
Abstract
We hypothesise that antimicrobial resistance (AMR) cannot be overcome by simply ‘drugging’ single biological targets, therefore, our focus is developing broad-spectrum therapeutics. Herein we present the synthesis of two novel spacer-linked, anthraquinone-triphenylphosphonium (AQ-TPP) conjugates (4) and (5) as early lead compounds in a new class of agent designed to penetrate the lipophilic barriers of the membranes of a diverse family of species; and establish bacterial growth inhibitory properties against methicillin-resistant Staphylococcus aureus (NCTC 13616) and Mycobacterium smegmatis [mc2155] (as a surrogate for Mycobacterium tuberculosis). The MIC and MBC values of (4) and (5) were determined against MRSA and found to be equipotent [MIC for each: 1 µg/mL (1.2 µM)]. Whereas the amide-linked conjugate (5) was determined to have 2-fold greater bactericidal potency [MBC 1 µg/mL (1.2 µM] than ester-linked (4). The anthraquinone-TPP conjugates (4) and (5) were active in vitro against Mycobacterium smegmatis (graphical abstract) and were equipotent as determined by their MIC values; in contrast to MRSA, the less hydrophobic (4) had 2-fold greater bactericidal potency than (5) as measured by their MBC values. Notably, both conjugates showed potent intracellular growth inhibitory activity in infected THP-1 macrophages. The conjugates are promising leads for the development of new antibacterial drugs.
Key policy highlights
Development of a novel class of antimicrobial compounds
Drug screen against a WHO ‘high’ priority drug-resistant pathogen that poses the greatest threat to human health (methicillin-resistant Staphylococcus aureus)
Drug screen against a surrogate for Mycobacterium tuberculosis, one of the leading causes of mortality worldwide.
GRAPHICAL ABSTRACT
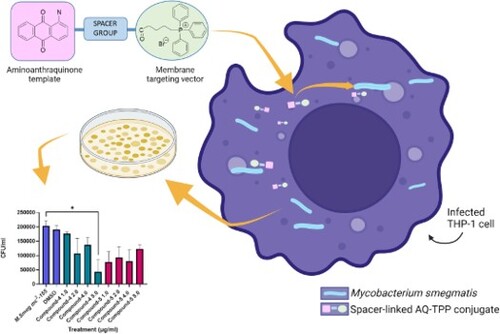
Introduction
The big problem
Less than a century after the discovery of antibiotics, antimicrobial resistance (AMR) has emerged as a significant global threat to public health. Many disease-causing bacteria are now resistant to antibiotics, with the most critical groups including multi-drug resistant ‘Super Bugs’. The WHO refers to ‘the silent pandemic of antimicrobial resistance’ with 1.27 M deaths per year due to AMR, which is expected to rise to 10M (Murray et al. Citation2022). The rate at which this problem is increasing poses potentially the greatest threat to human life and undermines the foundation of much of modern medicine (EClinicalMedicine Citation2021; Walsh et al. Citation2023), yet the paucity of drugs in the antimicrobial drug pipeline is of serious concern and urgently needs to be addressed (WHO Citation2020). Emerging from the COVID-19 pandemic has uncovered the lack of preparedness for a public health crisis on such a scale, yet this is what the world is facing if the AMR crisis is left unchallenged.
Our knowledge of the three classical mechanisms that lead to antibiotic resistance (reduction of intracellular antibiotic concentrations, modification of the antibiotic target, and inactivation of the antibiotic) has been enhanced through recent advances in the molecular basis of resistance (Darby et al. Citation2023). These include multilayered, complex, and dynamic changes such as Epistasis and Compensatory evolution where the fitness effect of a mutation can be affected by another mutation in a distant genetic location (Munita and Arias Citation2016; Hasan et al. Citation2022). A complicating factor is that strains carrying resistance mechanisms often coexist with other species in multi-species communities; responses of individual species to antibiotics can depend on resistance mechanisms circulating in their neighbours (Pathak et al. Citation2023).
Broad-spectrum therapeutics
Accepting that drug resistance is inevitable for all antibiotics, empirical observation of the cumulative data suggests that agents interacting with multiple biological targets have a lower tendency to acquire target-based resistance (Singh et al. Citation2017). The term broad-spectrum therapeutics (BSTs) has recently been introduced to describe antimicrobials with broad-spectrum activity directed at common, invariable, and essential components of different classes of microbes (National Institutes of Health Citation2011; Firth and Prathapan Citation2021). Drug repositioning for COVID-19 has certainly accelerated the move away from the ‘one drug-one target’ paradigm. There are no emergency treatments for pandemics, and everyone is at risk, should a pandemic originate from a bacterial pathogen resistant to all known treatments. The common essential component in the work we describe in this article is the cell membrane and its profitability as a BST target for new antibacterial drug design.
Common culprits
Mycobacterium tuberculosis (Mtb) is an obligate, acid-fast, intracellular bacillus, causing the human disease Tuberculosis (TB) and is a leading cause of human morbidity (WHO Citation2023a). Following the significant impact from the COVID-19 pandemic, there has been a 4.5% increase in global TB cases, up to 10.6M, with an estimated 4000 deaths per day (WHO Citation2023a). The global health community has pledged to lead the fight against this infection, which includes the development of novel compounds that can circumvent multi-drug resistance mechanisms, yet the outputs remain slow and sparse. Staphylococcus aureus is a gram-positive bacterium causing infections in many different parts of the body and is found in both community and hospital-acquired settings. The drug-resistant strain, methicillin-resistant Staphylococcus aureus (MRSA), is a global health-threatening organism and as such, a leading cause of morbidity and mortality worldwide (Hasanpour et al. Citation2023). According to the WHO, the proportion of bloodstream infections caused by MRSA nearly doubled between 2014 and 2020 (WHO Citation2023b) with the bacterium being responsible for an estimated 121,000 deaths per annum (Vos et al. Citation2020).
Structural biology as the driver of new drug design
Mitochondria evolved some billion years ago from α-proteobacteria and became endosymbionts later in eukaryotic cells, in accord with the theory proposed originally by Lynn Sagan in 1967 (Sagan Citation1967). The evolution of complex cellular systems of mitochondria has made it independent of host cells due to the presence of their own genomic material. Mitochondrial biological activities exhibit similar characteristics of bacterial ancestors of mitochondria (most evident in the connection between double-walled Gram-negative bacteria) energy production and the existence of their own DNA (Sullivan and Chandel Citation2014).
The structural and evolutionary features around the relationship between human intracellular mitochondria and their bacterial progenitors, and notably the mitochondrial membrane potential (ΔΨm) provides the basis for rational design of compounds that can negotiate the ‘waxy’ Mtb cell envelope to gain access to their bacterial target(s). This structural biology approach driving drug design has been applied to cancer therapy but has received scant attention for direct-acting antibacterial drug design. In cancer therapeutics, there is considerable interest in developing drugs to target mitochondria selectively as a potentially efficient way to eliminate cancer cells by acting at these critical cellular organelles that have central roles in cell metabolism and apoptosis (Jeena et al. Citation2020).
