Abstract
The archetypal status of α1‐antitrypsin in biology and medicine grew from the finding, thirty years ago, by Carl‐Bertil Laurell, of the association of its deficiency with emphysema. In biology, α1‐antitrypsin now provides the model for both the structure and the remarkable mechanism of the serpin protease inhibitors that control the key proteolytic pathways of the body. In medicine, the plasma deficiency of α1‐antitrypsin has drawn attention to protease‐antiprotease imbalance as a contributory cause of chronic obstructive pulmonary disease. But even more significantly, the finding that the common genetic deficiency of α1‐antitrypsin was also associated with the development of liver cirrhosis introduced the new entity of the conformational diseases. The proposal that the same general mechanism was responsible for the best known of the conformational diseases, the common late‐onset dementias, was controversial. It was vindicated however by the recent finding that a mutation, which results in the liver aggregation of α1‐antitrypsin, also results in a typical late‐onset dementia when it occurs in a brain‐specific homologue of α1‐antitrypsin. The extensive development of such diverse fields of studies, each based on α1‐antitrypsin, is a measure of the encouragement Laurell gave to younger colleagues in the field. It also reflects the great advantage of linked contributions from clinical as well as basic sciences. Time after time, scientific controversies and deadlocks have been solved by landmark clinical cases, which have revealed unexpected findings and insights, within and beyond the fields of study.
Introduction
The unique contribution of clinical medicine to biological science is its ability to provide totally unexpected leads that open new horizons in understanding and research. Such was the case 30 years ago Citation[1] when the Swedish medical biochemist, Carl‐Bertil Laurell, observed the absence of the alpha‐1 protein band in plasma from two patients in a respiratory‐disease hospital (). The change in the electrophoretic pattern was quickly identified as being due to a deficiency of a plasma protease inhibitor of previously uncertain significance Citation[2], called α1‐antitrypsin. This finding of the deficiency of a protease inhibitor not only revealed a prime mechanism underlying the development of emphysema Citation[1]Citation[3], but also gave a handle that led to the identification of the function and dysfunctions of a whole new family of protease inhibitors—the serpins Citation[4]Citation[5]Citation[6]. The special significance of this family to medicine Citation[7] is that its members, all of which bear a close identity in structure and function to that of α1‐antitrypsin Citation[8], control the crucial proteolytic cascades of the plasma. So the impetus provided by Laurell's observation has directly contributed to the understanding we now have of the way antithrombin controls coagulation, of how heparin modulates this activity and of how mutations in antithrombin result in familial thrombosis. Likewise we can now see how the plasminogen activator inhibitor PAI‐1 controls fibrinolysis, how C1‐inhibitor controls complement activation and similarly how other intracellular as well as extracellular pathways are controlled by the many other members of the serpin family that have now been identified. But the special bonus from Laurell's discovery was the later realisation that the deficiency of α1‐antitrypsin was due to its misfolding and aggregation at its site of synthesis in the liver. The way in which this aggregation in the hepatocytes leads on to liver damage and eventual cirrhosis provided the first detailed model for a whole new category of disorders—the conformational diseases Citation[9]. With this came the implication that the same process, affecting other proteins, would underlie the common dementias such as Alzheimer's and Parkinson's diseases. This prediction was vindicated by the recent finding, in a similar late‐onset dementia, of mutations in a brain‐specific serpin, which are identical in nature and consequence to those in α1‐antitrypsin resulting in cirrhosis Citation[10]Citation[11]. The universality of Laurell's finding to biology as a whole is highlighted by the most recent finding: that a disease phenotype in the fruit‐fly Drosophila Citation[12] results from a mutation precisely identical to that causing the common deficiency of α1‐antitrypsin in people of European descent. The story of the way all of these understandings developed from the original seminal observation by Laurell, is a fascinating one. It is told here from the perspective of a participant in the story who viewed much of the events from opposite poles of the earth—in New Zealand till 1986 and after that in Cambridge, England.
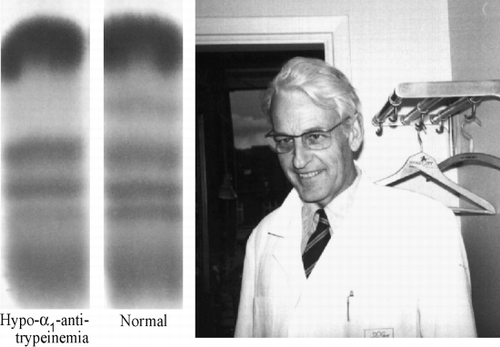
Figure 1. C‐B Laurell (1919–2001). Photographed in Malmö in 1972. Left, the original paper electrophoretic strips from 1962 showing the almost complete absence of the alpha‐1 band from the plasma of a respiratory disease patient.
