INTRODUCTION
The ABO blood group antigens (A, B and H antigens) are composed of complex oligosaccharides linked to membrane proteins and lipids [Citation[1], Citation[2]] and abundant on red blood cell membranes. Each antigen has a unique terminal immunodominant monosaccharide [Citation[3]]. For example, the terminal immunodominant monosaccharide of the A antigen is N-acetyl-α-D-galactosamine. Individuals lacking this antigen produce potent, naturally occurring, highly specific, complement-fixing antibodies to this epitope [Citation[4]]. These naturally occurring antibodies can cause fatal hemolytic transfusion reactions when incompatible blood is transfused. The H antigen, blood type O, is the precursor of the blood type A and B antigens. Blood type O is weakly immunogenic, and red blood cells expressing only this antigen are universally transfusable [Citation[5]].
Exoglycosidases are enzymes that modify carbohydrate epitopes on glycoproteins and glycolipids by cleaving specific monosaccharide units from the non-reducing ends, thereby modulating the immune response. Some exoglycosidases modify the A, B and H antigens by hydrolysis of the terminal immunodominant residue [Citation[6-10]]. Hydrolysis of the terminal N-acetyl-α-D-galactosamine residue of A2 antigens on erythrocyte membranes converts the epitope to H antigen (blood type O) [Citation[11], Citation[12]]. Although α-N-acetylgalactosaminidases (αNAG) (E. C. 3.2.1.49) with activity against blood group A antigen have been characterized by others, those enzymes have low pH optima, are inefficient or are multimeric enzymes [Citation[6-9]]. Unlike αNAG purified from other sources, Clostridium perfringens α-N-acetylgalactosaminidase is monomeric and has a pH optimum in the range of 6.5 to 7.2 with no α-galactosidase activity [Citation[13]]. These are desirable properties if the enzyme is to be used in enzymatic conversion biotechnology. In preliminary studies, recombinant α-N-acetylgalactosaminidase (rαNAG) from Clostridium perfringens showed similar properties to native αNAG(nαNAG).
There are quantitative and qualitative differences in type A1 and A2 red blood cells. On A1 red blood cells membranes there are approximately 1,200,000 A epitopes, but there are only 240,000 epitopes on A2 cells [Citation[14]]. There are more binding sites for the lectin Dolichos biflorus on A1 red blood cells than on A2 cells [Citation[15]]. Of particular note is the repetitive A epitope (type 3 chain A) unique to A2 red blood cells [Citation[16]]. A epitopes on A2 red blood cells are mostly expressed in straight oligosaccharide chains, while on A1 cells they are more complex and branched [Citation[15]]. Goldstein postulated that degradation of this epitope might require incubation of A1 red blood cells with α-L-fucosidase, β-D-galactosidase and α-N-acetylgalactosaminidase [Citation[17]]. It is apparent that the A epitopes present on the A2 cell glycocalyx are less complex and thought to be amenable to enzymatic conversion. In addition, type A2 red blood cell units constitute 25% of all type A units available for enzymatic conversion.
It is possible that treatment of type A2 erythrocytes with an appropriate exoglycosidase could produce universally transfusable type O erythrocytes, a technology that could increase the compatible blood supply while decreasing waste of type A red blood cell units and the risk of transfusion reactions [Citation[18]]. Preliminary studies of rαNAG from Clostridium perfringens have been performed in our laboratory, demonstrating hydrolysis of A2 epitopes from erythrocyte membranes [Citation[19]]. However, conditions for deantigenation were not determined. This article describes an enzyme-linked immunosorbent assay (ELISA) with type A2 erythrocyte membranes used to study the blood group A2 degrading activity of rαNAG from Clostridium perfringens. Conditions for enzymatic conversion of blood type A2 to O were investigated.