Our approach has been to attach a mitochondria-directing vector to a suitable functional group in experimental anthraquinone-based templates to augment intracellular drug accumulation (Figure a). The anthraquinone (anthracene-9,10-dione) ring system has provided a fruitful source of biologically active compounds; emodin (1,3,8-trihydroxy-6-methyl-anthraquinone) (Figure b) is a simple anthraquinone derivative from various plants that have attracted interest for their spectrum of biological activities; these include anticancer (Haque et al. Citation2018), anti-inflammatory (J.-W. Han et al. Citation2015) and antibacterial activities (May Zin et al. Citation2017). However, its practical usage is severely limited due to poor solubility in water and low oral bioavailability caused by extensive glucuronidation in liver and intestine glucuronidation in liver and intestine (Shia et al. Citation2010). A chloro-derivative of emodin was shown to have improved antibacterial potency against Gram-positive strains, including MRSA that was attributed in part to increased membrane permeability in addition to direct actions on DNA (Duan et al. Citation2017). Notably, the core structure of the anthracycline class of antibiotics, represented by some of the most clinically used anticancer drugs, including doxorubicin and daunorubicin, are too toxic to be applied to the treatment of bacterial infections despite sharing a common mechanism of targeting DNA- topoisomerase 2, the human equivalent of bacterial DNA-gyrase (Marinello et al. Citation2018).
Figure 1. (a) Bacteria-Targeting Concept: Spacer-linked Anthraquinone-TPP (AQ-TPP) Conjugates. (b) Anthraquinone natural product: emodin.
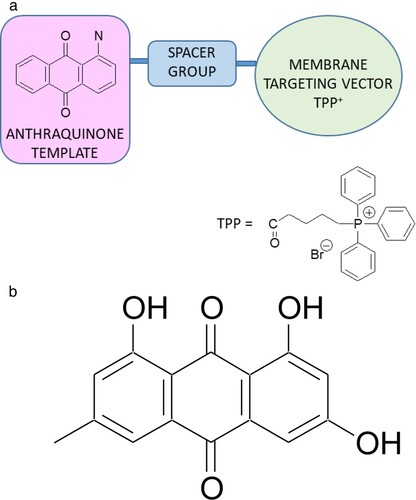
One of the most widely studied examples of a mitochondrial-targeting vector is the triphenylphosphonium (TPP) cation. TPP is a highly lipophilic, resonance-stabilised cation because of extensive delocalisation of its single positive charge over the three phenyl rings bonded to the phosphorus atom (Zielonka et al. Citation2017). The anthracycline anticancer drug doxorubicin (Dox) when conjugated to TPP was shown to selectively accumulate in breast cancer cell mitochondria of both sensitive wild-type and Dox-resistant cell lines with the potential to reverse drug resistance (Fujiwara et al. Citation1985; M. Rabbani et al. Citation2005; Han et al. Citation2014).
Mtb is a facultative intracellular pathogen with an exceptional ability to survive in nutrient-sparse environments within the human host. A key factor is the highly complex mycobacterial cell envelope that sets Mtb apart from other prokaryotic organisms. The unique make-up of the mycobacterial envelope gives a survival advantage over other bacterial species. It is a complex, physically resilient, layered structure made predominantly from sugars and lipids (Smith Citation2011). The core structure is a cross-linked peptidoglycan network covalently attached to an arabinogalactan polysaccharide that in sequence is covalently attached to a long chain, β-hydroxy fatty acids (mycolic acids); additional lipids are interspersed in the tightly packed mycolic acid layer to form what has been described as an outer ‘mycomembrane’ (Brennan Citation2003) that presents a substantial permeability barrier, preventing many molecules from reaching the cytoplasm and is a major contributor to the intrinsic resistance of mycobacteria to most antibiotics. It can be speculated that for a therapeutic agent to traverse the mycomembrane and reach the cytoplasm of Mtb, the molecule should incorporate structural features that include polar, hydrophilic and hydrophobic, lipophilic residues to reflect the components of the permeability barrier of Mtb.
Several drugs are used for treating MRSA infections (e.g. vancomycin, daptomycin and linezolid) all of which carry safety concerns (Lian et al. Citation2021), and the bacterium is resistant to the β-lactamases, one of the most prescribed drug classes. As with the cell structure of Mtb, the peptidoglycan layer is remarkably thick, and surrounds the cytoplasmic membrane.
Aims
In response to the pressing need for new agents that have antibacterial activity against drug-resistant pathogens, this work is motivated by the evolutionary relationship between mitochondria and bacteria.
The initial aim of this work was to develop a pilot series of a low molecular mass novel class of compounds by molecular hybridisation and fragment-based drug design methods, which combine two or more active scaffolds in a single structure. Specifically, the compounds incorporate cationic, lipophilic groups that promote cell uptake, that do not target the nucleus of human cells (non-genotoxicity). Subsequently, the second aim was to screen these compounds against two resistant strains of bacteria that cause serious human disease; MRSA and Mycobacterium smegmatis (a surrogate for Mtb) and obtain critical MBC and MIC data.
Methods and materials
*Abbreviations: AQ, anthraquinone; DCC, N,N′-dicyclohexylcarbodiimide; DCM, dichloromethane; DIPEA, N,N-diisopropylethylamine; DMF, N,N-dimethylformamide; DMSO, dimethylsulfoxide; HOBt, 1-hydroxybenzotriazole; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PMA, phorbol 12-myristate 13-acetate; PyBop, benzotriazole-1-yl-oxy-tris-pyrrolidinophosphonium hexafluorophosphate; TFA, trifluoroacetic acid; TPP, (4-carboxybutyl)triphenylphosphonium bromide.
Synthetic chemistry
Common solvents (including HPLC grade) were purchased from Fisher Scientific (Loughborough, UK) and used without further purification. Aluminium plates (Kieselgel 60 F254 silica plates) were used for thin-layer chromatography (TLC) analysis. Davisil Kieselgel LC60A 40–63 µm silica was used for column chromatography (Fisher Scientific; UK). All other reagents were obtained from Sigma-Aldrich Merck (Gillingham, UK) or Tokyo Chemical Industry UK Ltd, UK. Purity of compounds was assessed as > 95% by RP-HPLC on a Phenomenex Synergi Polar RP18 column [4 µM; (30 mm × 4.6 mm) with flow rate 1 mL min−1] using mobile phase 55% – ACN 45% H2O. UV-Visible detection at 520 nm, for compound (4). RP-HPLC was on a waters XBridge C18 column [3.5 µM; (50 mm × 4.6 mm) with flow rate 1.1 mL min−1] using mobile phase 70% – ACN 30% H2O. UV-Visible detection at 540 nm, for compound (5).
1H NMR spectra were recorded on a Bruker AC300 NMR spectrometer at 25°C at 300.1 MHz from samples dissolved either in deuterated DMSO or deuterated chloroform. High resolution mass spectra with electrospray ionisation (HRMS-ESI) were recorded using a ThermoFisher LTQ Orbitrap XL instrument.
Full characterisation data for all new compounds, including 1H NMR spectra; UV/visible absorption spectra; and HPLC characterisation chromatograms are presented in the accompanying Supplementary Information File.
