2009. Tissue micro array analysis of ganglioside N-glycolyl GM3 expression and signal transducer and activator of transcription (STAT)-3 activation in relation to dendritic cell infiltration and microvessel density in non-small cell lung cancer. BMC Cancer. 9:180. doi:10.1186/1471-2407-9-180.
- Vandamme-Feldhaus V, Schauer R. 1998. Characterization of the enzymatic 7-O-acetylation of sialic acids and evidence for enzymatic O-acetyl migration from C-7 to C-9 in bovine submandibular gland. J Biochem. 124(1):111–121. doi:10.1093/oxfordjournals.jbchem.a022069.
- Vander Heiden MG, Cantley LC, Thompson CB. 2009. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 324(5930):1029–1033. doi:10.1126/science.1160809.
- van de Wall S, Santegoets KCM, van Houtum EJH, Büll C, Adema GJ. 2020. Sialoglycans and Siglecs can shape the tumor immune microenvironment. Trends Immunol. 41(4):274–285. doi:10.1016/j.it.2020.02.001.
- van Elsas MJ, van Hall T, van der Burg SH. 2020. Future challenges in cancer resistance to immunotherapy. Cancers. 12(4):935. doi:10.3390/cancers12040935.
- van Houtum EJH, Büll C, Cornelissen LAM, Adema GJ. 2021. Siglec signaling in the tumor microenvironment. Front Immunol. 12:790317. doi:10.3389/fimmu.2021.790317.
- van Meerten T, van Rijn RS, Hol S, Hagenbeek A, Ebeling SB. 2006. Complement-induced cell death by rituximab depends on CD20 expression level and acts complementary to antibody-dependent cellular cytotoxicity. Clin Cancer Res. 12(13):4027–4035. doi:10.1158/1078-0432.CCR-06-0066.
- Varambally S, Bar-Dayan Y, Bayry J. 2004. Natural human polyreactive IgM induce apoptosis of lymphoid cell lines and human peripheral blood mononuclear cells. Int Immunol. 16(3):517–524. doi:10.1093/intimm/dxh053.
- Varki A. 2001. Loss of N-glycolylneuraminic acid in humans: mechanisms, consequences, and implications for hominid evolution. Am J Phys Anthropol. 33:54–69. doi:10.1002/ajpa.10018.abs.
- Varki A. 2008. Sialic acids in human health and disease. Trends Mol Med. 14(8):351–360. doi:10.1016/j.molmed.2008.06.002.
- Varki A. 2017. Biological roles of glycans. Glycobiology. 27(1):3–49. doi:10.1093/glycob/cww086.
- Varki A, Cummings RD, Aebi M. 2015a. Symbol nomenclature for graphical representations of glycans. Glycobiology. 25(12):1323–1324. doi:10.1093/glycob/cwv091.
- Varki A, Etzler ME, Cummings RD, Esko JD. 2009. Discovery and classification of glycan-binding proteins. In: Varki A, Cummings RD, Esko JD, et al., editors. Essentials of glycobiology. Chapter 26. 2nd ed. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press. https://www.ncbi.nlm.nih.gov/books/NBK1923/.
- Varki A, Kannagi R, Toole B. 2015b. Glycosylation changes in cancer. In: Varki A, Cummings RD, Esko JD, editor. Essentials of glycobiology. Harbor: Cold Spring; p. 597–609.
- Varki A, Schauer R. 2009. Sialic acids. In: Varki A, Cummings RD, Esko JD, et al., editor. Essentials of glycobiology. Chapter 14. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press. https://www.ncbi.nlm.nih.gov/books/NBK1920/.
- Varki A, Schnaar RL, Schauer R. 2017. Sialic acids and other nonulosonic acids. In: Varki A, Cummings RD, Esko JD, editor. Essentials of glycobiology. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; p. 2015–2017. Available from: https://www.ncbi.nlm.nih.gov/books/NBK453082/. doi:10.1101/glycobiology.3e.015
- Vasta GR, Feng C, González-Montalbán N. 2017. Functions of galectins as ‘self/non-self’-recognition and effector factors. Pathog Dis. 75(5):ftx046. doi:10.1093/femspd/ftx046.
- Veerman AJ, Hogeman PH, Huismans DR, Van Zantwijk CH, Bezemer PD. 1985. Peanut agglutinin, a marker for T-cell acute lymphoblastic leukemia with a good prognosis. Cancer Res. 45(4):1890–1893.