MATERIALS AND METHODS
Murine monoclonal anti-A antibody was obtained from Ortho Diagnostic, Raritan, NJ. Goat anti-mouse µ-chain specific alkaline phosphatase conjugate was purchased from Calbiochem, Lajolla, CA. Murine monoclonal anti-H antibody was obtained from Dako Corporation, Carpinteria, CA. Human plasma and erythrocytes were obtained from the American Red Cross, St. Louis, MO. Type A2 packed red blood cells used for membrane preparation were collected in a standard formulation of citrate/phosphate/dextrose (CPD) and were less than 21 days old. Lack of agglutination by Dolichos biflorus anti-A1 lectin was used to select type A2 red blood cells [Citation[20]]. Type A fresh frozen plasma was collected in CPD, stored at − 65°C and was less than 1 year old prior to thawing and use. Other red cell preservative solutions were prepared using standard formulas [Citation[15]]. Immulon 4 flat bottom microtiter plates were purchased from Dynatech Laboratories, Chantilly, VA. Dextrose, glucose, dithiothreitol (DTT), trisodium citrate, citric acid, adenine, mannitol, NaCl, NaH2PO4 were obtained from Fisher Scientific, Pittsburgh, PA. Bovine serum albumin (BSA) and p-nitrophenol-N-acetyl-α-D-galactosaminide were purchased from Sigma Chemical Company, St. Louis, MO. Protein concentrations were quantified with the Advanced Protein Assay Reagent obtained from Cytoskeleton, Denver, CO, using BSA as a standard.
Enzymatic activity was determined by quantifying hydrolysis of p-nitrophenol from p-nitrophenol-N-acetyl-α-D-galactosaminide. Enzyme aliquots were incubated in 40 mM NaH2PO4 buffer, pH 6.5, containing 1.0 mg mL−1 BSA, 1.0 mM DTT and 1.0 mM p-nitrphenol-N-acetyl-α-D-galactosaminide, at 37°C. One unit (U) of activity was defined as hydrolysis of 1.0 mmol of substrate per min. rαNAG from C. perfringens was purified by the method of Hsieh et al. with slight modification [Citation[13]]. The preparations were homogeneous by sodium dodecyl sulfate polyacrylamide gels (SDS-PAGE) with a specific activity of 84.11 U mg−1.
The procedure for erythrocyte membrane preparation, the plate-coating technique and the ELISA method were described by Hobbs et al. [Citation[21]] with differences being the use of A2 erythrocyte membranes and anti-A monoclonal antibody as described by Hsieh and Smith [Citation[12]]. Briefly, microtiter wells were coated with 0.66 µg mL−1 of A2 erythrocyte membranes in 0.02 M NaHCO3, pH 9.6. This coating-membrane concentration was determined to produce a desirable signal to noise ratio. After membrane coating of the plates, they were blocked with 0.1% (w/v) BSA in 0.05 M Tris-HCl and 0.15 mM NaCl, pH 7.4 (TBS buffer), washed and stored at − 70°C until used. At time of use, the plates were thawed and rehydrated with TBS buffer. The Microtiter wells were treated with 90 µL of enzyme, washed and developed with 75 µL of primary anti-A IgM monoclonal antibody (1:100 dilution) in TBS buffer. After washing of the wells to remove unbound primary antibody, they were developed with 75 µL of secondary conjugate, goat anti-mouse µ-chain-specific alkaline phosphatase conjugate (1:800 dilution) in TBS buffer. The wells were washed and developed with substrate as described by Hsieh and Smith [Citation[12]]. Final substrate volume in each microtiter well was 100 µL. The absorbance (OD405) of each well was determined at 405 nm. Degradation of A2 epitopes decreased binding of the primary and subsequent secondary conjugate, resulting in less absorbance when developed with substrate. Thus, the ΔOD405, the difference in the mean absorbance for the control (enzyme-untreated) and test (enzyme-treated) wells, was a function of enzyme concentration. For all experiments, data points were performed in duplicate and each experiment repeated in triplicate. Microtiter wells for all assays were washed and developed with primary monoclonal antibody, secondary conjugate and substrate as described above. The microtiter wells were treated with enzyme for 4 hours at 37°C at an enzyme concentration of 0.4 U mL−1 unless otherwise stated. Undiluted monoclonal anti-H antibody was substituted for anti-A monoclonal antibody for the studies of H antigen expression.
