
Abstract
Hypertrophic cardiomyopathy (HCM) is an inherited heart disease characterized by left ventricular hypertrophy. Although sarcomeric gene mutations can explain many HCM cases, the genetic basis of approximately half of HCM cases remains elusive. Here, we report the case of a 30-year-old woman with HCM after imaging and pathological examinations. We identified a novel mutation in a pathogenic gene from the HCM patient through whole-exome sequencing. A heterozygous missense mutation in exon 1 of the α-tropomyosin (TPM1) gene (NM_000366.5:exon1:c.71A > G) was found based on patient sequencing data. The mutation resulted in glutamine replacing arginine (p.Gln24Arg). This mutation was expected to cause significant and deleterious changes in the structure of the TPM1 protein. Therefore, according to pathogenicity assessment guidelines, the mutation was considered likely pathogenic. We found that most of these mutations are related to cardiomyopathy and majority of them are in exon 1 and are missense mutations. TPM1 mutations are thus a common cause of HCM and other congenital heart defects. Thus, a novel missense variant, p.Gln24Arg, in the TPM1 gene is associated with the autosomal dominant disease HCM. These results expand the knowledge spectrum of TPM1 gene function.
1. Introduction
Hypertrophic cardiomyopathy (HCM) is an inherited heart disease characterized by left ventricular hypertrophy. It is associated with multiple potential clinical outcomes, including impaired diastolic function, heart failure, and sudden cardiac death (SCD) (Tuohy et al. Citation2020). HCM is rare in the pediatric population, but is present in more than 1 in 500 in the general adult population, which is higher than that of any other single-gene-associated heart disease (Marian, Citation2021; Sabater-Molina et al. Citation2018; Maron and Maron Citation2013). It is also one of the most common causes of SCD in young athletes (de Noronha et al. Citation2009; Maron et al. Citation2009).
Recently, whole-exome sequencing (WES) has been increasingly used to detect the genetic basis of diseases through the sequencing of exomes that encode proteins. Genetic screening for very rare Mendelian diseases is an accurate and cost-effective method. In addition, compared with the traditional sequencing methods, prior assumptions related to the cause of the disease are not needed for WES (Petrovski et al. Citation2019). Especially in the cardiovascular field, many congenital heart diseases can be diagnosed through WES (Kalayinia et al. Citation2018).
In this study, WES was performed to identify mutations associated with HCM in a patient. The proband was a middle-aged woman. The patient was diagnosed with HCM one year prior and was admitted to the hospital for the first time due to chest tightness and chest pain during exercise. A heterozygous missense mutation was found in TPM1 (OMIM: 191010) based on patient sequencing data. Accordingly, genetic testing could help determine the genetic cause of HCM, provide accurate genetic counseling for patients with HCM in the future, and expand the spectrum of mutations that cause HCM for further research on its pathogenesis.
2. Materials and methods
2.1. Subjects
This study was reviewed and approved by the Ethics Committee of the Second Xiangya Hospital of Central South University. Written informed consent was obtained from the patient for the publication of the details. Blood was obtained from the affected proband. The subject was studied based on a cardiology examination, thorough family history, medical record review, electrocardiogram, echocardiography, and magnetic resonance imaging (MRI).
2.2. DNA extraction
Genomic DNA was extracted from the peripheral blood lymphocytes of the patient. Genomic DNA was prepared using a DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA).
2.3. Whole-exome sequencing
The main processes for WES were performed by the Berrygenomics Bioinformatics Institute (Beijing, China). The exomes were captured using Agilent SureSelect Human All Exon V6 kits, and the high-throughput sequencing platform was the Illumina HiSeq X-10. The basic bioinformatics analyses, including reads, mapping, variant detection, filtering, and annotation, were also performed by the Berrygenomics Bioinformatics Institute. Quality control is an essential step and is applied to guarantee a meaningful downstream analysis. The data processing steps were as follows: (1) discard paired reads if either read contains adapter contamination (>10 nucleotides aligned to the adapter, allowing ≤10% mismatches); (2) discard paired reads if more than 10% of bases are uncertain in either read; (3) discard paired reads if the proportion of low quality (Phred quality <5) bases is greater than 50% in either read. Verita Trekker was used to perform variant calling and identify SNPs and InDels.