- Vergez F, Green AS, Tamburini J. 2011. High levels of CD34+CD38low/-CD123+blasts are predictive of an adverse outcome in acute myeloid leukemia: a Groupe Ouest-Est des Leucemies Aigues et Maladies du Sang (GOELAMS) study. Haematologica. 96(12):1792–1798. doi:10.3324/haematol.2011.047894.
- Vimr ER, Kalivoda KA, Deszo EL, Steenbergen SM. 2004. Diversity of microbial sialic acid metabolism. Microbiol Mol Biol Rev. 68(1):132–153. doi:10.1128/MMBR.68.1.132-153.2004.
- Visser EA, Moons SJ, Timmermans SBPE, de Jong H, Boltje TJ, Büll C. 2021. Sialic acid O-acetylation: from biosynthesis to roles in health and disease. J Biol Chem. 297(2):100906. doi:10.1016/j.jbc.2021.100906.
- Vlad AM, Muller S, Cudic M. 2002. Complex carbohydrates are not removed during processing of glycoproteins by dendritic cells: processing of tumor antigen MUC1 glycopeptides for presentation to major histocompatibility complex class II-restricted T cells. J Exp Med. 196(11):1435–1446. doi:10.1084/jem.20020493.
- Vollmers HP, Brändlein S. 2009. Natural antibodies and cancer. N Biotechnol. 25(5):294–298. doi:10.1016/j.nbt.2009.03.016.
- von Itzstein M. 2007. The war against influenza: discovery and development of sialidase inhibitors. Nat Rev Drug Discov. 6(12):967–974. doi:10.1038/nrd2400.
- Vorup-Jensen T, Petersen SV, Hansen AG. 2000. Distinct pathways of mannan-binding lectin (MBL)- and C1-complex autoactivation revealed by reconstitution of MBL with recombinant MBL-associated serine protease-2. J Immunol. 165(4):2093–2100. doi:10.4049/jimmunol.165.4.2093.
- Vorup-Jensen T, Sørensen ES, Jensen UB. 2001. Recombinant expression of human mannan-binding lectin. Int Immunopharmacol. 1(4):677–687. doi:10.1016/s1567-5769(00)00052-7.
- Waldman AD, Fritz JM, Lenardo MJ. 2020. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 20(11):651–668. doi:10.1038/s41577-020-0306-5.
- Walter RB. 2014. The role of CD33 as therapeutic target in acute myeloid leukemia. Expert Opin Ther Targets. 18(7):715–718. doi:10.1517/14728222.2014.909413.
- Walter RB, Gooley TA, van der Velden VH. 2007. CD33 expression and P-glycoprotein-mediated drug efflux inversely correlate and predict clinical outcome in patients with acute myeloid leukemia treated with gemtuzumab ozogamicin monotherapy. Blood. 109(10):4168–4170. doi:10.1182/blood-2006-09-047399.
- Walz G, Aruffo A, Kolanus W, Bevilacqua M, Seed B. 1990. Recognition by ELAM-1 of the sialyl-Lex determinant on myeloid and tumor cells. Science. 250(4984):1132–1135. doi:10.1126/science.1701275.
- Wang B, Yang C, Jin X. 2020. Regulation of antibody-mediated complement-dependent cytotoxicity by modulating the intrinsic affinity and binding valency of IgG for target antigen. MAbs. 12(1):1690959. doi:10.1080/19420862.2019.1690959.
- Wang D, Wu L, Liu X. 2017. Glycan markers as potential immunological targets in circulating tumor cells. Adv Exp Med Biol. 994:275–284. doi:10.1007/978-3-319-55947-6_15.
- Wang DY, Johnson DB, Davis EJ. 2018. Toxicities associated With PD-1/PD-L1 blockade. Cancer J. 24(1):36–40. doi:10.1097/PPO.0000000000000296.
- Wang QS, Wang Y, Lv HY. 2015b. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. 23(1):184–191. doi:10.1038/mt.2014.164.
- Wang W, Li Q, Wu J. 2021. Lentil lectin derived from Lens culinaris exhibit broad antiviral activities against SARS-CoV-2 variants. Emerg Microbes Infect. 10(1):1519–1529. doi:10.1080/22221751.2021.1957720.