For the A2 deantigenation time course, enzyme was diluted into PBS buffer (20 mM NaH2PO4, pH 7.0, containing 130 mM NaCl) to a concentration of 1.0 U mL−1 and incubated in microtiter wells for 0, 2, 4, 8 and 24 hours. Data for this experiment only were expressed as OD405 to demonstrate the reciprocal relationship between anti-A and anti-H binding to erythrocyte membranes. The effect of temperature on deantigenation was studied in dilute CPD solution (10% (v/v) CPD in 150 mM NaCl), pH 7.0, at 4, 22 and 37°C. The effect of standard red blood cell preservative solutions on enzymatic activity in CPD, CPDA-1 and AS-1, both undiluted and diluted (10% (v/v) preservative solution in PBS buffer, pH 7.0), were compared to a PBS control. The pH optimum in dilute CPD solution (10% (v/v) CPD in 150 mM NaCl) was determined between pH 6.25 to 7.0. The effect of plasma on enzymatic activity in the ELISA was determined in 9.35 and 93.54% (v/v) type A plasma in dilute CPD (10% (v/v) CPD in 150 mM NaCl), pH 7.0 and compared with dilute CPD alone.
RESULTS
Degradation of the A2 epitope by the clostridial enzyme increased with increasing enzyme concentration in a curvilinear relationship. At an enzyme concentration of 4.0 U mL−1, 93.70% hydrolysis of the A epitope from A2 membranes was achieved. 50% hydrolysis was observed at 0.5 U mL−1 (). To study deantigenation as a function of time, hydrolysis of A2 epitopes and expression of H epitopes was determined at 0, 2, 4, 8 and 24 hours (). It was evident that by 24 hours approximately 85.6% of the A2 epitopes were hydrolyzed by an enzyme concentration of 1.0 U mL−1. Monoclonal anti-A binding decreased, while the binding of anti-H increased with enzymatic treatment. To conserve enzyme stock, an enzyme concentration of 0.4 U mL−1 was chosen for subsequent ELISA studies. From 4 to 37°C, hydrolysis of A2 epitope increased with increasing reaction temperature (). The enzyme hydrolyzed A2 epitopes at 4°C with 20% of the efficiency compared to 37°C. The effect of undiluted preservative solutions on the enzyme activity was characterized in CPD, CPDA-1 and AS-1. The enzyme in CPD and CPDA-1 showed a 41.12% and 19.42% increase in activity, respectively, and also a 1.86% increase in AS-1 when compared to the PBS buffer control (). However, in the study with dilute preservative solutions, there was little effect on enzyme activity of any of the dilute preservative solutions when compared to the PBS control (). There was a nominal effect on the hydrolysis of the A2 epitopes in dilute CPD solution in the pH range of 6.25 to 7.0 although there was a modest decrease at pH 7.0 (). At pH 7.0, 74% hydrolysis of A2 epitopes was observed when compared with pH 6.25. The effect of type A plasma on enzymatic activity is shown in . When enzyme reactions were run in 93.54% (v/v) plasma there was a 17.8% decrease in activity, and when enzyme was incubated in 9.35% (v/v) plasma in dilute CPD, there was a 33.8% decrease in activity, compared to the dilute CPD control.
Figure 1 Degradation of A2 epitopes as a function of enzyme concentration. Enzyme at the designated concentration in PBS buffer (20 mM NaH2PO4 + 130 mMNaCl, pH 7.0) was incubated at 37°C for 4 hr. All the data points are the mean of three independent duplicate determinations. Error bars represent the mean + / − S.D.
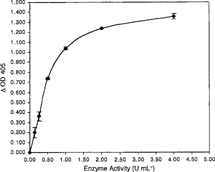
Figure 2 Binding of monoclonal anti-A and anti-H as a function of reaction time. Reactions were run at a final enzyme concentration of 1 U mL−1 in PBS buffer, pH 7.0, at 37°C for the designated time. All data points are the mean of two independent duplicate determinations. Error bars represents the mean + / − S.D.
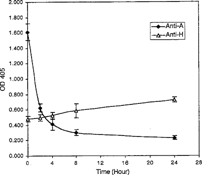
Figure 3 Degradation of A2 epitopes as a function of temperature. Enzyme, at a final concentration of 0.4 U mL−1 in PBS buffer, pH 7.0, was incubated at the indicated temperature for 4 hr. All data points are the mean of three independent duplicate determinations. Error bars represent the mean + / − S.D.
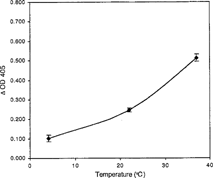
Figure 4 Degradation of A2 epitopes in undiluted preservative solution. Enzyme, at a final concentration of 0.4 U mL−1 in the designated preservative solution or buffer was incubated at 37°C for 4 hr. All data points are the mean of three independent duplicate determinations. Error bars represent the mean + / − S.D.