1-((4-Hydroxybutyl)amino)anthracene-9,10-dione (2). 1-Chloroanthraquinone (1 g, 4.1 mmol) was suspended in DMSO (15 mL), 4-amino-1-butanol (3.7 g, 42 mmol) was added and the mixture was heated for 30 min over a boiling water bath. The solution was cooled and added to a large excess of water (500 mL). The precipitated solid was filtered off, dried and purified by silica-gel column chromatography [CHCl3 and EtOAc (4:1) plus MeOH (1%)]. The fractions containing the major product were combined, filtered and evaporated in vacuo to dryness to give (2) as red solid (0.7 g, 57%). 1H NMR (CDCl3 300 MHz): δ 9.78 (t, 1H, Ar-NH), 8.25 (m, 2H, H-5 and 8), 7.75 (m, 2H, H-6 and 7), 7.55 (m, 2H, H-3 and H-4), 7.06 (dd, 1H, H-2), 3.76 (t, 2H, -CH2-OH), 3.38 (m, 2H, -NH-CH2-), 1.85 (m, 4H, -NH-CH2-CH2-CH2). TLC (chloroform: methanol, 9:1 + 1% glacial acetic acid): Rf 0.5. HRMS (ESI) (+) m/z: 296.1284 (100%) [M + H]+. Calculated for [C18H17O3NH]+ 296.1281; Found 296.1284.
1-[4-(3-Piperidin-4-yl-propyl)-piperidin-1-yl]-anthraquinone trifluoroacetate (3). 1-Chloroanthraquinone (1 g, 4.1 mmol) was suspended in DMSO (50 mL), 1,3-di-4-piperidylpropane (8.67 g, 41 mmol) was added and the mixture was heated for 30 min over a boiling water bath. The solution was cooled and added to a large excess of water (500 mL). The precipitated solid was filtered off, dried and purified by silica-gel column chromatography [chloroform: methanol (3:2) plus DIPEA (0.25%)]. The fractions containing the major product were combined, filtered and evaporated, dissolved in TFA and evaporated in vacuo to dryness. The residue was triturated with diethyl ether to give compound (3) as a pale red solid (0.22 g, 10%). TLC (chloroform: methanol 4:1): Rf 0.1. HRMS (ESI) (+) m/z: 417.2527 (100%) [C27H33N2O2]+. Calculated for [C27H33N2O2]+ 417.2537. Found 417.2527.
(5-(4-((9,10-Dioxo-9,10-dihydroanthracen-1-yl)amino)butoxy)-5-oxopentyl)triphenylphosphonium (4). (4-Carboxybutyl)triphenylphosphonium bromide (0.50 g, 1.1 mmol), DCC (0.35 g, 1.70 mmol) and DMAP (10 mg) were dissolved in CH2Cl2 (10 mL) at rt for 15 min then added to 1-[(4-hydroxybutyl)amino]anthraquinone (2) (0.25 g, 0.85 mmol) in CH2Cl2 (5 mL), stirred for 1 h then washed three times with water. The organic layer was dried with Na2SO4, filtered, concentrated and applied to a silica gel chromatography column, using CH2Cl2 initially and then CH2Cl2: ethyl acetate and then CH2Cl2: ethyl acetate + 2% – 7% methanol. Fractions containing the pure product were combined, filtered and evaporated in vacuo to give (4) as a red solid (0.21 g, 39% yield). 1H NMR (DMSO-d6, 300 MHz): δ (ppm) 9.68 (t, 1H, AQ-NH, J 9 Hz), 8.10–8.21 (m, 2H, AQ-H5 and H8), 7.72–7.94 (m, 17H, HAR of AQ and TPP), 7.65 (d, 1H, AQ-H3, J 15 Hz), 7.43 (d,1H, AQ-H4, J 15 Hz), 7.26 (d,1H, AQ-H2, J 18 Hz), 4.06 (s, 2H, CH2O), 3.62 (t, 2H, CH2P, J 30 Hz), 3.36 (m, 2H, AQ-NH-CH2), 2.41 (t, 2H, CH2-C = O, J 15 Hz), 1.50–1.80 (m, 8H, NHCH2CH2CH2CH2O and OCOCH2CH2CH2CH2P). TLC (chloroform: methanol, 9:1 + 1% glacial acetic acid): Rf 0.15. HRMS (ESI) (+) m/z: 640.2600 (100%) [C41H39O4NP]+. Calculated for [C41H39NO4P]+ 640.2611; Found 640.2600. HPLC tR 4.27 min.
[5-(4-{3-[1-(9,10-Dioxo-9,10-dihydro-anthracen-1-yl)-piperidin-4-yl]-propyl}-piperidin-1-yl)-5-oxo-pentyl]-triphenyl-phosphonium bromide (5). PyBOP (0.22 g, 0.42 mmol), HOBt (0.055 g, 0.41 mmol) and 4-(carboxybutyl)triphenylphosphonium bromide (0.19 g, 0.42 mmol) were dissolved in DMF (5 mL). DIPEA (0.3 mL, 1.7 mmol) was added and the reaction mixture was left at rt for 15 min before the addition of 1-[4-(3-piperidin-4-yl-propyl)-piperidin-1-yl]-anthraquinone trifluoroacetate (3) (0.15 g, 0.28 mmol) in DMF (2 mL). The reaction mixture was left at rt for 1 h before the addition of water and extraction with chloroform. The combined organic layers were dried over Na2SO4, filtered, concentrated and applied to a silica gel chromatography column using [chloroform: ethyl acetate (4:1) plus MeOH (1%)] to give (5) as a dark purple-red solid (0.16 g, 74%). HRMS (ESI) (+) m/z: 761.3863 (100%) [C50H54N2O3P]+. Calculated for [C50H54N2O3P]+ 761.3867; Found 761.3863. HPLC tR 8.548 min.
Bacterial strains and growth conditions
Mycobacterium smegmatis mc2155 (M. smeg mc2155) was used in this study as a surrogate organism for Mtb. M. smegmatis is widely used a model system to study Mtb (Mohan et al. Citation2015; Lelovic et al. Citation2020). M. smeg mc2155 was maintained in Mueller-Hinton broth (Merck; 70192-500G) supplemented with 0.05% Tween-80 (SLS; P1754-500ML) and plated on Mueller-Hinton agar (Merck; 70191-500G). Broth cultures were incubated at 37°C (200 rpm) for 72 h and agar plates were incubated statically at 37°C for 72 h. Staphylococcus aureus (NCTC 13616) liquid cultures were maintained in Mueller-Hinton broth at 37°C (200 rpm) for 24 h and plated on Mueller-Hinton agar incubated statically at 37°C for 24 h.
Determination of minimum inhibitory concentration (MIC)
MIC was carried out using a standard broth microdilution method (Andrews Citation2001) on untreated flat bottomed 96-well microtiter plates (SLS; 3788). Synthesised compounds anthraquinone-TPP conjugates 4 and 5 were prepared at a stock concentration of 10 mg/ml in DMSO (Fisher Scientific; BP231-100) and subjected to an on-plate two-fold dilution series in Mueller-Hinton broth (256 µg/mL - 0.5 µg/mL; 2.5% - 0.004% final DMSO concentration per well). M. smegmatis inoculum was prepared by back diluting a 72 h broth culture to an OD600nm equivalent of 1 × 106 CFU/mL in Mueller-Hinton broth supplemented with 0.1% Tween-80. S. aureus inoculum was prepared by back diluting a 24 h broth culture to an OD600nm equivalent of 1 × 106 CFU/mL in un-supplemented Mueller-Hinton. The total volumes per well following the on-plate dilution of both compound and inoculum was 200 µL. Sterility, growth and DMSO toxicity controls were included in all 96-well test plates. M. smeg mc2155 microtiter plates were incubated at 37°C (150 rpm) for 72 h and S. aureus microtiter plates were incubated at 37°C (150 rpm) for 24 h. The MIC was considered the lowest compound concentration that inhibited bacterial growth after 72 h as judged by the unaided eye (EUCAST Citation2022).
