
ABSTRACT
Extracorporeal membrane oxygenation (ECMO) might be the last resort for some patients with cardiac or cardiorespiratory failure. Although ECMO has improved survival rates, it is associated with neurocognitive complications post-recovery. Neurocognitive complications, including verbal, learning, and memory deficits, decrease quality of life and increase burden on society and families. Enormous evidence has proven that neuronal plasticity of the central nervous system accounts for neurocognitive complications. However, cellular and molecular mechanisms underlying neuronal plasticity in ECMO remain unclear. Systemic inflammatory response syndrome (SIRS) is inevitable after ECMO therapy. SIRS in cardiopulmonary bypass (CPB) has been proven to be associated with nerve injury. Moreover, inflammatory mediators mediate the activation of microglia, astroglia, and other cells that affect synaptic plasticity. This review discusses the presumptive effects of upregulated inflammatory factors and activation in non-neuronal cells on neuronal structural and functional plasticity in ECMO. Additionally, the review improves our understanding of these neuronal mechanisms and identifies potential targets that can enhance the ability of prophylaxis and treatment for neurocognitive complications after ECMO therapy.
Introduction
Extracorporeal membrane oxygenation (ECMO) is an artificial circuit system that provides respiratory and cardiac support to patients whose heart and lungs are unable to provide an adequate amount of gas exchange or perfusion to sustain life. This reversible pathophysiological process allows time for resolution of the disease, either spontaneously or through interventions, such as drug therapy and operative treatment. In this process, deoxygenated venous blood is drained into a membrane oxygenator, oxygenated, and returned into venous or arterial circulation (Barton et al. Citation2019). With the aid of ECMO, patients can survive for several days without palpitations and breathlessness. Therefore, ECMO seems to be the last alternative for life support in such patients. After more than 40 years of development, ECMO has been greatly improved with regards to implementation methods and management strategies. Since the early 1990s, ECMO has been widely used with improved survival rates (Tian et al. Citation2017).
After ECMO therapy, associated neurocognitive complications have been reported to occur in approximately 41% of patients (Graber et al. Citation2015). Moreover, patients who undergo ECMO may develop some mental disorders, including anxiety and depression, after therapy (de la Torre Citation2012). These neurocognitive complications, which result from neuronal injuries due to ECMO, hinder the body's recuperative ability. Additionally, more than 50% of patients who develop such complications after ECMO therapy are unable to engage in economic activities, increasing the economic burden on the family and society (Short Citation2005). Therefore, clinicians should pay attention to possible ECMO-associated neurocognitive complications. Changes in neuronal plasticity after cardiac surgery might account for neurocognitive complications (Kapoor Citation2020). However, the cellular and molecular mechanisms underlying the changes in neuronal plasticity and neurocognitive complications after ECMO remain unclear. To develop better treatment strategies for patients with neurocognitive complications after ECMO therapy, the putative central mechanism of neuronal plasticity is discussed in this review.
Systemic inflammatory response syndrome (SIRS) in ECMO
The development of ECMO was based on the concept of cardiopulmonary bypass (CPB) (Millar et al. Citation2016). Several studies have previously demonstrated that CPB-induced neurocognitive dysfunctions are associated with SIRS and ischemia-reperfusion injury (Stinnett et al. Citation2017). During the therapeutic process, blood moves in a cyclical manner. Therefore, ischemia-reperfusion injury does not occur in ECMO. Further improvements in ECMO, such as pump and circuit design, were used to diminish the degree of SIRS. The innate immune system is activated when blood is exposed to the non-endothelial surface.
Activation of the complement and contact systems, intrinsic and extrinsic coagulation pathways, platelets, neutrophils, endothelium-derived pro-inflammatory factors, and chemokines in SIRS leads to dysfunction of the blood–brain barrier (BBB) (Millar et al. Citation2016). As a result, increased permeability of the BBB promotes or assists movement of innate immune cells, such as neutrophils, pro-inflammatory factors, and chemokines, into the brain parenchyma (Figure ). Neurocognitive functions are impaired in lipopolysaccharide (LPS)-induced systemic inflammation, increasing BBB permeability and promoting immune cell infiltration and inflammatory molecule transfer into brain parenchyma (Audoy-Remus et al. Citation2008). SIRS has been hypothesized to develop by ECMO, and eventually cause neurocognitive complications. Neuronal function and structure changes, neurogenesis, and degeneration during SIRS underlie the development of neurocognitive defects (Beyer et al. Citation2020; Dominguez-Rivas et al. Citation2021).