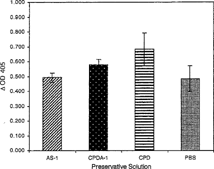
Figure 5 Degradation of A2 epitopes in dilute preservative solutions [10% (v/v) CPD, CPDA-1 or AS-1 preservative solution in PBS, pH 7.0] was compared to enzyme activity in PBS. Enzyme at 0.4 U mL−1 in the indicated solutions was incubated at 37°C for 4 hr. All data points are the mean of three independent duplicate determinations. Error bars represent the mean + / − S.D.
![Figure 5 Degradation of A2 epitopes in dilute preservative solutions [10% (v/v) CPD, CPDA-1 or AS-1 preservative solution in PBS, pH 7.0] was compared to enzyme activity in PBS. Enzyme at 0.4 U mL−1 in the indicated solutions was incubated at 37°C for 4 hr. All data points are the mean of three independent duplicate determinations. Error bars represent the mean + / − S.D.](/cms/asset/20f85100-0554-4d14-b627-47563cf30dae/ianb19_a_55904_f0005_b.gif)
Figure 6 Degradation of A2 epitopes as a function of pH in dilute CPD solution [10% (v/v) CPD in 150 mM NaCl, at the designated pH]. Enzyme in dilute CPD at final concentration of 0.4 U mL−1 was incubated at 37°C for 4 hr. All data points are the mean of three independent duplicate determinations. Error bars represent the mean + / − S.D.
![Figure 6 Degradation of A2 epitopes as a function of pH in dilute CPD solution [10% (v/v) CPD in 150 mM NaCl, at the designated pH]. Enzyme in dilute CPD at final concentration of 0.4 U mL−1 was incubated at 37°C for 4 hr. All data points are the mean of three independent duplicate determinations. Error bars represent the mean + / − S.D.](/cms/asset/fd0046a6-8064-4330-b2c1-745ccd62b487/ianb19_a_55904_f0006_b.gif)
Figure 7 Degradation of A2 epitopes as a function of plasma concentration. Final plasma concentration was 93.5, 9.4 and 0% in reactions containing 0.4 U mL−1 of enzyme in dilute CPD [10% (v/v) CPD, pH 7.0 in 150 mM NaCl]. All data points are the mean of three independent duplicate determinations. Error bars represent the mean + / − S.D.
![Figure 7 Degradation of A2 epitopes as a function of plasma concentration. Final plasma concentration was 93.5, 9.4 and 0% in reactions containing 0.4 U mL−1 of enzyme in dilute CPD [10% (v/v) CPD, pH 7.0 in 150 mM NaCl]. All data points are the mean of three independent duplicate determinations. Error bars represent the mean + / − S.D.](/cms/asset/bf108575-9d97-4458-a1f7-aa6a1b08667a/ianb19_a_55904_f0007_b.gif)
DISCUSSION
The recombinant Clostridium perfringens hydrolyzed the terminal N-acetyl-α-D-galactosamine residues from blood type A2 epitope on blood type A2 membranes. Hydrolysis of the A2 epitopes with the recombinant clostridial enzyme decreased binding of monoclonal anti-A with concomitant increased binding of monoclonal anti-H. At 1.0 U mL−1, the enzyme degraded 85.6% of A2 epitope after a 24-hour incubation. In comparison, when the same ELISA format was used to study deantigenation of the blood group B epitope with native Glycine max (soybean) α-D-galactosidase, 93% removal of the B epitope was observed at a higher enzyme concentration of 10 U mL−1 after a 4-hour incubation [Citation[22]]. The gene for the soybean enzyme was cloned and recombinant α-D-galactosidase expressed [Citation[23]]. This recombinant enzyme successfully deantigenated type B to O red cells in cell suspension studies [Citation[24]]. In those experiments, high concentrations of enzyme, 100 U mL−1, were required to achieve complete deantigenation at a 70% hematocrit. For this reason, it is reasonable to pursue seroconversion of type A2 red cells under similar conditions when adequate mass of recombinant clostridial enzyme is available.