2.4. Mutation validation
The mutation in the proband was validated using Sanger sequencing. The primer pair was designed by Integrated DNA Technologies (IDT) according to the input TPM1 genome sequence, and the polymerase chain reaction product was sequenced using an ABI 3100 Genetic Analyzer (ABI, Foster City, CA).
2.5. Multiple sequence alignment analysis
The sequences of the TPM1 protein from different species were download from Ensemble (https://asia.ensembl.org/index.html). Multiple sequence analysis was performed using ClustalX.
2.6. Model construction of target protein
The three-dimensional (3D) structure of TPM1 was calculated and designed after obtaining the amino acid sequence from the Uniprot Knowledge Database (ID: P09493). Therefore, a homology modeling method was used to predict the 3D structure of the wild-type and mutant TPM1 structures. Structural prediction of TPM1 was based on the automatic Swiss modeling method (https://swissmodel.expasy.org/) (Waterhouse et al. Citation2018).
2.7. Functional enrichment analysis of genes related to TPM1 in HCM
To clarify the gene function, we used the HitPredict online tool (http://www.hitpredict.org/) to predict the TPM1-related interaction network (Patil et al. Citation2011). Based on Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases, the Metascape online tool (https://metascape.org/) was used to perform functional enrichment analysis of the TPM1 genes mutated in HCM (Zhou et al. Citation2019).
2.8. Analysis of mutations in the TPM1 gene
The TPM1 gene mutation sites and reports related to the gene are compiled in a table in Supporting File 1, based on information as of August 20th, 2021. The transcript used for our statistical analysis was NM_001018005.2. We performed different types of analyses on the information related to this gene mutation.
3. Results
3.1. Case report
The proband was a 30-year-old female diagnosed with HCM at the age of 29. She had asymmetrical hypertrophy of the interventricular septum, with a maximum septum thickness of 20 mm. For 3 years, she had been experiencing chest tightness and chest pain during exercise, and this situation became increasingly serious in the year prior. Her mother was healthy, and her father had probably suffered from cardiomyopathy and died suddenly at 48 years of age. We considered that her father had also mostly likely died of hypertrophic cardiomyopathy.
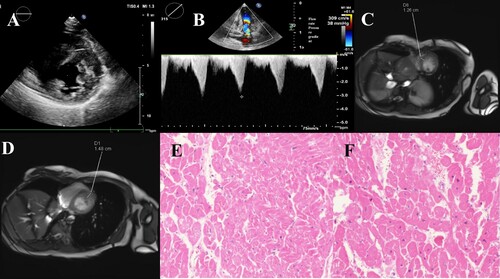
Echocardiography revealed hypertrophy of the anterior wall of the entire left ventricle. Local irregular echoes could be seen in the myocardium, and the middle part of the ventricular septum was 15 mm thick (Figure (a,b)). The electrocardiogram showed sinus bradycardia with arrhythmia, abnormal QS waves in multiple leads, and right ventricular hypertrophy (Supporting File 2). MRI revealed that the anterior wall thickness of the basal segment of the left ventricle was 14.8 mm. The thickness of the interventricular septum was 12.6 mm. The anterior and inferior walls of the left ventricle had thickened asymmetrically (Figure (c,d)). A pathological tissue biopsy revealed hypertrophy of myocardial cells, multifocal fibrous hyperplasia in the myocardium, and a small amount of adipose tissue (Figure (e,f)).
Figure 1. Heart imaging picture. A, B. Echocardiography revealed that the left anterior ventricular wall was mainly hypertrophy. C, D. MRI showed that the anterior wall thickness of the basal segment of the left ventricle was 14.8 mm, and the thickness of the interventricular septum was 12.6 mm. The anterior and inferior walls of the left ventricle are thickened asymmetrically. E, F. Pathological tissue biopsy revealed hypertrophy of myocardial cells, multifocal fibrous hyperplasia in the myocardium, and a small amount of adipose tissue.