Figure 1. Systemic inflammatory response syndrome (SIRS) in extracorporeal membrane oxygenation (ECMO). The schematic diagram of major equipment used in ECMO therapy is shown on the left. The centrifugal pump and oxygenator are the important components of ECMO. Drained venous blood (blue arrows) is oxygenated in the oxygenator. Thereafter, oxygenated blood (red arrows) returns for arterial circulation. As blood components and biomaterials interact, the activated contact system promotes the production of inflammatory factors and cytokines and the activation of platelets, endothelium, and neutrophils. A mass of inflammatory factors, cytokines, and activated neutrophils migrate into the brain parenchyma with increase in permeability and structural damage of the blood-brain barrier (BBB). Thereafter, under SIRS conditions, neuronal plasticity of the CNS due to heterogenous substances or activated neuroglia cells in the brain parenchyma account for the development of neurocognitive complications after ECMO.
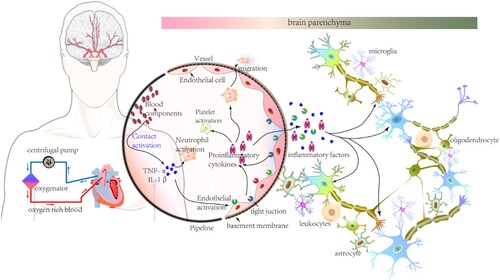
Blood–brain barrier dysfunction
The BBB consists of endothelial cells (ECs), basal lamina, and pericytes. ECs, which are linked with tight junction proteins and surrounded by the basal lamina, act as the major interface of blood. Pericytes surrounding the basal lamina possess smooth muscle-like properties. The basal lamina and pericytes are surrounded by the endfeet of astrocytes. Additionally, neuronal axons and resting microglia are found close to the surface of astrocytic endfeet (Figure ). The BBB is the barrier between interstitial fluid and blood. Inflammatory cascades are activated when blood interacts with foreign surfaces during ECMO. Upregulated complement-related proteins enhance the permeability of vascular tissues and facilitate inflammatory factor release during ECMO (Ricklin and Lambris Citation2013). Chemokines, which are overexpressed by activated microglia, astrocytes, and injured neurons increase BBB permeability and alter tight junction proteins (Bajetto et al. Citation2001; Lai and Todd Citation2006; Amantea et al. Citation2009). Astrocytic endfeet, which provide a strong association between ECs and pericytes, are disrupted by basal lamina degradation (del Zoppo Citation2010). Additionally, under inflammatory conditions in the central nervous system (CNS), ligands released from activated microglia, which bind to receptors located in ECs, increase BBB permeability further (Uehara and Uehara Citation2011; Gunduz et al. Citation2017). As a result, released inflammatory factors increase BBB permeability. The damaged BBB facilitates the migration of immune cells and potentially toxic molecules, resulting in the activation of microglia and astroglia. Activation of glial cells, such as M1 type microglia, releases inflammatory factors that impair neurocognitive functions, including spatial learning ability and memory (Hein et al. Citation2010; Alam et al. Citation2018). This complex relationship between BBB dysfunction and activation of neuroglial cells might exist under SIRS conditions after ECMO therapy.
Synaptic plasticity
Synaptic plasticity, including long-term potentiation (LTP) and spinal structural integrity, underlies the cellular mechanism of cognition (Nishijima et al. Citation2016). Neuroglial cells are essential for the maintenance of neuronal structure and function (Santello et al. Citation2019). Under pathological conditions, the role of neuroglial cells in synapses seems to change dramatically (Santello et al. Citation2019). Over-activated neuroglial cells, such as microglia and astrocytes, enhance the degree of inflammation (Liddelow et al. Citation2017). Inflammation occurring in central regions contributes to neurocognitive decline (Feng et al. Citation2017; Zhu et al. Citation2017). Cognitive decline is associated with synaptic abnormalities (Xu et al. Citation2013). Under SIRS conditions, changes in neuronal plasticity may contribute to neurocognitive complications after ECMO therapy.
Decrease in dendritic spines
Dendritic spines, which are thin-necked short protrusions from dendrites, are essential to neuronal plasticity. They are actin-rich dynamic structures that are adjusted for synaptic plasticity (Dunaevsky et al. Citation1999; Matus Citation2000). The structural and functional relationships between dendritic spines and synaptic plasticity was discovered by the induction of LTP or long-term depression (LTD) (Lang et al. Citation2004; Okamoto et al. Citation2004; Zhou et al. Citation2004). In the brain, the cognitive process, consisting of perception, emotion, and volition, requires the coordinated activity of several neurons in space and time (Lamme Citation2003; Sakamoto et al. Citation2008). Indeed, thousands of spines in a neuron can detect synchronous firings from several neurons scattered in different parts of the brain (Tanaka et al. Citation2008). Different spatial regions are gathered together by spines in one neuron with synchronous motilities at one time. These spines have been reported to perform critical roles in learning, memory formation, and cognition (Kasai et al. Citation2010).