The clostridial enzyme shows particular promise because it is active at pH 6.4–7.0, the pH range seen in stored red blood cell units [Citation[25]]. The exoglycosidase was stable and active in the temperature range found in freshly collected and stored red blood cell units, i.e. normal body temperature (37°C), at time of collection to storage temperatures of 1–6°C. Stability and activity of the enzyme in the thermal range 4–24°C is extremely useful for enzymatic conversion or erythrocytes. According to standard transfusion medicine protocol, it is important to keep erythrocytes at or below 24°C after collection and at 1–6°C during storage to decrease bacterial growth, if inadvertently contaminated, as well as to maintain cell function and survival [Citation[26]]. Enzyme activity in CPD increased approximately 40% and also increased in CPDA-1 and AS-1 when compared to PBS control. However, in dilute preservative solutions, there was little difference among them. The recombinant enzyme could possibly be added to undiluted preservative solutions, and at the time of collection, blood could be drawn into bags containing enzyme/preservative solution. It is noteworthy that the recombinant clostridial enzyme was active in CPD at the pH range commonly found in stored red cell units [Citation[25]].
Furthermore, the recombinant enzyme hydrolyzed the A2 epitope in the presence of type A plasma anticoagulated with CPD, however, activity was lower in both undiluted and diluted plasma compared to the PBS control. Additional studies will be pursued to elucidate the nature of this inhibition. Soluble blood group A substance normally present in plasma is possibly competing for the enzyme, or there is a non-competitive plasma inhibitor. Studies of hydrolysis of A2 epitopes as a function of enzyme concentration and time in packed red cells collected in CPD will be informative.
Current enzymatic biotechnology for converting type B to O red blood cells using native Coffea (coffee bean) α-D-galactosidase requires washing and equilibration of red blood cells with a low-pH buffer, addition of high concentrations of enzyme and back washing of cell suspensions to achieve neutral pH [Citation[18],Citation[27],Citation[28]]. Whole units of packed red blood cells have been converted and safely infused [Citation[28-30]]. Recombinant Coffee α-D-galactosidase has also been successfully employed [Citation[31]]. The extensive cell washing along with the high enzyme mass required are drawbacks to this technology, either with Coffea enzyme [Citation[28],Citation[31]] or with soybean enzyme [Citation[24]]. Use of eukaryotic α-N-acetylgalactosaminidases to achieve efficient deantigenation under similar condition has not been as successful [Citation[32],Citation[33]]. Hoskins et al. [Citation[7]] purified an α-N-acetylgalactosaminidase from Ruminococcus torques. The enzyme was active at neutral pH, and used to deantigenate the A1 epitope red blood cells [Citation[29],Citation[34],Citation[35]]. Erythrocytes treated with the Ruminococcus enzyme bound less the lectin from D. biflorus, which specifically binds to a subset of epitopes found on A1 erythrocytes [Citation[36]] than untreated A1 red blood cells. Significant ablation of the A epitopes was demonstrated by decreased binding of anti-A antibody; however, it was clear that these cells were not completely deantigenated and still expressed numerous A epitopes as evidenced by high hemagglutination titers [Citation[34]]. The Ruminococcus enzyme preparations were also used to deantigenate A2 red blood cells. The hemagglutination titer of monoclonal anti-A antibody decreased from 1024 to 64 on enzyme-treated cells [Citation[34]]. A hemagglutination titer of 64 is still high and cells expressing this much A epitope would not be transfusable. Furthermore, for A2 red blood cells, studies varying the incubation time, enzyme mass, and hematocrit were not described with the Ruminococcus enzyme.
In conclusion, recombinant C. perfringens α-N-acetylgalactosaminidase may be useful for enzymatic conversion of type A2 to universally transfusable type O red blood cells. It may be possible to add the enzyme directly to packed red blood cell units without washing and equilibration of red cells to lower the pH as required by the current technology used for seroconversion of B red cells [Citation[28]]. When adequate enzyme mass is obtained, red cell suspension studies will be pursued. If these studies demonstrate efficient deantigenation and conversion to blood type O with intact cell function, then deantigenation of type A2 red cells collected in enzyme/preservative solutions will be investigated. If the latter is successful, then in vivo survival studies of enzymatically converted cells with follow.