3.2. Genetic analysis
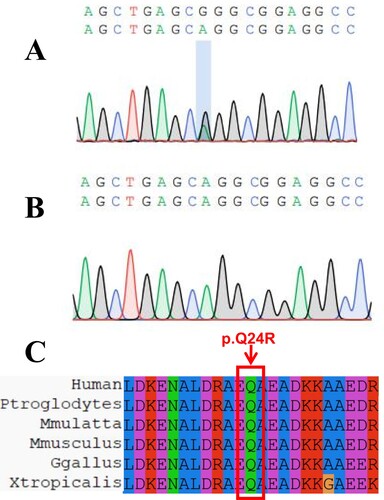
Results revealed 12,627,881,202 total clean bases, 86,481,966 clean reads, and 81,854,501 mapped reads (94.65%). Coverage of the target region was 97.05%. Total sequences on target comprised 5590.48 Mb. Average sequencing depth of the target was 92.47. We used WES to analyze patients and discovered a new mutation in the TPM1 gene. The mutation reference sequence depth was 62, the alt sequence depth was 29, the total depth was 91, and the alt rate was 0.32. Among the sequencing results, only TPM1 was found to be related to cardiomyopathy, and other genes related to cardiomyopathy were not found. NM_000366.5:exon1:c.71A > G was determined to be located in exon 1 of TPM1, in chr15:63,335,099 (genomic version: hg19), and its transcript is NM_000366.5. The mutation was determined to cause the replacement of arginine with glutamine. The mutation in the proband was further validated by performing Sanger sequencing (Figure (a,b)).
Figure 2. Sanger sequencing results and homology comparison. A. Sanger sequencing result of the proband. B. the result of normal human Sanger sequencing. C. Homology alignment of missense mutation sites
3.3. Pathogenicity assessment of mutations
According to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines, we evaluated the pathogenicity of this mutation. First, the mutation in TPM1 was verified to cause HCM. Then, we confirmed that the mutation was located in an important region of the Pleckstrin homology domain using the SMART (Simple Modular Architecture Research Tool) database. At the same time, the mutation was found in the region of chr5:63,334,766-63,335,432, which was determined to be a variation heatmap for mutations in the dbSNP database. Moreover, this mutation was not found in the normal control population using the gnomAD, 1000 g, and ExAC databases. Further, this mutation was predicted to be pathogenic based on the four commonly used databases for genetic mutation prediction (CADD, SIFT, Polyphen2, and MutationTaster). Finally, most variants on TPM1 were missense variants. We chose PM1, PM2, PP2, and PP3 to take the evidence to the evaluation level. Therefore, we determined that the mutation as likely pathogenic.
3.4. Multiple sequence alignment analysis
ClustalX analysis showed that glutamine and other amino acids in this domain are highly conserved in nature, and any mutations in this region might have a profound impact on the phenotypes of individuals (Figure (c)).
3.5. TPM1 protein structure analysis
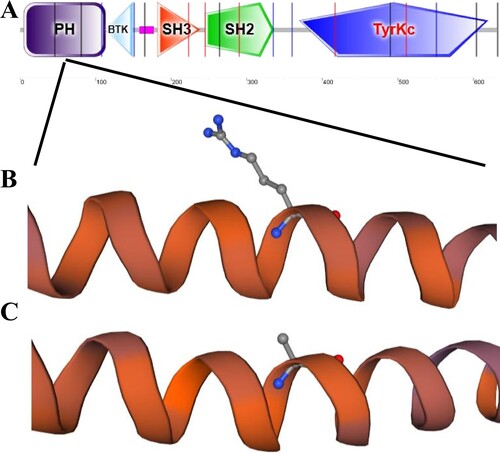
The template (7ko4.1.Q) was selected to have sequence identity (93.66%) to construct the model. Protein mutations were determined to lead to instability of the protein structure, which might cause abnormalities in patient. In the TPM1 protein, there was a difference in the sequence between wild-type and mutant proteins. Moreover, this region was located in the key region of the TPM1 protein (Figure ).