During immune response in the CNS, a decrease in learning-dependent dendritic spines of the primary motor cortex causes deficits in multiple learning tasks in mice (Garre et al. Citation2017). These impairments of neurocognitive functions are associated with upregulated tumor necrosis factor-α (TNF-α) of activated monocytes (Garre et al. Citation2017). Therefore, downregulation of the dendritic spine contributes to neurocognitive deficits under central inflammatory response. Drebrin is exclusively resident in dendritic spines and participates excitatory input reception. Additionally, drebrin is involved in the LTP process by its effect on clustering of postsynaptic density scaffold proteins, such as postsynaptic density 95 (PSD-95) (Figure ) (Fukazawa et al. Citation2003; Takahashi et al. Citation2003). The activation of transcriptional factors for drebrin expression is suppressed by arachidonic acid (AA). The expression of AA is modulated by TNF-α and IL-1β by microglia and astroglia (Calon et al. Citation2004; Ooe et al. Citation2004; Takahashi et al. Citation2006). As a result, decreased drebrin expression reduces dendritic spine density (Takahashi et al. Citation2006). Some studies have shown that cognitive deficits in patients with Alzheimer's disease are associated with decreased drebrin expression (Kobayashi et al. Citation2004; Counts et al. Citation2006; Kojima and Shirao Citation2007). Therefore, decreased dendritic spine density-induced cognitive decline might be associated with downregulation of drebrin.
Figure 2. The diagram of spine density damage under SIRS conditions after ECMO. In the box, elevated levels of major histocompatibility complex (MHC) class I molecules in astrocytes under SIRS conditions affect spine plasticity. Drebrin expression decreases when its transcriptional factors are inhibited by activation of arachidonic acid (AA). Additionally, PSD of dendrites decreases with SP reduction, resulting in a decrease in spine density. ATP, adenosine triphosphate; SP, synaptopodin; PSD, postsynaptic density; RYR, ryanodine receptors.
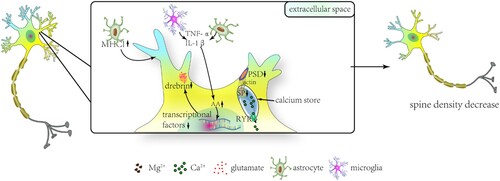
Neuroglial cells can directly influence dendritic spine plasticity. Major histocompatibility complex (MHC) class I molecules, which are found in astrocytes at a minimal degree, increase significantly in the medial prefrontal cortex (mPFC) in SIRS (Sobue et al. Citation2018). In a study, upregulated MHC class I molecules were associated with sociability and recognition memory deficits in mice by a reduction in dendritic spine density of the mPFC (Sobue et al. Citation2018). Under SIRS conditions, proteins in neurons and non-neuronal cells mediating dendritic spine structural plasticity might account for neurocognitive complications in ECMO. Synaptopodin (SP), an actin-modulating protein, is required for long-term spine stability by regulating spine Ca2+ transients and second messenger release (Mundel et al. Citation1997; Asanuma et al. Citation2005; Korkotian and Segal Citation2011). SP in hippocampal CA1 area pyramidal neurons were found to have decreased significantly under SIRS conditions (Strehl et al. Citation2014). This finding suggested that SIRS in ECMO might cause long-term neuronal plasticity through SP expression changes. The decrease in dendritic spine density during SIRS might result in neurocognitive complications after ECMO therapy (Figure ).
Long-term potentiation deficit
Glial cells are considered as the third element of synapses in the central nervous system. Cytokines, produced by glial cells, participate in inflammatory responses of the nervous system. Although these cytokines are expressed at low levels, even undetected, in the healthy brain (Rothwell and Luheshi Citation2000; Allan et al. Citation2005), they are necessary for the regulation of glial-neuronal connection with bidirectional communication and synaptic plasticity effects (Bezzi et al. Citation1998; Schneider et al. Citation1998; Bezzi et al. Citation2001; Avital et al. Citation2003).