Related Research Data
REFERENCES
- Schenkel-Brunner, H. (2001). Biochemical basis of the ABO system subgroups. Wien. Klin. Wochenschr. 113(20–21): 787–798. [PUBMED], [INFOTRIEVE]
- Roseman, S. (2001). Reflections on glycobiology. J. Biol. Chem. 276(45): 41327–41342. [CROSSREF]
- KoScielak, J. (2001). ABH blood group active glycoconjugates from human red cells. Transfus. Med. 11(4): 267–279. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Watkins, W.M. (2001). The ABO blood group system: historical background. Transfus. Med. 11(4): 243–265. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Goodnough, L.T., Brecher, M.E., Kanter, M.H., AuBuchon, J.P. (1999). Transfusion medicine. First of two parts-blood transfusion. N. Engl. J. Med. 340(6): 438–447. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Chien, S. (1986). Purification and characterization of α-N-acetylgalactosaminidase from shrimp hepatopancreas. J. Clin. Chem. Biochem. Soci. 15: 86–96. [CSA]
- Hoskins, L.C., Boulding, E.T., Larson, G. (1997). Purification and characterization of blood group A-degrading isoforms of α-N-acetylgalactosaminidase from Ruminococcus torques Strain IX-70. J. Biochem. Chem. 272(12): 7932–7939.
- Itoh, T., Uda, Y. (1984). α-N-Acetylgalactosaminidase from squid liver: purification and characterization of two enzymes. J. Biochem. 95: 959–970. [PUBMED], [INFOTRIEVE]
- Nakagawa, H., Asakawa, M., Enomoto, N. (1987). Purification and characterization of α-N-acetylgalactosaminidase from skipjack liver. J. Biochem. 101: 855–862. [PUBMED], [INFOTRIEVE]
- Goldstein, J. 1984. Preparation of transfusable red cells by enzymatic conversion, in The Red Blood Cell: Sixth Ann Arbor Conference, New York, G.J. Brewer, Ed., Alan R. Liss, Inc.: New York, pp. 139–157.
- Hata, J., Dhar, M., Mitra, M., Harmata, M., Haibach, F., Sun, P., Smith, D. (1992). Purification and characterization of N-Acetyl-α-D-Galactosaminidase from Gallus domesticus. Biochem. Int. 28: 77–86.
- Hsieh, H.-Y., Smith, D. (2003). Clostridium perfringens α-N-acetylgalactosaminidase blood group A2-degrading activity. Biotechnol. Appl. Biochem. 37: 157–163. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Hsieh, H.-Y., Mitra, M., Wells, D.C., Smith, D. (2000). Purification and characterization of α-N-acetylgalactosaminidase from Clostridium perfringens. IUBMB Life 50: 91–97. [CSA], [CROSSREF]
- Mollison, P.L., Engelfrietand, C.P., Contreras, M. 1997. ABO, Lewis, and P Groups and Ii Antigens, in Blood Transfusion in Clinical Medicine, 10th Ed., P.L. Mollison, C.P. Engelfrietand, M. Contreras, Eds., Blackwell Science Ltd.: Malden, Massachusetts, pp. 116–118
- Harmening, D.M. 1999. The ABO blood group system, in Modern Blood Banking and Transfusion Practices, 3rd ed., D. M. Harmening, Ed., F.A. Davis Company: Philadelphia, Pennsylvania, pp. 104–106.