Figure 3. Simulation of TPM1 protein important region and prediction of 3D structure. A. the region where the mutation site is located. B. Prediction of the 3D structure of the protein after mutation in the proband. C. Prediction of the 3D structure of the protein in the control group.
3.6. Functional enrichment analysis of genes related to TPM1 in HCM
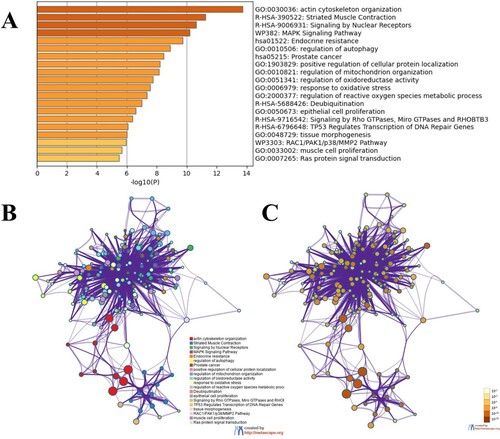
GO analysis showed that the functionally enriched terms of TPM1-related interacting genes were mainly associated with actin cytoskeleton organization. In addition, KEGG pathway analysis showed that most TPM1-related interacting genes are mainly involved in the biological processes of striated muscle contraction (Figure ).
Figure 4. Function and pathway enrichment analysis. A. GO function enrichment analysis of TPM1 related interaction genes. B, C. Enrichment analysis of KEGG pathway related to TPM1 interacting genes.
3.7. Summary and analysis of mutations in the TPM1 gene
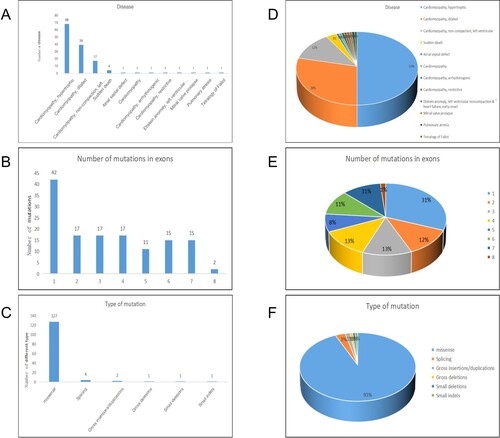
In total, 136 mutation reports related to TPM1 were identified. According to the classification of the disease type, 50% (68/136) of these indicated that the gene is related to HCM. Second, approximately 28.7% (39/136) suggested that this gene is associated with dilated cardiomyopathy, and the third most common classification was left ventricular insufficiency, accounting for 12.5% (39/136) of reports. Other diseases accounted for a small proportion. According to the exon classification of TPM1 gene mutation sites, most were located in exon 1, accounting for 30.9% (42/136). The ratio of mutations in exons 2–7, but not exon 8, was relatively similar owing to the shorter sequence. According to a classification of mutation types, approximately 93.4% (127/136) of mutation types were missense mutations, and only a few were splicing mutations and InDels (Figure ).
Figure 5. Summary and analysis of all mutations. A, D are classified according to the type of disease, the number and proportion of mutations in different diseases. B, E are classified according to the exons where the mutation sites are located, and the number and proportion of mutations in different exons. C, F are classified by mutation type, the number and proportion of mutations of different mutation types.