Determination of minimum bactericidal concentration (MBC)
MBC was determined by spotting a 10 μL suspension from all no visible growth (synthesised compounds: anthraquinone-TPP conjugates 4 & 5) and controls wells onto Mueller-Hinton agar. Agar plates were incubated at 37°C for 72 h for M. smeg mc2155 and 37°C for 24 h for S. aureus. The MBC was determined as the lowest drug concentration exhibiting no visible growth on Mueller-Hinton agar after the respective incubation periods.
Intracellular mycobacteria infections in THP-1 cells
THP-1 human monocytic cells purchased from ECACC/PHE (Catalog no. 88081201) were routinely cultured in T75 flasks in Roswell Park Memorial Institute (RPMI; Sigma)-1640 supplemented with L-glutamine, NaHCO3, 10% heat-inactivated fetal bovine serum (FBS), and 100 U/mL pen/strep, at 37°C with 5% CO2. THP-1 cells were seeded at 2 × 104 cell/well in a 96 well plate and differentiated for 24 h with 20 ng/ml phorbol 12-myristate 13-acetate (PMA; Sigma) in media (RPMI – supplemented with L-glutamine, NaHCO3, 10% FBS, and 100 U/mL pen/strep) to prime the THP-1 monocytes into macrophage-like cells. Cells were washed and left for a further 24 h in fresh complete media.
M. smeg mc2155, was maintained as described previously. Cultures were maintained in the mid-log phase, and concentrations were adjusted for infection based on growth curves (Supplementary Information File, entry no.11). Differentiated THP-1 cells were infected with M. smeg mc2155 at a multiplicity of infection (MOI) of 1:5 in antibiotic-free complete RPMI media for 4 h, followed by 2× cells wash with warm PBS. After each wash step, infected cells were suspended in antibiotic-free media for 24 h prior to treatments.
Compounds (4) and (5) were resuspended in DMSO to obtain 10 mg/mL stock concentrations. Serial dilutions were performed in sterile ultrapure water (Omnipure) to the following concentrations: 0, 0.5, 1.0, 2.0, 4.0, 8.0, 16.0 and 32.0 µg/mL for 4, 24 and 48 h. The following controls were used: DMSO (vehicle control), 0 µg/mL concentration also contained ultrapure water (another vehicle control), media only (negative control) and media with no cells (blank). Following treatments, THP-1 cells were lysed using 1% Triton × (Thermo Fisher Scientific) for 10 min. Lysate was collected and serial dilutions 10-1–10-8 were made. The serial dilutions were spotted on to Mueller Hinton agar plates using the Miles and Misra method. Colony counts were measured following the growth of M. smeg mc2155, on solid agar after incubation for 48 h.
Confocal microscopy studies in HCT-116 colon carcinoma cells
For live cell imaging, HCT-116 cells (1.5 × 105) were seeded in a 35 mm glass-bottomed micro well dish (Ibidi/Thistle Scientific, Glasgow, UK) and cultured for 24 h. For lysosome co-staining, cells were washed × 3 with PBS. LysoTrackerTM Green DND 26 [(60 nM) Invitrogen, Paisley, UK] was added to the cell culture medium and incubated at 37°C for 30 min prior to the addition of compound (4) (2 µM) and imaging. For mitochondria co-staining, cells were washed × 3 with PBS, MitoTrackerTM Green FM [(50 nM) Invitrogen, Paisley, UK] was added to the cell culture medium and incubated at 37°C for 30 min prior to the addition of compound (4) (2 µM) and imaging. Cells were imaged using a Zeiss LSM800 confocal laser scanning microscope with a HCX PL APO lambda blue 63.0 × 1.4 oil objective, using Diode (405 nm), Argon (458, 488, 514 nm), HeNe (543 nm), HeNe (594 nm) and HeNe (633 nm) lasers. Image analyses were performed using Image J software.
Cytotoxicity testing (MTT assay)
THP-1 cells were seeded at 2 × 104 cell/well in a 96 well plate and differentiated for 24 h with 20 ng/mL PMA in media (RPMI – supplemented with L-glutamine (1%), Pen/Strep (1%) and FBS (10%)). After 24 h, cells were washed and left for a further 24 h rest period in fresh media.
Compounds were prepared as stated previously, at a concentration range of 0.5–32.0 µg/mL. Each plate corresponded to a different timepoint 4, 24, 48 and 72 h, with both compounds tested, n = 3. Following incubation with the compound, media was removed and replaced with fresh RPMI (70 µL per well) and then MTT was added (50 µL) to give the desired final concentration. The plate was covered in foil to protect it from light and incubated (37°C, 5% CO2) for 3 h. The reagent was then removed and crystals dissolved in DMSO, the plate was left for 30 min at RT and then the plate was read using a Tecan Magellan Sunrise absorbance reader at 540 nm with a reference wavelength of 620 nm. Data was presented as percentage cell viability. All calculations were performed after background absorbance correction and blank absorbance subtraction.
Statistical analysis
All data were obtained from at least three experimental replicates and presented as mean ± SD. Data analysis was performed using Graphpad Prism 9.3.1 (GraphPad Software, CA). The specific statistical test used is indicated in figure legends. The significance level was set as P < 0.05.
Results and discussion
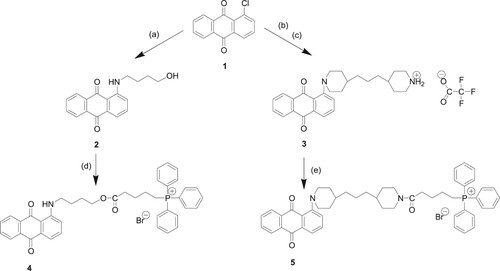
Despite refocused attention to the proper use of antibiotics, the urgent need to curb the proliferation of antibacterial-resistant bacteria, coupled with a decline in the approval of new antibacterials has resulted in the current ‘antibiotic crisis’. We present here novel synthetic compounds (4) and (5) with potent antibacterial activity that constitute a new class of chemicals – spacer-linked anthraquinone-TPP conjugates, designed as membrane-targeting agents (Scheme 1).
Scheme 1. Reagents and conditions: (a) 4-amino-1-butanol, DMSO, 95°C; (b) 1,3-di-4-piperidylpropane, DMSO, 95°C; (c) TFA, diethyl ether; (d) (4-carboxybutyl)triphenylphosphonium bromide, DCC, DMAP, CH2Cl2; (e) 4-(carboxybutyl)triphenylphosphonium bromide, PyBOP, HOBt, DIPEA, DMF.
Synthetic chemistry
Scheme 1 shows the synthetic procedures for the preparation of two novel anthraquinone-triphenylphosphonium (AQ-TPP) conjugates (4) and (5) from commercially available 1-chloroanthraquinone (1) commencing with nucleophilic aromatic substitution of the chlorine atom in (1) by either 4-amino-1-butanol or 1,3-di-4-piperidylpropane to afford intermediate anthraquinone-spacer compounds (2) and (3) respectively. Subsequently, reaction of the primary hydroxy group in (2) with 4-(carboxybutyl)triphenylphosphonium bromide under base-catalysed coupling conditions with dicyclohexylcarbodiimide gave the ester-linked AQ-TPP conjugate (4). Separately, the free secondary amino group of anthraquinone-spacer intermediate (3) was reacted with (4-carboxybutyl)triphenylphosphonium bromide, in which the carboxylic acid group was preactivated under peptide-coupling conditions with PyBop and HOBt, to afford the amide-linked AQ-TPP conjugate (5).