- Wang Y, Fukuda T, Isaji T. 2015a. Loss of α1,6-fucosyltransferase inhibits chemical-induced hepatocellular carcinoma and tumorigenesis by down-regulating several cell signaling pathways. FASEB J. 29(8):3217–3227. doi:10.1096/fj.15-270710.
- Wang Z, Wang Z, Li B, Wang S, Chen T, Ye Z. 2019. Innate immune cells: a potential and promising cell population for treating osteosarcoma. Front Immunol. 10:1114. doi:10.3389/fimmu.2019.01114.
- Watanabe Y, Bowden TA, Wilson IA, Crispin M. 2019. Exploitation of glycosylation in enveloped virus pathobiology. Biochim Biophys Acta Gen Subj. 1863(10):1480–1497. doi:10.1016/j.bbagen.2019.05.012.
- Wdowiak K, Francuz T, Gallego-Colon E. 2018. Galectin targeted therapy in oncology: current knowledge and perspectives. Int J Mol Sci. 19(1):210. doi:10.3390/ijms19010210.
- Weiner GJ. 2010. Rituximab: mechanism of action. Semin Hematol. 47(2):115–123. doi:10.1053/j.seminhematol.2010.01.011.
- Weiner LM, Surana R, Wang S. 2010. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 10(5):317–327. doi:10.1038/nri2744.
- Werther JL, Tatematsu M, Klein R. 1996. Sialosyl-Tn antigen as a marker of gastric cancer progression: an international study. Int J Cancer. 69(3):193–199. doi:10.1002/(SICI)1097-0215(19960621)69:3<193::AID-IJC8>3.0.CO;2-V.
- Wherry EJ, Kurachi M. 2015. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 15(8):486–499. doi:10.1038/nri3862.
- Wielgat P, Rogowski K, Niemirowicz-Laskowska K, Car H. 2020. Sialic acid-Siglec axis as molecular checkpoints targeting of immune system: smart players in pathology and conventional therapy. Int J Mol Sci. 21(12):4361. doi:10.3390/ijms21124361.
- Wiersma VR, de Bruyn M, Helfrich W, Bremer E. 2013. Therapeutic potential of galectin-9 in human disease. Med Res Rev. 33(Suppl. 1):E102–E126. doi:10.1002/med.20249.
- Willasch AM, Salzmann-Manrique E, Krenn T. 2017. Treatment of relapse after allogeneic stem cell transplantation in children and adolescents with ALL: the Frankfurt experience. Bone Marrow Transplant. 52(2):201–208. doi:10.1038/bmt.2016.224.
- Winkler IG, Barbier V, Nowlan B. 2012. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat Med. 18(11):1651–1657. doi:10.1038/nm.2969.
- Wolchok J. 2018. Putting the immunologic brakes on cancer. Cell. 175(6):1452–1454. doi:10.1016/j.cell.2018.11.006.
- Wong HK, Wilson AJ, Gibson HM. 2006. Increased expression of CTLA-4 in malignant T-cells from patients with mycosis fungoides – cutaneous T cell lymphoma. J Invest Dermatol. 126(1):212–219. doi:10.1038/sj.jid.5700029.
- Wu SC, Paul A, Ho A. 2021. Generation and use of recombinant galectins. Curr Protoc. 1(3):e63. doi:10.1002/cpz1.63.
- Xerri L, Devilard E, Hassoun J, Olive D, Birg F. 1997. In vivo expression of the CTLA4 inhibitory receptor in malignant and reactive cells from human lymphomas. J Pathol. 183(2):182–187. doi:10.1002/(SICI)1096-9896(199710)183:2<182::AID-PATH918>3.0.CO;2-I.
- Xie P, Shen YF, Shi YP. 2008. Overexpression of glucosylceramide synthase in associated with multidrug resistance of leukemia cells. Leuk Res. 32(3):475–480. doi:10.1016/j.leukres.2007.07.006.
- Xu Y, Zhang P, Zhang K, Huang C. 2021. The application of CA72-4 in the diagnosis, prognosis, and treatment of gastric cancer. Biochim Biophys Acta Rev Cancer. 1876(2):188634. doi:10.1016/j.bbcan.2021.188634.