4. Discussion
HCM is a global disease that is considered one of the most common genetic heart diseases (Maron and Maron Citation2013). The prevalence of HCM in China is 0.08% (Zou et al. Citation2004). In clinical practice, the diagnosis of HCM is suggested based on electrocardiographic changes and is based on the finding of increased wall thickness of the left ventricle through echocardiography or cardiovascular MRI. When a myocardial biopsy is performed, enlarged myocytes with bizarre shapes and pleiotropic nuclei are observed, with the normal parallel alignment of myocytes being lost. For the diagnosis of cardiomyopathy, the etiology is very important. Endomyocardial biopsy is an important means to determine the etiology. Previous studies have shown that endomyocardial biopsy can provide an etiological diagnosis for a variety of cardiomyopathies (Tian et al. Citation2010). However, many functional changes in HCM, including changes in calcium circulation and sensitivity, disturbances in pressure perception, and impaired cardiac energy homeostasis, are not clinically detectable (Elliott et al. Citation2014; Hensley et al. Citation2015; Maron Citation2018; Maron and Maron Citation2013). In addition, because HCM shows age-related penetrance, many mutation carriers do not have typical symptoms or left ventricular hypertrophy, based on imaging findings, early in life. Therefore, genetic testing is useful for screening susceptible relatives of families affected by HCM. Moreover, genetic testing represents a major advancement in the understanding of the pathogenesis of HCM (Sabater-Molina et al. Citation2018).
HCM is inherited in an autosomal dominant pattern and exhibits marked genetic heterogeneity. At least 12 pathogenic genes have been identified to date. Most of these encode cardiac sarcomeric proteins, such as β-myosin heavy chain (MYH7), myosin binding protein C (MYBPC3), cardiac troponin T (TNNT2), troponin I (TNNI3), cardiac α-actin (ACTC), and α-tropomyosin (TPM1). Therefore, it is important to identify new mutations and genes that cause HCM (Marian, Citation2021; Maron et al. Citation2012; Sabater-Molina et al. Citation2018; Teekakirikul et al. Citation2019). It is also important to provide detection, treatment, and prevention strategies for HCM patients and families in the future.
TPM1 mutations account for approximately 3% of HCM cases (Nakajima-Taniguchi et al. Citation1995; Watkins et al. Citation1995). The TPM1 gene is considered one of the common causes of HCM, dilated cardiomyopathy, left ventricular dysfunction, and congenital heart disease (Marian, Citation2021). TPM1 is essential for normal heart development and contractile functions (England et al. Citation2017). There is also evidence that familial hypertrophic cardiomyopathy 3 (CMH3, Phenotype MIM number: 115196) is caused by a heterozygous mutation in TPM1 on chromosome 15q22 (Thierfelder et al. Citation1993). Our summary and analysis suggest that most mutations in the TPM1 gene are related to cardiomyopathy. At the same time, most mutation sites were determined to occur in exon 1, which indicates that this region is a hotspot for mutations. In addition, most mutations were found to be missense mutations, indicating that majority of the mutations that cause disease in this gene are point mutations. This also shows that TPM1 is very important, and most point mutations are sufficient to change traits. The collected information is crucial for future research on disease pathogenicity and diagnosis, as well as the mechanism underlying the function of the TPM1 gene. The patients reported in this study are also included in the highest proportion of the three classification criteria.
GO functional enrichment analysis showed that the TPM1-related interacting genes were mainly related to the organization of actin cytoskeleton. In addition, the KEGG pathway analysis showed that associated pathways were mainly involved in the biological process of striated muscle contraction. GO and KEGG enrichment analysis can be used to infer the pathogenesis when generating animal models harboring the TPM1 point mutation in the future, to identify the pathogenic mechanisms associated with the TPM1 point mutation.
TPM1 is a highly conserved and widely distributed actin-binding protein and member of the tropomyosin family (England et al. Citation2017). Further, it is involved in the muscle contraction system and the cytoskeleton of non-muscle cells. A parallel double-stranded α-helical coiled-coil protein, which binds to the long groove of actin, is essential for the regulation and stability of actin (Brown et al. Citation2005). Its function is also related to the troponin complex, which responds to Ca2+ signals (Jagatheesan et al. Citation2010). In recent years, it has been discovered that a variety of TPM1 gene mutation sites are related to HCM (Carlus et al. Citation2020).
In this study, the mutation was not found in ExAC, gnomAD, and 1000 g databases; missense mutations occurred in exon regions of the genome, and the four tools used to predict the pathogenicity of mutations considered it a pathogenic mutation. After screening and genetic pattern analysis, only the mutation Gln24Arg in the TPM1 gene was identified as the HCM-causing mutation. Moreover, after being assessed according to the ACMG pathogenicity criteria, it was considered likely pathogenic.