The structures of the test compounds (4) and (5) were confirmed by combinations of high-resolution mass spectrometry, NMR spectroscopy and HPLC methods.
Biological evaluation
The MIC and MBC values of AQ-TPP conjugates (4) and (5) were experimentally determined against extracellular M. smeg mc2155 and MRSA (NCTC 13616) and are shown in Table . As shown, the anthraquinone-TPP conjugates (4) and (5) are highly effective antibacterial compounds with bactericidal activity at low micromolar concentrations and favourable MBC and MIC ratios between 1.0 and 2.0.
Table 1. Minimum inhibitory and bactericidal concentrations of compounds (4) and (5) screened against M. smeg mc2155 and MRSA (NCTC 13616)*.
There is a family of ATP-dependent transporters, known collectively as the ATP-binding cassette (ABC) which is involved in the transporting of nutrients and other molecules across human cell membranes. The ABC transporters are composed of two cytoplasmic domains that bind to ATP and two transmembrane domains. Classical multi-drug resistance (MDR) results from the efflux of a wide variety of drugs by well-studied members of a superfamily: P-glycoprotein (ABCB1), breast cancer resistance protein (BCRP/MXR/ ABC-P/ABCG2) and multi-drug resistance-associated proteins (Katayama et al. Citation2014; Mansoori et al. Citation2017). The hydrophilic regions share homology with peripheral membrane components of bacterial active transport systems (Chen et al. Citation1986). In early development of anthracycline antibiotics, it was shown that increasing the lipophilicity of the salt forms partially evaded MDR phenomena (Lampidis et al. Citation1997). As a prelude to intracellular studies of antibacterial activity in infected THP1 cells, we selected a colon cancer cell line as a representative human cell expressing P-glycoprotein to investigate cell uptake of compound (4).
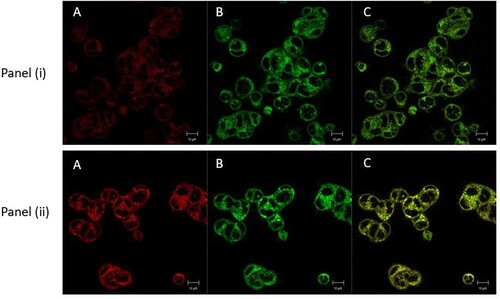
Using confocal microscopy, we determined that compound (4) does not accumulate in the nucleus or in lysosomes but exclusively in the mitochondria, with a significant co-localisation with the tracer compound MitoTracker GreenFM and thus offers the prospect of non-genotoxicity to human cells. This observation is consistent with the presence of the predicted TPP vector-mediated targeting and accumulation in these organelles (Figure ).
Figure 2. Confocal microscopy live cell imaging of HCT-116 colon cells, treated with 2 μM of compound (4) for 2 h. Panel (i) (A) Compound (4) stained with LysoTrackerTM Green DND26 (60 nM), (B) LysoTrackerTM Green DND26 (60 nM), and (C) merged image showing no colocalization. Panel (ii) (A) Compound (4) stained with MitoTrackerTM Green FM (50 nM), (B) MitoTrackerTM Green FM (50 nM), and (C) merged image showing clear colocalization. Areas where compound (4) and mitochondria colocalize appear as yellow/orange (C). Captured at 63 × magnification, scale bar 10 μm.
Antibacterial drugs designed to kill intracellular pathogens are required to enter and be retained in the host cell; the evident cellular uptake (Figure ) of TPP-conjugate (4) provided the motivation for treating THP-1 macrophages infected with the Mtb surrogate M. Smeg with a range of concentrations of the compounds.
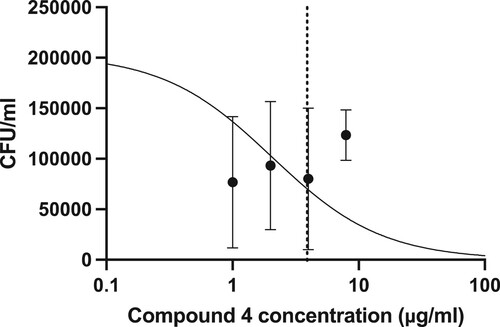
Before undertaking infection studies, THP-1 cells were infected at a range of MOIs to monitor cell viability. An MOI 1:5 was selected, as there was no significant impact on THP-1 cell viability attributable to M. smeg bacteria alone (Figure ). Upon determining a suitable MOI, THP-1 cells were subsequently infected with M. smeg (mc2155) (MOI 1:5), then treated with a range of concentrations of each compound for 24 h, then lysed and plated out on Mueller Hinton agar plates to determine CFU/mL (Figure ). Over the concentration range screened, it was possible to determine an intracellular LC50 of 3.8 µg/mL for compound (4) (Figure ). Based on the concentration range screened, it was not possible to determine the LC50 for compound (5).
Figure 3. Cytotoxic effect of different MOIs of M. smeg mc2155 infection on differentiated THP-1 cells over 24 h, determined by the MTT assay. Results are mean ± SD of three independent determinations. NTC = no treatment control, 0.1% Triton-X-100 was the positive control. Statistical Analysis was performed using Graph Pad Prism 9.3.1. One-way ANOVA using Dunnett’s multiple comparison test, where **p < 0.01.
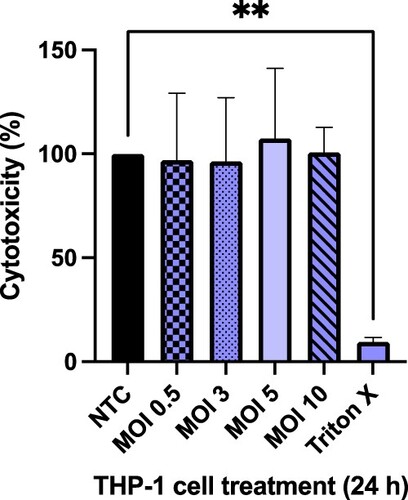
Figure 4. Colony counts on Mueller Hinton agar plates following treatment of intracellular infections. THP-1 cells were infected with M. smeg mc2155 for 4 h at an MOI of 1:5, then washed and treated for 24 h. Colony counts were measured following the growth of M. smeg mc2155 on solid agar after incubation for 72 h. Results are mean ± SD of three independent determinations. Statistical Analysis was performed using Graph Pad Prism 9.3.1. One-way ANOVA using Dunnett’s multiple comparison test, where *p < 0.05.
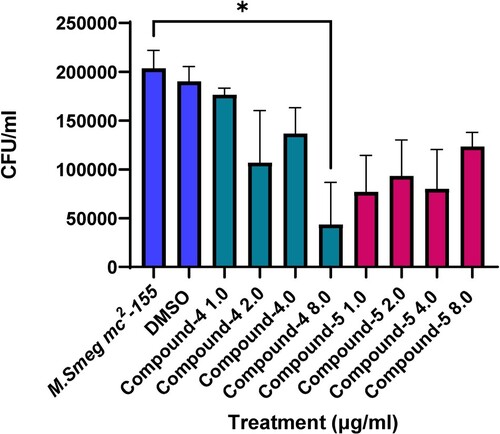
Figure 5. Colony counts on Mueller Hinton agar plates following treatment of intracellular infections. THP-1 cells were infected with M. smeg mc2155 for 4 h at an MOI of 1:5, then washed and treated for 24 h. Colony counts were measured following the growth of M. smeg mc2155 on solid agar after incubation for 72 h. In vitro intracellular LC50 determination for compound (4) was calculated to be 3.8 µg/mL.