Under SIRS conditions, cytokines and inflammatory factors affect LTP by regulating transmitter release and receptor excitability. Under pathological conditions, high levels of interleukin-1 (IL-1) are released from astrocytes, glial cells, neurons, and vascular endothelial cells (Figure ). Studies have shown that IL-1 stimulates the activities of both c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (p38MAPK). These activated kinases might inhibit glutamate release and lead to LTP deficit (Vereker et al. Citation2000). Additionally, IL-1 promotes the expression of inflammation mediators, such as IL-6, nitric oxide (NO), and prostaglandin E2 (PGE2), through the activation of MAPK and nuclear factor-kappa B (NF-κB) signaling pathways (Di Filippo et al. Citation2008). Using cerebral cortex slices of rats, the inhibitory effect of IL-6 on glutamate release was discovered (D'Arcangelo et al. Citation2000). The inhibitory effect of IL-6 on LTP might be related to the MAPK/ERK pathway (Tancredi et al. Citation2000). Furthermore, NO and PGE2 affect the induction of LTP directly by increasing GABA levels and decreasing Ca2+ influx, respectively, during neuronal inflammation (Chen et al. Citation2002; Calabrese et al. Citation2007; Di Filippo et al. Citation2008). In the hippocampus, IL-18, a member of the IL-1 family, impairs the induction of LTP by modulating metabotropic glutamate receptors (mGluRs) negatively (Cumiskey et al. Citation2007). This effect of IL-18 is associated with downstream cyclooxygenase 2 and inducible nitric oxide synthase (iNOS) (Cumiskey et al. Citation2007). As described above, IL-1 is necessary for LTP induction but disturbs neurotransmitter release and impairs LTP at a higher concentration under SIRS conditions (Figure ). Notably, other cytokines have been shown to contribute to LTP formation physiologically but, at higher levels, defect LTP under SIRS conditions.
Figure 3. The imaginary model for LTP defect in SIRS after ECMO. In the normal state (black lines), glutamate released from presynaptic neurons acts on a-amino-3-hydroxy-5-methyl-isoxazole-4-propionic acid receptors (AMPARs), leading to an excitatory depolarization of the postsynaptic membrane. Under activity-induced depolarization (such as high-frequency stimulation in experiments), voltage-dependent magnesium (Mg2+) blockage for N-methyl-D-aspartate receptors (NMDARs) is removed. Calcium ion (Ca2+) influx occurs through NMDARs, metabotropic glutamate receptors (mGLuRs), and muscarinic receptors (MRs) and activates several signal transduction systems, including a-calcium–calmodulin-dependent protein kinase II (CaMKII), mitogen-activated protein kinase (MAPK), and extracellular signal-regulated kinase (ERK), which increase the expression of AMPARs in the membrane and activation of nuclear transcription factors (NTFs) in the nucleus. Therefore, the long-term potentiation of glutamate-mediated neurotransmission occurs. Under SIRS conditions (red lines), glutamate is released, and signaling pathways through NMDARs, MRs, and AMPARs are inhibited by inflammatory factors and cytokines (such as IL-1 and ATP), which are released from neurons and neuroglial cells. Additionally, IL-1 promotes the production of IL-6, NO, and PGE2 through the MAPK and NF-κB pathways. NO and PGE2 can directly influence the induction of LTP. Reactive oxygen species (ROS) can influence LTP directly or indirectly by activating JNK and p38. Moreover, Ca2+ influx through M1R is modulated by GABA. The decreased SP in SIRS weakens the connection between PSD and SP, reducing the release of Ca2+ through RYR. The decrease in intracellular free Ca2+ levels suppresses the activity of kinases (such as ERK), which is required for LTP. This results in LTP deficit (red lines). ATP, adenosine triphosphate; IL-1, interleukin-1; IL-6, interleukin-6; PGE2, prostaglandin E2; MAPKs, mitogen-activated protein kinases; NO, nitric oxide; JNK, c-Jun N-terminal kinase; GABA, γ-aminobutyric acid; SP, synaptopodin; PSD, postsynaptic density; RYR, ryanodine receptors; M1R, muscarinic receptor 1.