- Yamada K, Mitsui Y, Kakoi N, Kinoshita M, Hayakawa T, Kakehi K. 2012. One-pot characterization of cancer cells by the analysis of mucin-type glycans and glycosaminoglycans. Anal Biochem. 421(2):595–606. doi:10.1016/j.ab.2011.12.017.
- Yamaguchi K, Shiozaki K, Moriya S. 2012. Reduced susceptibility to colitis-associated colon carcinogenesis in mice lacking plasma membrane-associated sialidase. PLoS One. 7(7):e41132. doi:10.1371/journal.pone.0041132.
- Yamashiro S, Ruan S, Furukawa K. 1993. Genetic and enzymatic basis for the differential expression of GM2 and GD2 gangliosides in human cancer cell lines. Cancer Res. 53(22):5395–5400.
- Yamazaki T, Galluzzi L. 2017. Blinatumomab bridges the gap between leukemia and immunity. Oncoimmunology. 6(11):e1358335. doi:10.1080/2162402X.2017.1358335.
- Yan X, Lin Y, Liu S, Aziz F, Yan Q. 2015. Fucosyltransferase IV (FUT4) as an effective biomarker for the diagnosis of breast cancer. Biomed Pharmacother. 70:299–304. doi:10.1016/j.biopha.2014.12.048.
- Yanada M, Konuma T, Yamasaki S. 2021. Relapse of acute myeloid leukemia after allogeneic hematopoietic cell transplantation: clinical features and outcomes. Bone Marrow Transplant. 56(5):1126–1133. doi:10.1038/s41409-020-01163-z.
- Yanada M, Takami A, Yamasaki S. 2020. Allogeneic hematopoietic cell transplantation for adults with acute myeloid leukemia conducted in Japan during the past quarter century. Ann Hematol. 99(6):1351–1360. doi:10.1007/s00277-020-04051-0.
- Yang JM, Byrd JC, Siddiki BB. 1994. Alterations of O-glycan biosynthesis in human colon cancer tissues. Glycobiology. 4(6):873–884. doi:10.1093/glycob/4.6.873.
- Yang WH, Nussbaum C, Grewal PK, Marth JD, Sperandio M. 2012. Coordinated roles of ST3Gal-VI and ST3Gal-IV sialyltransferases in the synthesis of selectin ligands. Blood. 120(5):1015–1026. doi:10.1182/blood-2012-04-424366.
- Yao Y, Li F, Huang J, Jin J, Wang H. 2021. Leukemia stem cell-bone marrow microenvironment interplay in acute myeloid leukemia development. Exp Hematol Oncol. 10(1):39. doi:10.1186/s40164-021-00233-2.
- Yeh SC, Wang PY, Lou YW. 2016. Glycolipid GD3 and GD3 synthase are key drivers for glioblastoma stem cells and tumorigenicity. Proc Natl Acad Sci USA. 113(20):5592–5597. doi:10.1073/pnas.1604721113.
- Yi B, Zhang M, Schwartz-Albiez R, Cao Y. 2011. Mechanisms of the apoptosis induced by CD176 antibody in human leukemic cells. Int J Oncol. 38(6):1565–1573. doi:10.3892/ijo.2011.992.
- Yoneyama T, Ohyama C, Hatakeyama S. 2014. Measurement of aberrant glycosylation of prostate specific antigen can improve specificity in early detection of prostate cancer. Biochem Biophys Res Commun. 448(4):390–396. doi:10.1016/j.bbrc.2014.04.107.
- Yoshida S, Fukumoto S, Kawaguchi H, Sato S, Ueda R, Furukawa K. 2001. Ganglioside G(D2) in small cell lung cancer cell lines: enhancement of cell proliferation and mediation of apoptosis. Cancer Res. 61(10):4244–4252.
- Yoshida S, Kawaguchi H, Sato S, Ueda R, Furukawa K. 2002. An anti-GD2 monoclonal antibody enhances apoptotic effects of anti-cancer drugs against small cell lung cancer cells via JNK (c-Jun terminal kinase) activation. Jpn J Cancer Res. 93(7):816–824. doi:10.1111/j.1349-7006.2002.tb01324.x.
- Yurkiewicz IR, Muffly L, Liedtke M. 2018. Inotuzumab ozogamicin: a CD22 mAb-drug conjugate for adult relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Drug Des Devel Ther. 12:2293–2300. doi:10.2147/DDDT.S150317.