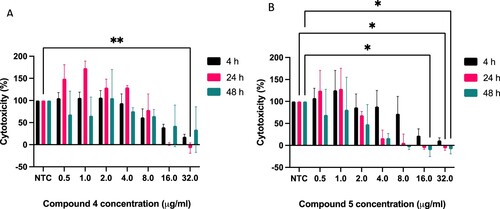
Our next objective was to determine the cytotoxic effects of the compounds on THP-1 macrophage cells over a period of 48 h, and this was assessed using the MTT assay with the negative control (NTC) being THP-1 cells in compound free culture media (Figure ). The difference in cell viability over time was only found to be statistically significant at 32 µg/mL after 24 h exposure for compound (4), and at 16 µg/mL (48 h post treatment) and ≥ 24 h exposure from 32 µg/mL for compound (5) (Figure ).
Figure 6. Cytotoxic effect of Compounds 4 (A) & 5 (B) on differentiated THP-1 cells over 4, 24 and 48 h. Results are mean ± SD of three independent determinations (each repeat done in triplicate), NTC = no treatment control. Statistical Analysis was performed using Graph Pad Prism 9.3.1. Two-way ANOVA using Tukey’s multiple comparison test, where *p < 0.05 and **p < 0.01
The MIC and MBC values of AQ-TPP conjugates (4) and (5) were experimentally determined against MRSA (strain NCTC 13616) and were found to be equipotent as determined by measurement of their MIC values [each 1 µg/mL (1.2 μM)] (Table ). Whereas the amide-linked conjugate (5) was determined to have 2-fold greater bactericidal potency [MBC 1 µg/mL (1.2 μM)] than (4). We speculate this may be related to its increased hydrophobicity and, or the intrinsic hydrolytic and proteolytic stability of its tertiary amide bond to TPP, compared to the potentially more labile corresponding ester bond in conjugate (4). An ester-linked TPP conjugate of ciprofloxacin has been reported to have greater antibacterial potency than the free drug in some MRSA strains, which was attributed in good part to subversion of the site of action from DNA gyrase and topoisomerase IV to membrane damaging effects (Kang et al. Citation2020); essentially side-stepping resistance. The MICs of emodin against a patient-derived isolate (numbered as MRSA 133630) and MSSA (S. aureus ATCC 29213) were reported as 15.63 and 0.19 μg/mL, respectively, an 80-fold difference, implying a much higher concentration is needed to suppress MRSA (May Zin et al. Citation2017). In contrast, in our study, compounds (4) and (5) both had equipotent MIC values in both the resistant strain S. aureus (NCTC 13616) and in a methicillin sensitive S. aureus (MSSA) strain (NCTC 6571) (data not shown for the latter).
The use of bactericidal rather than bacteriostatic agents as first-line therapy has been recommended because the destruction of microorganisms curtails, although not avoids, the development of bacterial resistance (Stratton Citation2003). Bactericidal potency achieved through a rapid mechanism of action is a preferred choice for curbing resistance-conferring mutations.
The bacterial cell membrane is vital for survival and multiplication, and functions as a selectively permeable barrier to preserve cellular homeostasis (Van Bambeke et al. Citation2008). The selection for resistance to membrane-active antibacterial agents would require an alteration of the charge of membrane lipids or to the membrane constituents, which is incompatible with bacterial survival. Consequently, the induction of membrane permeabilization and depolarisation is a promising strategy for the design of novel agents against drug-resistant bacteria. Antimicrobial peptides (AMPs) that kill bacteria much more rapidly than conventional antibiotics, act on bacterial membranes (Yeaman and Yount Citation2003) and have been predicted to have a lower propensity to induce resistance (Rai and Mehra Citation2021). We propose that low molecular mass agents with structural features capable of penetrating the lipid barriers of bacteria and effecting membrane permeabilization can emulate their (high mass) AMP counterparts. MRSA possesses an exceptionally thick, rigid cell wall arising from a highly cross-linked peptidoglycan structure in addition to a single cytoplasmic membrane which a drug must negotiate (Ambade et al. Citation2023). The key structural features for new drug design require an appropriate balance between hydrophobic and polar components (providing transient binding interactions with matching functional groups in the composition of the outer bacterial membranes in order to traverse). These structural features are invested in the amphipathic composition of conjugates (4) and (5).
M. smeg is a non-pathogenic bacterium that is widely used as a tool for genetic analysis research on infection and has found application reliably as a surrogate to identify new drug leads against Mtb (Lelovic et al. Citation2020). The anthraquinone-TPP conjugates (4) and (5) were active in vitro against the M. smeg mc2155 strain and were equipotent as determined by their MIC values; whereas, and in contrast to MRSA, the less hydrophobic (4) had 2-fold greater bactericidal potency than (5) as measured by their MBC values.
Mycobacteria are a diverse group of organisms but share extremely complex molecular architectures that play important roles in withstanding stresses, modulating the immune system, and mediate drug resistance. Unlike most bacteria, the peptidoglycan structure of mycobacteria is atypical, having a large number of unique chemical linkages and unique modifications. These characteristics doubtless are determinants of virulence and pathogenesis and are pertinent to drug design; however, the complete chemical structure and supra-structure of mycobacterial peptidoglycan is not totally known, and thus presents notable challenges for de novo antimycobacterial drug design. That said, recent studies (Rimal et al. Citation2022) have made some progress in using high resolution LC-MS to analyse the peptidoglycan ultrastructure of M. smeg. That conjugates (4) and (5) have bactericidal activity against M. smeg provided the motivation for studying their potential for intracellular activity (Lelovic et al. Citation2020; Riaz et al. Citation2020). Whilst a primary mechanism of resistance in Mycobacterium is mutations in drug target genes, many drug-resistant clinical strains reported do not have mutations in (known) target genes. Intended resistance, often a substantial increase, suggests alternative non-gene-related mechanisms operate notably in clinical strains and expression of drug efflux pumps has a significant role. Indeed, drug tolerance to Mtb residing in macrophages has been attributed to macrophage-induced multiple efflux pumps (Adams et al. Citation2011).
In respect of circumventing efflux pumps, the cationic, lipophilic TPP vector is a design feature of (4) and (5) to improve cellular uptake. As a prelude, we have shown that in accord with predictions, that (4) was taken up by human cells and accumulated in the mitochondria of the colon carcinoma cell line known to overexpress the p-glycoprotein (Pgp or P170) transmembrane efflux pump; this would imply that the conjugate can circumvent this efflux pump and then is translocated to the mitochondria by exploiting the negative membrane potential to gain entry. Interestingly, some reports indicate that the therapeutic window of some antibacterial drugs could be maximised by imparting mitochondrial sequestration via peptide conjugation; whereby the mitochondria act as a repository for the antibacterial agent that can shuttle to the intracellular bacterial cells to exert their action (Pereira and Kelley Citation2011).