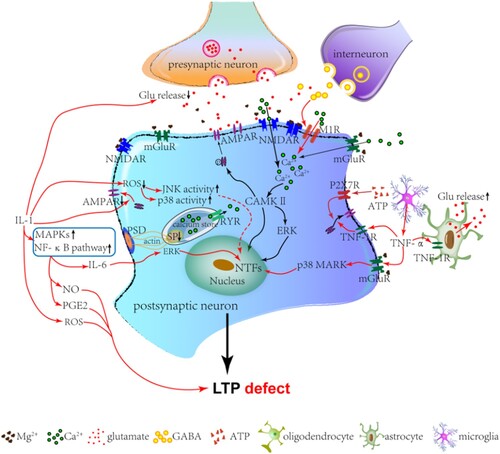
Glutamate has been established to primarily trigger LTP formation through a-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (AMPARs). Trafficking of AMPARs was thought to contribute to synaptic plasticity (Malinow and Malenka Citation2002). Surface levels and function of AMPARs are modulated by inflammatory factors (Figure ). In the physiological state, cytokines, such as tumor necrosis factor-α (TNF-α), are produced by neurons and glia (Beattie et al. Citation2002). TNF-α facilitates synaptic plasticity by increasing surface AMPARs through TNF-1 receptors (Beattie et al. Citation2002). However, TNF-α, which is produced by microglia, exhibits an inhibitory effect on LTP through the group I metabotropic glutamate receptor (mGluRI) and p38 MAPK during the inflammatory response (Butler et al. Citation2004; Cumiskey et al. Citation2007). Otherwise, extracellular free adenosine triphosphate (ATP) connected with the postsynaptic P2X7 receptor (an ionotropic ATP-gated receptor) modulates the recruitment of surface AMPARs (Man et al. Citation2003; Opazo et al. Citation2003).
Except for the effect of IL-1 on neurotransmitter release, the sensitized process of NMDARs is facilitated by IL-1 with the upregulation of tyrosine phosphorylation (Viviani et al. Citation2006). Thereafter, NMDAR-mediated current is magnified by IL-1 in a dose-dependent manner (Yang et al. Citation2005). Additionally, the recruitment of AMPARs to the membrane surface is influenced by IL-1 (Lai et al. Citation2006). During inflammation of the CNS, the impairment of hippocampal LTP causes a selective decrease in NMDAR NR2B subunit levels and an increase in IL-1β levels (Di Filippo et al. Citation2013). Therefore, IL-1β modulates hippocampal LTP by reducing the number of NMDARs. As shown above, the neurotransmitter system and receptor activity are both affected by SIRS.
Conclusion and future perspectives
Over the past four decades, ECMO has become an important treatment for salvaging patients with serious respiratory and cardiac diseases. With the advancement of the ECMO technology, survival of several patients has improved. However, the high incidence rate of neurocognitive complications after ECMO hinders recovery. Therefore, clinicians should attach importance to understanding the neuronal mechanism underlying neurocognitive complications occurring after ECMO therapy. Few putative mechanisms account for neurocognitive complications after ECMO. In SIRS, neuronal synaptic plasticity might be associated with neurocognitive complications after ECMO. Several cellular mechanisms underlying structural and functional changes causing neurocognitive complications during SIRS remain unclear. These hypotheses on mechanisms underlying neurocognitive complications under SIRS conditions after ECMO therapy will be investigated further. New therapeutic targets for treating neurocognitive complications after ECMO will be found. Therefore, clinical interventions, such as novel medicines, will be needed to prevent SIRS and decrease the incidence of neurocognitive complications after ECMO therapy.
Contributors
Ying Zhang: organization, literature review, and execution of the first draft of manuscript. Ming Zhang: review and critique of the manuscript; preparing paper’s figures.
Acknowledgment
The authors would like to appreciate the Servier Medical Art (https://smart.servier.com) for providing diagrams of the heart and the brain arteries in the Figure .
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Additional information
Funding
References
- Alam A, Hana Z, Jin Z, Suen KC, Ma D. 2018. Surgery, neuroinflammation and cognitive impairment. EBioMedicine. 37(2018):547–556.
- Allan SM, Tyrrell PJ, Rothwell NJ. 2005. Interleukin-1 and neuronal injury. Nat Rev Immunol. 5(8):629–640.
- Amantea D, Nappi G, Bernardi G, Bagetta G, Corasaniti MT. 2009. Post-ischemic brain damage: pathophysiology and role of inflammatory mediators. FEBS J. 276(1):13–26.
- Asanuma K, Kim K, Oh J, Giardino L, Chabanis S, Faul C, Reiser J, Mundel P. 2005. Synaptopodin regulates the actin-bundling activity of α-actinin in an isoform-specific manner. J Clin Invest. 115(5):1188–1198.
- Audoy-Remus J, Richard JF, Soulet D, Zhou H, Kubes P, Vallieres L. 2008. Rod-Shaped monocytes patrol the brain vasculature and give rise to perivascular macrophages under the influence of proinflammatory cytokines and angiopoietin-2. J Neurosci. 28(41):10187–10199.
- Avital A, Goshen I, Kamsler A, Segal M, Iverfeldt K, Richter-Levin G, Yirmiya R. 2003. Impaired interleukin-1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus. 13(7):826–834.