In summary, a novel missense mutation in the TPM1 gene (NM_000366.5:exon1:c.71A > G: p.Q24R) was identified as a likely pathogenic mutation in the Chinese patient with HCM using WES. This study expands the spectrum of TPM1 mutations that cause HCM. This could be used to provide appropriate genetic counseling for mutation carriers who are still not clinically affected.
Abbreviations
Hypertrophic cardiomyopathy (HCM), whole-exome sequencing (WES); Magnetic Resonance Imaging (MRI); American College of Medical Genetics and Genomics (ACMG); Gene Ontology (GO); Kyoto Encyclopedia of Genes and Genomes (KEGG); three-dimensional (3D)
Conflicts of interest
The authors declare there is no conflict of interest.
Consent for publication
Written informed consent for publication of their details was obtained from the patient.
Author contributions
Ke Gong, Qin Wu: conception and design, analysis and interpretation of the data, the drafting of the paper, the final approval of the version to be published; Ting Xie, Yong Luo, Hui Guo, Zhiping Tan, Jinlan Chen, Yifeng Yang, Li Xie: conception and design, revising it critically for intellectual content, the final approval of the version to be published. And that all authors agree to be accountable for all aspects of the work.
Acknowledgements
We would like to acknowledge our patient.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability
Additional supporting data is not publicly available to protect patient privacy.
Additional information
Funding
References
- Brown JH, Zhou Z, Reshetnikova L, Robinson H, Yammani RD, Tobacman LS, Cohen C. 2005. Structure of the mid-region of tropomyosin: bending and binding sites for actin. Proc Natl Acad Sci U S A. 102(52):18878–18883. doi:10.1073/pnas.0509269102.
- Carlus SJ, Almuzaini IS, Karthikeyan M, Loganathan L, Al-Harbi GS, Carlus FH, Al-Mazroea AH, Morsy MM, Abo-Haded HM, Abdallah AM, Al-Harbi KM. 2020. A novel homozygous TPM1 mutation in familial pediatric hypertrophic cardiomyopathy and in silico screening of potential targeting drugs. Eur Rev Med Pharmacol Sci. 24(14):7732–7744. doi:10.26355/eurrev_202007_22299.
- de Noronha SV, Sharma S, Papadakis M, Desai S, Whyte G, Sheppard MN. 2009. Aetiology of sudden cardiac death in athletes in the United Kingdom: a pathological study. Heart. 95(17):1409–1414. doi:10.1136/hrt.2009.168369.
- Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, et al. 2014. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. Eur Heart J. 35(39):2733–2779. doi:10.1093/eurheartj/ehu284.
- England J, Granados-Riveron J, Polo-Parada L, Kuriakose D, Moore C, Brook JD, Rutland CS, Setchfield K, Gell C, Ghosh TK, et al. 2017. Tropomyosin 1: Multiple roles in the developing heart and in the formation of congenital heart defects. J Mol Cell Cardiol. 106:1–13. doi:10.1016/j.yjmcc.2017.03.006.
- Hensley N, Dietrich J, Nyhan D, Mitter N, Yee MS, Brady M. 2015. Hypertrophic cardiomyopathy: a review. Anesth Analg. 120(3):554–569. doi:10.1213/ANE.0000000000000538.
- Jagatheesan G, Rajan S, Ahmed RP, Petrashevskaya N, Boivin G, Arteaga GM, Tae HJ, Liggett SB, Solaro RJ, Wieczorek DF. 2010. Striated muscle tropomyosin isoforms differentially regulate cardiac performance and myofilament calcium sensitivity. J Muscle Res Cell Motil. 31(3):227–239. doi:10.1007/s10974-010-9228-3.
- Kalayinia S, Goodarzynejad H, Maleki M, Mahdieh N. 2018. Next generation sequencing applications for cardiovascular disease. Ann Med. 50(2):91–109. doi:10.1080/07853890.2017.1392595.
- Marian AJ. 2021. Molecular genetic basis of hypertrophic cardiomyopathy. Circ Res. 128(10):1533–1553. doi:10.1161/CIRCRESAHA.121.318346.