- Clausen, H., Levery, S.B., Nudelman, E., Tsuchiya, S., Hakomori, S. (1985). Repetitive A epitope (Type 3 Chain A) defined by blood group A1-specific monoclonal antibody Th-1: chemical basis of qualitative A1 and A2 distinction. Proc. Natl. Acad. Sci. U.S.A. 82: 1199–1203. [PUBMED], [INFOTRIEVE], [CSA]
- Goldstein, J. (1989). Conversion of ABO blood groups. Transfus. Med. Rev. 3(3): 206–212. [PUBMED], [INFOTRIEVE]
- Lublin, D.M., Universal RBCs. (2000). Transfusion 40(11): 1285–1289. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Hsieh, H.-Y., Calcutt, M.J., Chapman, L.F., Mitra, M., Smith, D. (2003). Purification and characterization of a recombinant α-N-Acetylgalactosaminidase from Clostridium perfringens. Protein Expr. Purif. 32(2): 309–316. [CSA], [CROSSREF]
- Hoskins, L.C., Larson, G., Naff, G.B. (1995). Blood group A immunodeterminants on human red cells differ in biologic activity and sensitivity to α-N-Acetylgalactosaminidase. Transfusion 35: 813–821. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Hobbs, L., Phillips, R., Smith, D. (1993). An elisa for blood group specific exoglycosidases. J. Immunol. Methods 160: 261–266. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Hobbs, L., Mitra, M., Phillips, R., Smith, D. (1996). The activity of a blood type B specific exoglycosidase from Glycine max. Clin. Chim. Acta. 247: 7–21. [CSA], [CROSSREF]
- Davis, M.O., Hata, J., Johnson, S., Walker, J., Smith, D. (1996). Cloning, expression and characterization of a blood group B active recombinant α-D-Galactosidase from soybean (Glycine max). Biochem. Mol. Biol. Int. 39(3): 471–485. [PUBMED], [INFOTRIEVE], [CSA]
- Vosnidou, N.C., Johnson, S.A., Mitra, M., Wells, D.C., Li, C.Q., Evans, M.L., Harmata, M.A., Walker, J.C., Smith, D.S. (1998). Seroconversion of Type B to O erythrocytes using recombinant Glycine max α-D-Galactosidase. Biochem. Mol. Biol. Int. 46(1): 175–186. [PUBMED], [INFOTRIEVE], [CSA]
- American Association of Blood Banks. 1999. Blood component preparation, storage, shipping, and transportation, in American Association of Blood Banks Technical Manual, 14th Ed., V. Vengelen-Tyler, Ed., American Association of Blood Banks Publication: Bethesda, Maryland, p. 164.
- Code of Federal Regulations, Food and Drugs, Title 21, Parts 600–799, 1994.
- Goldstein, J., Siviglia, G., Hurst, R., Lenny, L.L.L.R. (1982). Group B erythrocytes enzymatically converted to group O survive normally in A, B, and O individuals. Science 215: 168–170. [PUBMED], [INFOTRIEVE]
- Lenny, L.L., Hurst, R., Goldstein, J., Benjamin, L.J., Jones, R.L. (1991). Single-unit transfusion of RBC enzymatically converted from Group B to Group O to A and O normal volunteers. Blood 77: 1383–1388. [PUBMED], [INFOTRIEVE]
- Kruskall, M.S., AuBuchon, J.P., Anthony, K.Y., Herschel, L., Pickard, C., Biehl, R., Horowitz, M., Brambilla, D.J., Popovsky, M.A. (2000). Transfusion to blood group A and O patients of group B RBCs that have been enzymatically converted to group O. Transfusion 40: 1290–1298. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Lenny, L.L., Hurst, R., Goldstein, J., Galbraith, R.A. (1994). Transfusions to Group O subjects of 2 units of red cells enzymatically converted from group B to Group O. Transfusion 34: 209–214. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Zhu, A., Leng, L., Monahan, C., Zhang, Z., Hurst, R. (1996). Characterization of recombinant α-Galactosidase for use in seroconversion from blood group B to O of human erythrocytes. Arch. Biochem. Biophys. 327(2): 324–329. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Sung, S.J., Sweeley, C.C. (1980). Purification and partial characterization of porcine liver α-N-Acetylgalactosaminidase. J. Biochem. Chem. 255(14): 6589–6594.
- Izumi, K., Yamamoto, K., Tochikura, T., Hirabayashi, Y. (1992). Serological study using α-N-Acetylgalactosaminidase from Acremonium Sp. Biochim. Biophys. Acta. 1116: 72–74. [PUBMED], [INFOTRIEVE]
- Hoskins, L.C., Boulding, E.T. (2001). Changes in immunologic properties of Group A RBCs during treatment with an A-Degrading Exo-α-N-Acetylgalactosaminidase. Transfusion 35: 813–812. [CSA], [CROSSREF]
- Hoskins, L.C., Boulding, E.T. Erratum. (2002). Transfusion 42: 662, [CSA]
- Issitt, P.D., Anstee, D.J. 1998. The ABO and H blood group systems, in Applied Blood Group Serology P.D. Issitt, D.J. Anstee, Eds., Montgomery Scientific Publications: Durham, North Carolina, p. 203.