- Bajetto A, Bonavia R, Barbero S, Florio T, Schettini G. 2001. Chemokines and their receptors in the central nervous system. Front Neuroendocrinol. 22(3):147–184.
- Barton R, Ignjatovic V, Monagle P. 2019. Anticoagulation during ECMO in neonatal and paediatric patients. Thromb Res. 173:172–177.
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. 2002. Control of synaptic strength by glial TNFα. Science. 295(5563):2282–2285.
- Beyer MMS, Lonnemann N, Remus A, Latz E, Heneka MT, Korte M. 2020. Enduring changes in neuronal function upon systemic inflammation Are NLRP3 inflammasome dependent. J Neurosci. 40(28):5480–5494.
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A. 1998. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 391(6664):281–285.
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A. 2001. CXCR4-activated astrocyte glutamate release via TNFα: amplification by microglia triggers neurotoxicity. Nat Neurosci. 4(7):702–710.
- Butler MP, O'Connor JJ, Moynagh PN. 2004. Dissection of tumor-necrosis factor-α inhibition of long-term potentiation (LTP) reveals a p38 mitogen-activated protein kinase-dependent mechanism which maps to early—but not late—phase LTP. Neuroscience. 124(2):319–326.
- Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM. 2007. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 8(10):766–775.
- Calon F, Lim GP, Yang F, Morihara T, Teter B, Ubeda O, Rostaing P, Triller A, Salem N, Ashe J, et al. 2004. Docosahexaenoic acid protects from dendritic pathology in an Alzheimer's disease mouse model. Neuron. 43(5):633–645.
- Chen C, Magee JC, Bazan NG. 2002. Cyclooxygenase-2 regulates prostaglandin E2 signaling in hippocampal long-term synaptic plasticity. J Neurophysiol. 87(6):2851–2857.
- Counts SE, Nadeem M, Lad SP, Wuu J, Mufson EJ. 2006. Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J Neuropathol Exp Neurol. 65(6):592–601.
- Cumiskey D, Butler MP, Moynagh PN, O'Connor JJ. 2007. Evidence for a role for the group I metabotropic glutamate receptor in the inhibitory effect of tumor necrosis factor-α on long-term potentiation. Brain Res. 1136(1):13–19.
- Cumiskey D, Pickering M, O'Connor JJ. 2007. Interleukin-18 mediated inhibition of LTP in the rat dentate gyrus is attenuated in the presence of mGluR antagonists. Neurosci Lett. 412(3):206–210.
- D'Arcangelo G, Tancredi V, Onofri F, D'Antuono M, Giovedi S, Benfenati F. 2000. Interleukin-6 inhibits neurotransmitter release and the spread of excitation in the rat cerebral cortex. Eur J Neurosci. 12(4):1241–1252.
- de la Torre JC. 2012. Cardiovascular risk factors promote brain hypoperfusion leading to cognitive decline and dementia. Cardiovasc Psychiatry Neurol. 2012(2012):367516.
- del Zoppo GJ. 2010. The neurovascular unit in the setting of stroke. J Intern Med. 267(2):156–171.
- Di Filippo M, Chiasserini D, Gardoni F, Viviani B, Tozzi A, Giampa C, Costa C, Tantucci M, Zianni E, Boraso M, et al. 2013. Effects of central and peripheral inflammation on hippocampal synaptic plasticity. Neurobiol Dis. 52(2013):229–236.
- Di Filippo M, Sarchielli P, Picconi B, Calabresi P. 2008. Neuroinflammation and synaptic plasticity: theoretical basis for a novel, immune-centred, therapeutic approach to neurological disorders. Trends Pharmacol Sci. 29(8):402–412.
- Dominguez-Rivas E, Avila-Munoz E, Schwarzacher SW, Zepeda A. 2021. Adult hippocampal neurogenesis in the context of lipopolysaccharide-induced neuroinflammation: A molecular, cellular and behavioral review. Brain Behav Immun. 97:286–302.
- Dunaevsky A, Tashiro A, Majewska A, Mason C, Yuste R. 1999. Developmental regulation of spine motility in the mammalian central nervous system. Proc Natl Acad Sci USA. 96(23):13438–13443.
- Feng X, Valdearcos M, Uchida Y, Lutrin D, Maze M, Koliwad SK. 2017. Microglia mediate postoperative hippocampal inflammation and cognitive decline in mice. JCI Insight. 2(7):e91229.
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. 2003. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 38(3):447–460.
- Garre JM, Silva HM, Lafaille JJ, Yang G. 2017. CX3CR1+ monocytes modulate learning and learning-dependent dendritic spine remodeling via TNF-α. Nat Med. 23(6):714–722.