Active efflux of drugs across membranes is a fundamental survival strategy and a major mycobacterial efflux pump (equivalent to the human cell Pgp efflux pump), highly conserved across fast and slower growing mycobacteria species is EfpA; the gene from Mtb shares 80% identity with that of M. smeg (Rai and Mehra Citation2021). Overexpression of EfpA in M. smeg leads to dramatic fold-increases in drug tolerance, and the increase in MICs correlated with higher levels of EfpA expression. We have yet to determine whether or not conjugates (4) and (5) are substrates for EfpA but the bactericidal potency and retention of bactericidal intracellular activity against M. smeg is very promising.
Mtb can enter and localise into alveolar macrophages thereby gaining some protection from some antibiotics. For the development of new drugs for any intracellular pathogen, one very important factor is whether the candidate drug can work inside the cell or not. Our results show that (4) and (5) had clear and potent M. smeg killing activity in infected macrophages at low concentrations over short drug exposure times; encouraging further investigation into mechanisms of action and development of these formative members of the series of spacer-linked anthraquinone-TPP conjugates.
Conclusions
We designed positively charged TPP-conjugated anthraquinones (4) and (5) to target anionic lipids of the bacterial membrane to induce collapse of the membrane architecture by interaction of the hydrophobic anthraquinone-spacer segment with lipophilic moieties on the bacterial membrane. We envision the conjugates ‘walk’ through membrane-embedded lipoprotein networks to gain access to membranes in a manner analogous to how TPP vectors haul their drug cargo into negatively charged sub-cellular organelles. The planned focus is deciphering the effect of the conjugates on membrane integrity and bacterial cell entry. In conclusion, the prototype spacer-linked anthraquinone-TPP conjugates (4) and (5) of this new class possess intracellular bactericidal effects, and are likely to engage multiple targets, predicted commonly to include cell membranes; thus, provide promising early leads for developing novel antibacterial drugs.
Supplemental Figures
Download MS Word (1.6 MB)Acknowledgements
We thank the National Mass Spectrometry Facility (NMSF) at Swansea University, Swansea, UK, for high resolution mass spectrometric analyses.
Authors contributions
DJM, SD and DM acquired the funding for this work and analysed and interpreted the data. AS, AT, AF, FFG & MN performed the experiments. DJM, AT and SD conceived the concept and wrote the manuscript. All authors have read, contributed and approved the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its supplementary materials. Raw data has been shared to https://figshare.com/ at https://doi.org/10.6084/m9.figshare.25335139
.Additional information
Funding
References
- Adams KN, Takaki K, Connolly LE, Wiedenhoft H, Winglee K, Humbert O, Edelstein PH, Cosma CL, Ramakrishnan L. 2011. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell. 145(1):39–53. doi:10.1016/j.cell.2011.02.022.
- Ambade SS, Gupta VK, Bhole RP, Khedekar PB, Chikhale RV. 2023. A review on five and six-membered heterocyclic compounds targeting the Penicillin-Binding Protein 2 (PBP2A) of Methicillin-Resistant Staphylococcus aureus (MRSA). Molecules. 28(20):7008. doi:10.3390/molecules28207008.
- Andrews JM. 2001. Determination of minimum inhibitory concentrations. J Antimicrob Chemother. 48(suppl_1):5–16. doi:10.1093/jac/48.suppl_1.5.
- Brennan PJ. 2003. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis. 83(1–3):91–97. doi:10.1016/S1472-9792(02)00089-6.
- Chen C, Chin JE, Ueda K, Clark DP, Pastan I, Gottesman MM, Roninson IB. 1986. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell. 47(3):381–389. doi:10.1016/0092-8674(86)90595-7.
- Darby EM, Trampari E, Siasat P, Gaya MS, Alav I, Webber MA, Blair JMA. 2023. Molecular mechanisms of antibiotic resistance revisited. Nat Rev Microbiol. 21(5):280–295. doi:10.1038/s41579-022-00820-y.
- Duan F, Xin G, Niu H, Huang W. 2017. Chlorinated emodin as a natural antibacterial agent against drug-resistant bacteria through dual influence on bacterial cell membranes and DNA. Sci Rep. 7(1):12721. doi:10.1038/s41598-017-12905-3.
- EClinicalMedicine. 2021. Antimicrobial resistance: a top ten global public health threat. EClinicalMedicine. 41:101221. doi:10.1016/j.eclinm.2021.101221.
- EUCAST. 2022. EUCAST reading guide for broth microdilution. V4. https://www.eucast.org/ast_of_bacteria/previous_versions_of_documents.
- Firth A, Prathapan P. 2021. Broad-spectrum therapeutics: a new antimicrobial class. Current Research in Pharmacology and Drug Discovery. 2:100011. doi:10.1016/j.crphar.2020.100011.
- Fujiwara A, Hoshino T, Westley JW. 1985. Anthracycline antibiotics. Crit Rev Biotechnol. 3(2):133–157. doi:10.3109/07388558509150782.
- Han J-W, Shim D-W, Shin W-Y, Heo K-H, Kwak S-B, Sim E-J, Jeong J-H, Kang T-B, Lee K-H. 2015. Anti-Inflammatory effect of emodin via attenuation of NLRP3 inflammasome activation. Int J Mol Sci. 16(12):8102–8109. doi:10.3390/ijms16048102.
- Han M, Vakili MR, Soleymani Abyaneh H, Molavi O, Lai R, Lavasanifar A. 2014. Mitochondrial delivery of doxorubicin via triphenylphosphine modification for overcoming drug resistance in MDA-MB-435/DOX Cells. Mol Pharm. 11(8):2640–2649. doi:10.1021/mp500038g.
- Haque E, Kamil M, Irfan S, Sheikh S, Hasan A, Nazir A, Mir SS. 2018. Blocking mutation independent p53 aggregation by emodin modulates autophagic cell death pathway in lung cancer. Int J Biochem Cell Biol. 96:90–95. doi:10.1016/j.biocel.2018.01.014.
- Hasan CM, Dutta D, Nguyen ANT. 2022. Revisiting antibiotic resistance: mechanistic foundations to evolutionary outlook. Antibiotics. 11(1):40. doi:10.3390/antibiotics11010040.
- Hasanpour AH, Sepidarkish M, Mollalo A, Ardekani A, Almukhtar M, Mechaal A, Hosseini SR, Bayani M, Javanian M, Rostami A. 2023. The global prevalence of methicillin-resistant Staphylococcus aureus colonization in residents of elderly care centers: a systematic review and meta-analysis. Antimicrob Resist Infect Control. 12(1):4. doi:10.1186/s13756-023-01210-6.
- Jeena MT, Kim S, Jin S, Ryu J-H. 2020. Recent progress in mitochondria-targeted drug and drug-free agents for cancer therapy. Cancers (Basel). 12(1):4. doi:10.3390/cancers12010004.
- Kang S, Sunwoo K, Jung Y, Hur JK, Park K-H, Kim JS, Kim D. 2020. Membrane-targeting Triphenylphosphonium functionalized ciprofloxacin for Methicillin-Resistant Staphylococcus aureus (MRSA). Antibiotics. 9(11):758. doi:10.3390/antibiotics9110758.
- Katayama K, Noguchi K, Sugimoto Y. 2014. Regulations of P-Glycoprotein/ABCB1/ MDR1 in human cancer cells. New J Sci. 2014:1–10. doi:10.1155/2014/476974.