- Zajonc DM, Kronenberg M. 2009. Carbohydrate specificity of the recognition of diverse glycolipids by natural killer T cells. Immunol Rev. 230(1):188–200. doi:10.1111/j.1600-065X.2009.00802.x.
- Zell S, Geis N, Rutz R, Schultz S, Giese T, Kirschfink M. 2007. Down-regulation of CD55 and CD46 expression by anti-sense phosphorothioate oligonucleotides (S-ODNs) sensitizes tumour cells to complement attack. Clin Exp Immunol. 150(3):576–584. doi:10.1111/j.1365-2249.2007.03507.x.
- Zhan L, Chen L, Chen Z. 2018. Knockdown of FUT3 disrupts the proliferation, migration, tumorigenesis and TGF-β induced EMT in pancreatic cancer cells. Oncol Lett. 16(1):924–930. doi:10.3892/ol.2018.8738.
- Zhang D, Zhang G, Hayden MS. 2004. A toll-like receptor that prevents infection by uropathogenic bacteria. Science. 303(5663):1522–1526. doi:10.1126/science.1094351.
- Zhang W, Peumans WJ, Barre A. 2000. Isolation and characterization of a jacalin-related mannose-binding lectin from salt-stressed rice (Oryza sativa) plants. Planta. 210(6):970–978. doi:10.1007/s004250050705.
- Zhang Z, Liu S, Zhang B, Qiao L, Zhang Y, Zhang Y. 2020. T cell dysfunction and exhaustion in cancer. Front Cell Dev Biol. 8:17. doi:10.3389/fcell.2020.00017.
- Zhang Z, Wuhrer M, Holst S. 2018. Serum sialylation changes in cancer. Glycoconj J. 35(2):139–160. doi:10.1007/s10719-018-9820-0.
- Zhao J, Simeone DM, Heidt D, Anderson MA, Lubman DM. 2006. Comparative serum glycoproteomics using lectin selected sialic acid glycoproteins with mass spectrometric analysis: application to pancreatic cancer serum. J Proteome Res. 5(7):1792–1802. doi:10.1021/pr060034r.
- Zhao X, Singh S, Pardoux C. 2010. Targeting C-type lectin-like molecule-1 for antibody-mediated immunotherapy in acute myeloid leukemia. Haematologica. 95(1):71–78. doi:10.3324/haematol.2009.009811.
- Zheng B, Yu SF, Del Rosario G. 2019b. An anti-CLL-1 antibody-drug conjugate for the treatment of acute myeloid leukemia. Clin Cancer Res. 25(4):1358–1368. doi:10.1158/1078-0432.CCR-18-0333.
- Zheng Y, Feng W, Wang YJ, Sun Y, Shi G, Yu Q. 2019a. Galectins as potential emerging key targets in different types of leukemia. Eur J Pharmacol. 844:73–78. doi:10.1016/j.ejphar.2018.11.019.
- Zhou H, Li Y, Liu B. 2017. Downregulation of miR-224 and let-7i contribute to cell survival and chemoresistance in chronic myeloid leukemia cells by regulating ST3GAL IV expression. Gene. 626:106–118. doi:10.1016/j.gene.2017.05.030.
- Zhou X, Hu W, Qin X. 2008. The role of complement in the mechanism of action of rituximab for B-cell lymphoma: implications for therapy. Oncologist. 13(9):954–966. doi:10.1634/theoncologist.2008-0089.
- Ziller F, Macor P, Bulla R, Sblattero D, Marzari R, Tedesco F. 2005. Controlling complement resistance in cancer by using human monoclonal antibodies that neutralize complement-regulatory proteins CD55 and CD59. Eur J Immunol. 35(7):2175–2183. doi:10.1002/eji.200425920.
- Zwaan CM, Reinhardt D, Jürgens H. 2003. Gemtuzumab ozogamicin in pediatric CD33-positive acute lymphoblastic leukemia: first clinical experiences and relation with cellular sensitivity to single agent calicheamicin. Leukemia. 17(2):468–470. doi:10.1038/sj.leu.2402749.
- Zwaan CM, Reinhardt D, Zimmerman M. 2010. Salvage treatment for children with refractory first or second relapse of acute myeloid leukaemia with gemtuzumab ozogamicin: results of a phase II study. Br J Haematol. 148(5):768–776. doi:10.1111/j.1365-2141.2009.08011.x.