- Graber LC, Quillinan N, Marrotte EJ, McDonagh DL, Bartels K. 2015. Neurocognitive outcomes after extracorporeal membrane oxygenation. Best Pract Res Clin Anaesthesiol. 29(2):125–135.
- Gunduz D, Tanislav C, Schluter KD, Schulz R, Hamm C, Aslam M. 2017. Effect of ticagrelor on endothelial calcium signalling and barrier function. Thromb Haemostasis. 117(2):371–381.
- Hein AM, Stasko MR, Matousek SB, Scott-McKean JJ, Maier SF, Olschowka JA, Costa AC, O'Banion MK. 2010. Sustained hippocampal IL-1β overexpression impairs contextual and spatial memory in transgenic mice. Brain Behav Immun. 24(2):243–253.
- Kapoor MC. 2020.. Neurological dysfunction after cardiac surgery and cardiac intensive care admission: A narrative review part 2: Cognitive dysfunction after critical illness; potential contributors in surgery and intensive care; pathogenesis; and therapies to prevent/treat perioperative neurological dysfunction. Ann Card Anaesth. 23(4):391–400.
- Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Noguchi J. 2010. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 33(3):121–129.
- Kobayashi R, Sekino Y, Shirao T, Tanaka S, Ogura T, Inada K, Saji M. 2004. Antisense knockdown of drebrin A, a dendritic spine protein, causes stronger preference, impaired pre-pulse inhibition, and an increased sensitivity to psychostimulant. Neurosci Res. 49(2):205–217.
- Kojima N, Shirao T. 2007. Synaptic dysfunction and disruption of postsynaptic drebrin-actin complex: a study of neurological disorders accompanied by cognitive deficits. Neurosci Res. 58(1):1–5.
- Korkotian E, Segal M. 2011. Synaptopodin regulates release of calcium from stores in dendritic spines of cultured hippocampal neurons. J Physiol. 589(Pt 24):5987–5995.
- Lai AY, Swayze RD, El-Husseini A, Song C. 2006. Interleukin-1 beta modulates AMPA receptor expression and phosphorylation in hippocampal neurons. J Neuroimmunol. 175(1-2):97–106.
- Lai AY, Todd KG. 2006. Microglia in cerebral ischemia: molecular actions and interactions. This paper is one of a selection of papers published in this Special Issue, entitled Young Investigator's Forum. Can J Physiol Pharmacol. 84(1):49–59.
- Lamme VA. 2003. Why visual attention and awareness are different. Trends Cogn Sci. 7(1):12–18.
- Lang C, Barco A, Zablow L, Kandel ER, Siegelbaum SA, Zakharenko SS. 2004. Transient expansion of synaptically connected dendritic spines upon induction of hippocampal long-term potentiation. Proc Natl Acad Sci USA. 101(47):16665–16670.
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, et al. 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 541(7638):481–487.
- Malinow R, Malenka RC. 2002. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 25(1):103–126.
- Man HY, Wang Q, Lu WY, Ju W, Ahmadian G, Liu L, D'Souza S, Wong TP, Taghibiglou C, Lu J, et al. 2003. Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron. 38(4):611–624.
- Matus A. 2000. Actin-based plasticity in dendritic spines. Science. 290(5492):754–758.
- Millar JE, Fanning JP, McDonald CI, McAuley DF, Fraser JF. 2016. The inflammatory response to extracorporeal membrane oxygenation (ECMO): a review of the pathophysiology. Crit Care. 20(1):387.
- Mundel P, Heid HW, Mundel TM, Kruger M, Reiser J, Kriz W. 1997. Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes. J Cell Biol. 139(1):193–204.
- Nishijima T, Torres-Aleman I, Soya H. 2016. Progress in brain research. Prog Brain Res. 225:243–268.
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. 2004. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 7(10):1104–1112.
- Ooe N, Saito K, Mikami N, Nakatuka I, Kaneko H. 2004. Identification of a novel basic helix-loop-helix-PAS factor, NXF, reveals a Sim2 competitive, positive regulatory role in dendritic-cytoskeleton modulator drebrin gene expression. Mol Cell Biol. 24(2):608–616.
- Opazo P, Watabe AM, Grant SG, O'Dell TJ. 2003. Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J Neurosci. 23(9):3679–3688.
- Ricklin D, Lambris JD. 2013. Complement in immune and inflammatory disorders: pathophysiological mechanisms. The Journal of Immunology. 190(8):3831–3838.
- Rothwell NJ, Luheshi GN. 2000. Interleukin 1 in the brain: biology, pathology and therapeutic target. Trends Neurosci. 23(12):618–625.