- Lampidis TJ, Kolonias D, Podona T, Israel M, Safa AR, Lothstein L, Savaraj N, Tapiero H, Priebe W. 1997. Circumvention of P-GP MDR as a function of anthracycline lipophilicity and charge. Biochemistry. 36(9):2679–2685. doi:10.1021/bi9614489.
- Lelovic N, Mitachi K, Yang J, Lemieux MR, Ji Y, Kurosu M. 2020. Application of Mycobacterium smegmatis as a surrogate to evaluate drug leads against Mycobacterium tuberculosis. J Antibiot. 73(11):780–789. doi:10.1038/s41429-020-0320-7.
- Lian X, Xia Z, Li X, Karpov P, Jin H, Tetko IV, Xia J, Wu S. 2021. Anti-MRSA drug discovery by ligand-based virtual screening and biological evaluation. Bioorg Chem. 114:105042. doi:10.1016/J.BIOORG.2021.105042.
- Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. 2017. The different mechanisms of cancer drug resistance: a brief review. Adv Pharm Bull. 7(3):339–348. doi:10.15171/apb.2017.041.
- Marinello J, Delcuratolo M, Capranico G. 2018. Anthracyclines as topoisomerase II poisons: from early studies to new perspectives. Int J Mol Sci. 19(11):3480. doi:10.3390/ijms19113480.
- May Zin WW, Buttachon S, Dethoup T, Pereira JA, Gales L, Inácio Â, Costa PM, Lee M, Sekeroglu N, Silva AMS, et al. 2017. Antibacterial and antibiofilm activities of the metabolites isolated from the culture of the mangrove-derived endophytic fungus Eurotium chevalieri KUFA 0006. Phytochemistry. 141:86–97. doi:10.1016/j.phytochem.2017.05.015.
- Mohan A, Padiadpu J, Baloni P, Chandra N. 2015. Complete genome sequences of a Mycobacterium smegmatis laboratory strain (MC2155) and isoniazid-resistant (4XR1/R2) mutant strains. Genome Announc. 3(1):e01520–14. doi:10.1128/genomeA.01520-14.
- Munita JM, Arias CA. 2016. Mechanisms of antibiotic resistance. Vir Mech Bact Patho. 2021:481–511. doi:10.1128/9781555819286.ch17.
- Murray CJL, Ikuta KS, Sharara F, Swetschinski L, Robles Aguilar G, Gray A, Han C, Bisignano C, Rao P, Wool E, … Naghavi M. 2022. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet. 399(10325):629–655. doi:10.1016/S0140-6736(21)02724-0.
- National Institutes of Health. 2011. NIH funds development of new broad-spectrum therapeutics. https://www.nih.gov/news-events/news-releases/nih-funds-development-new-broad-spectrum-therapeutics.
- Pathak A, Angst DC, León-Sampedro R, Hall AR. 2023. Antibiotic-degrading resistance changes bacterial community structure via species-specific responses. ISME J. 17(9):1495–1503. doi:10.1038/s41396-023-01465-2.
- Pereira MP, Kelley SO. 2011. Maximizing the therapeutic window of an antimicrobial drug by imparting mitochondrial sequestration in human cells. J Am Chem Soc. 133(10):3260–3263. doi:10.1021/ja110246u.
- Rabbani A, Finn RM, Ausió J. 2005. The anthracycline antibiotics: antitumor drugs that alter chromatin structure. BioEssays. 27(1):50–56. doi:10.1002/bies.20160.
- Rai D, Mehra S. 2021. The Mycobacterial Efflux Pump EfpA can induce high drug tolerance to many antituberculosis drugs, including moxifloxacin, in Mycobacterium smegmatis. Antimicrob Agents Chemother. 65(11). doi:10.1128/AAC.00262-21.
- Riaz MS, Kaur A, Shwayat SN, Behboudi S, Kishore U, Pathan AA. 2020. Dissecting the mechanism of intracellular Mycobacterium smegmatis growth inhibition by platelet activating Factor C-16. Front Microbiol. 11:1046. doi:10.3389/fmicb.2020.01046.
- Rimal B, Senzani S, Ealand C, Lamichhane G, Kana B, Kim SJ. 2022. Peptidoglycan compositional analysis of Mycobacterium smegmatis using high-resolution LC–MS. Sci Rep. 12(1):11061. doi:10.1038/s41598-022-15324-1.
- Sagan L. 1967. On the origin of mitosing cells. J Theor Biol. 14(3):225–274. doi:10.1016/0022-5193(67)90079-3.
- Shia C-S, Hou Y-C, Tsai S-Y, Huieh P-H, Leu Y-L, Chao P-DL. 2010. Differences in pharmacokinetics and ex vivo antioxidant activity following intravenous and oral administrations of emodin to rats**Chi-Sheng Shia and Yu-Chi Hou contributed equally to this work. J Pharm Sci. 99(4):2185–2195. doi:10.1002/jps.21978.
- Singh SB, Young K, Silver LL. 2017. What is an “ideal” antibiotic? Discovery challenges and path forward. Biochem Pharmacol. 133:63–73. doi:10.1016/j.bcp.2017.01.003.
- Smith J. 2011. Nanoparticle delivery of anti-tuberculosis chemotherapy as a potential mediator against drug-resistant tuberculosis. Yale J Biol Med. 84:361–369.
- Stratton CW. 2003. Dead bugs don’t mutate: susceptibility issues in the emergence of bacterial resistance. Emerg Infect Dis. 9(1):10–16. doi:10.3201/eid0901.020172.
- Sullivan LB, Chandel NS. 2014. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2(1):17. doi:10.1186/2049-3002-2-17.
- Van Bambeke F, Mingeot-Leclercq M-P, Struelens MJ, Tulkens PM. 2008. The bacterial envelope as a target for novel anti-MRSA antibiotics. Trends Pharmacol Sci. 29(3):124–134. doi:10.1016/j.tips.2007.12.004.
- Vos T, Lim SS, Abbafati C, Abbas KM, Abbasi M, Abbasifard M, Abbasi-Kangevari M, Abbastabar H, Abd-Allah F, Abdelalim A, … Murray CJL. 2020. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 396(10258):1204–1222. doi:10.1016/S0140-6736(20)30925-9.
- Walsh TR, Gales AC, Laxminarayan R, Dodd PC. 2023. Antimicrobial resistance: addressing a global threat to humanity. PLoS Med. 20:e1004264. doi:10.1371/journal.pmed.1004264.
- WHO. 2020. World Health Organization. 10 global health issues to track in 2021.
- WHO. 2023a. Global tuberculosis report 2023. https://iris.who.int/bitstream/handle/10665/373828/9789240083851-eng.pdf?sequence = 1.
- WHO. 2023b. Proportion of bloodstream infection due to methicillin-resistant Staphylococcus aureus (MRSA). https://data.who.int/indicators/i/5DD9606.
- Yeaman MR, Yount NY. 2003. Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev. 55(1):27–55. doi:10.1124/pr.55.1.2.
- Zielonka J, Joseph J, Sikora A, Hardy M, Ouari O, Vasquez-Vivar J, Cheng G, Lopez M, Kalyanaraman B. 2017. Mitochondria-targeted Triphenylphosphonium-based compounds: syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem Rev. 117(15):10043–10120. doi:10.1021/acs.chemrev.7b00042.