- Maron BJ. 2018. Clinical course and management of hypertrophic cardiomyopathy. N Engl J Med. 379(7):655–668. doi:10.1056/NEJMra1710575.
- Maron BJ, Doerer JJ, Haas TS, Tierney DM, Mueller FO. 2009. Sudden deaths in young competitive athletes: analysis of 1866 deaths in the United States, 1980-2006. Circulation. 119(8):1085–1092. doi:10.1161/CIRCULATIONAHA.108.804617.
- Maron BJ, Maron MS. 2013. Hypertrophic cardiomyopathy. Lancet. 381(9862):242–255. doi:10.1016/S0140-6736(12)60397-3.
- Maron BJ, Maron MS, Semsarian C. 2012. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol. 60(8):705–715. doi:10.1016/j.jacc.2012.02.068.
- Nakajima-Taniguchi C, Matsui H, Nagata S, Kishimoto T, Yamauchi-Takihara K. 1995. Novel missense mutation in α-tropomyosin gene found in Japanese patients with hypertrophic cardiomyopathy. J Mol Cell Cardiol. 27(9):2053–2058. doi:10.1016/0022-2828(95)90026-8.
- Patil A, Nakai K, Nakamura H. 2011. HitPredict: a database of quality assessed protein-protein interactions in nine species. Nucleic Acids Res. 39(Database issue):D744–D749. doi:10.1093/nar/gkq897.
- Petrovski S, Aggarwal V, Giordano JL, Stosic M, Wou K, Bier L, Spiegel E, Brennan K, Stong N, Jobanputra V, et al. 2019. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet. 393(10173):758–767. doi:10.1016/S0140-6736(18)32042-7.
- Sabater-Molina M, Perez-Sanchez I, Hernandez Del Rincon JP, Gimeno JR. 2018. Genetics of hypertrophic cardiomyopathy: A review of current state. Clin Genet. 93(1):3–14. doi:10.1111/cge.13027.
- Teekakirikul P, Zhu W, Huang HC, Fung E. 2019. Hypertrophic cardiomyopathy: an overview of genetics and management. Biomolecules. 9(12). doi:10.3390/biom9120878.
- Thierfelder L, MacRae C, Watkins H, Tomfohrde J, Williams M, McKenna W, Bohm K, Noeske G, Schlepper M, Bowcock A, et al. 1993. A familial hypertrophic cardiomyopathy locus maps to chromosome 15q2. Proc Natl Acad Sci U S A. 90(13):6270–6274. doi:10.1073/pnas.90.13.6270.
- Tian Z, Zeng Y, Cheng KA, Gao P, Zhao DC, Cui QC, Jiang XC, Chen LF, Fang Q. 2010. Importance of endomyocardial biopsy in unexplained cardiomyopathy in China: a report of 53 consecutive patients. Chin Med J. 123(7):864–870.
- Tuohy CV, Kaul S, Song HK, Nazer B, Heitner SB. 2020. Hypertrophic cardiomyopathy: the future of treatment. Eur J Heart Fail. 22(2):228–240. doi:10.1002/ejhf.1715.
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. 2018. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46(W1):W296–W303. doi:10.1093/nar/gky427.
- Watkins, H., Anan, R., Coviello, D.A., Spirito, P., Seidman, J.G., Seidman, C.E., 1995. A De novo mutation in α-tropomyosin that causes hypertrophic cardiomyopathy. Circulation 91(9), 2302-2305. doi:10.1161/01.CIR.91.9.2302.
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. 2019. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 10(1):1523. doi:10.1038/s41467-019-09234-6.
- Zou Y, Song L, Wang Z, Ma A, Liu T, Gu H, Lu S, Wu P, Zhang dagger Y, Shen dagger L, et al. 2004. Prevalence of idiopathic hypertrophic cardiomyopathy in China: a population-based echocardiographic analysis of 8080 adults. Am J Med. 116(1):14–18. doi:10.1016/j.amjmed.2003.05.009.