- Sakamoto K, Mushiake H, Saito N, Aihara K, Yano M, Tanji J. 2008. Discharge synchrony during the transition of behavioral goal representations encoded by discharge rates of prefrontal neurons. Cereb Cortex. 18(9):2036–2045.
- Santello M, Toni N, Volterra A. 2019. Astrocyte function from information processing to cognition and cognitive impairment. Nat Neurosci. 22(2):154–166.
- Schneider H, Pitossi F, Balschun D, Wagner A, del Rey A, Besedovsky HO. 1998. A neuromodulatory role of interleukin-1β in the hippocampus. Proc Natl Acad Sci USA. 95(13):7778–7783.
- Short BL. 2005. The effect of extracorporeal life support on the brain: a focus on ECMO. Semin Perinatol. 29(1):45–50.
- Sobue A, Ito N, Nagai T, Shan W, Hada K, Nakajima A, Murakami Y, Mouri A, Yamamoto Y, Nabeshima T, et al. 2018. Astroglial major histocompatibility complex class I following immune activation leads to behavioral and neuropathological changes. Glia. 66(5):1034–1052.
- Stinnett GR, Lin S, Korotcov AV, Korotcova L, Morton PD, Ramachandra SD, Pham A, Kumar S, Agematsu K, Zurakowski D, et al. 2017. Microstructural alterations and oligodendrocyte dysmaturation in white matter after cardiopulmonary bypass in a juvenile porcine model. J Am Heart Assoc. 6(8):1–27.
- Strehl A, Lenz M, Itsekson-Hayosh Z, Becker D, Chapman J, Deller T, Maggio N, Vlachos A. 2014. Systemic inflammation is associated with a reduction in Synaptopodin expression in the mouse hippocampus. Exp Neurol. 261:230–235.
- Takahashi H, Mizui T, Shirao T. 2006. Down-regulation of drebrin A expression suppresses synaptic targeting of NMDA receptors in developing hippocampal neurones. J Neurochem. 97(Suppl 1):110–115.
- Takahashi H, Sekino Y, Tanaka S, Mizui T, Kishi S, Shirao T. 2003. Drebrin-dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density-95 and dendritic spine morphogenesis. J Neurosci. 23(16):6586–6595.
- Tanaka J, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GC, Kasai H. 2008. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science. 319(5870):1683–1687.
- Tancredi V, D'Antuono M, Cafe C, Giovedi S, Bue MC, D'Arcangelo G, Onofri F, Benfenati F. 2000. The inhibitory effects of interleukin-6 on synaptic plasticity in the rat hippocampus are associated with an inhibition of mitogen-activated protein kinase ERK. J Neurochem. 75(2):634–643.
- Tian F, Morriss C, Chalak L, Venkataraman R, Ahn C, Liu H, Raman L. 2017. Impairment of cerebral autoregulation in pediatric extracorporeal membrane oxygenation associated with neuroimaging abnormalities. Neurophotonics. 4(4):01.
- Uehara K, Uehara A. 2011. P2Y1, P2Y6, and P2Y12 receptors in rat splenic sinus endothelial cells: an immunohistochemical and ultrastructural study. Histochem Cell Biol. 136(5):557–567.
- Vereker E, O'Donnell E, Lynch MA. 2000. The inhibitory effect of interleukin-1β on long-term potentiation Is coupled with increased activity of stress-activated protein kinases. J Neurosci. 20(18):6811–6819.
- Viviani B, Gardoni F, Bartesaghi S, Corsini E, Facchi A, Galli CL, Di Luca M, Marinovich M. 2006. Interleukin-1β released by gp120 drives neural death through tyrosine phosphorylation and trafficking of NMDA receptors. J Biol Chem. 281(40):30212–30222.
- Xu X, An L, Mi X, Zhang T. 2013. Impairment of cognitive function and synaptic plasticity associated with alteration of information flow in theta and gamma oscillations in melamine-treated rats. PLoS One. 8(10):e77796.
- Yang S, Liu ZW, Wen L, Qiao HF, Zhou WX, Zhang YX. 2005. Interleukin-1β enhances NMDA receptor-mediated current but inhibits excitatory synaptic transmission. Brain Res. 1034(1-2):172–179.
- Zhou Q, Homma KJ, Poo MM. 2004. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 44(5):749–757.
- Zhu S, Wang J, Zhang Y, He J, Kong J, Wang JF, Li XM. 2017. The role of neuroinflammation and amyloid in cognitive impairment in an APP/PS1 transgenic mouse model of Alzheimer's disease. CNS Neurosci Ther. 23(4):310–320.