Finding of the Deficiency
Serendipity leads to discovery when it presents to a receptive and prepared mind. Laurell was 42 years old when he first observed the deficiency of α1‐antitrypsin in 1962. By this time he had already made distinguished contributions to protein research, with the finding and naming in his doctoral thesis of transferrin Citation[13], and subsequently with the recognition of caeruloplasmin Citation[14] and the defining of the function of haptoglobin Citation[15]. Technically, his outstanding contribution was the use of plasma protein electrophoresis as a tool for clinical investigations. This originated with the use of paper electrophoresis following his visit to the laboratory of Tiselius in 1952 and then, in 1961, in the introduction of the much more selective agarose gel electrophoresis Citation[16]. The power of these techniques is well illustrated by their usage by a haematological physician, J Waldenström, whose office was adjacent to Laurell's laboratory in Malmö. Together they described Citation[17] the finding of ‘M’ proteins in myeloma leading to the recognition of the eponymously named Waldenström's macroglobulinaemia. Other medical investigators in Malmö took advantage of the new tool of electrophoresis and in 1961 a newly appointed senior respiratory physician introduced its routine use for all his patients. Laurell personally checked every electrophoretic result and noted the absence of the alpha‐1 band in two samples, both of which had originated from the specialist respiratory hospital. Laurell then contacted a young physician‐investigator, Sten Eriksson, to take part in a joint clinical laboratory survey. Eriksson did this with enthusiasm and thoroughness. Two further examples were found on a survey of past electrophoretic strips and then two further new examples were identified. The critical finding was that four of these first six cases of alpha 1 deficiency had emphysema. Laurell and Eriksson published their findings Citation[1]Citation[3] in 1963, with Eriksson giving the first comprehensive account of the clinical syndrome associated with the electrophoretic deficiency, in his thesis of 1965 Citation[18].
The achievements of Eriksson underline a special strength of Carl‐Bertil Laurell, in not only being able to inspire others but also in providing an initial impetus to their work and then stepping aside. The next decade was a time of extraordinary productivity in Malmö. Ohlsson and Ganrot and then Jeppsson joined Laurell in the biochemical investigation of the deficiency of α1‐antitrypsin. In addition two early clinical studies were published that are landmarks in the field—Larsson provided what is still the definitive description Citation[19] of the interaction of smoking with the deficiency and Sveger commenced the study of affected newborns that now defines the natural history of the syndrome Citation[20]. Young investigators from abroad also came to Malmö (), and two of these, Magne Fagerhöl from Norway and Diane Cox from Canada, led the work that clarified the complexities of the genetic variants of α1‐antitrypsin Citation[21]Citation[22]. It is necessary to recall now the difficulties they faced. In the 1960's haemoglobin provided the only defined molecular model for protein variation in human blood. But unlike haemoglobin, α1‐antitrypsin is a glycoprotein and as such moves as a series of bands on electrophoresis quite unlike the clear single change in mobility observed with the abnormal haemoglobins. So in the first years following the discovery of the deficiency of α1‐antitrypsin it was thought that the underlying abnormality was one of glycosylation, rather than of a single amino acid substitution as seen with Hb S and the other variants of haemoglobin. Furthermore, although as we now know, the Z variant of α1‐antitrypsin is responsible for its severe deficiency, the European also carries, at even greater frequency, the milder deficiency S variant. Fagerhöl and Cox systematically defined the different genetic variants and their frequencies of occurrence by introducing the Pi system of electrophoretic analysis Citation[23]Citation[24].
Figure 2. Laurell's initial finding soon inspired international research. Here at the Congress of Human Genetics in Paris in 1972 are, on the right Magne Fagerhol from Norway and Diane Cox from Canada, and in the foreground the author from New Zealand. The need at that time was to explain the complex electrophoretic banding of antitrypsin (below) which was categorised by Fagerhol and Cox with the Pi nomenclature Citation[22] and later shown Citation[109] with Jeppsson Citation[50] and others Citation[66]Citation[110], to be due to glycoforms and post‐translational changes as depicted to left.
![Figure 2. Laurell's initial finding soon inspired international research. Here at the Congress of Human Genetics in Paris in 1972 are, on the right Magne Fagerhol from Norway and Diane Cox from Canada, and in the foreground the author from New Zealand. The need at that time was to explain the complex electrophoretic banding of antitrypsin (below) which was categorised by Fagerhol and Cox with the Pi nomenclature Citation[22] and later shown Citation[109] with Jeppsson Citation[50] and others Citation[66]Citation[110], to be due to glycoforms and post‐translational changes as depicted to left.](/cms/asset/8db02afd-1bf2-4b2b-bd51-fc4bd84a45fa/icop_a_11176889_uf0002_b.gif)
The Alpha‐1 Syndrome
The recent suggestion by John Humphries, Robert Stockley and others, that the clinical manifestations accompanying the common homozygous Z state be referred to as a syndrome rather than a deficiency, deserves encouragement. It is too late to change the name of α1‐antitrypsin itself, which although a mouthful and a misnomer (it will be also referred to here as antitrypsin), has become irrevocably established in the literature. But the description of the clinical abnormality as ‘α1‐antitrypsin deficiency’ continues to be misleading as well as confusing. Although the plasma deficiency of the Z homozygote is directly related to the predisposition to obstructive lung disease, the label of α1‐antitrypsin deficiency obscures what is a major pathological consequence of the ZZ state—a variably progressive loss of hepatocytes with the potential development of liver cirrhosis Citation[25]. The early studies of the Malmö group noted the relationship of α1‐antitrypsin deficiency with liver disease but the essential link between the two became apparent with the report in 1971 by a US paediatrician, Harvey Sharp Citation[26], of the presence of inclusions of antitrypsin in the livers of children, homozygous for the Z mutation, who developed a rapidly progressive cirrhosis Citation[27]. Once again it was studies from Malmö that put this finding into context. Sveger, in his comprehensive study of newborn Z homozygotes Citation[20]Citation[28], showed that near 10% developed a neonatal cholestasis with progression, in some 1–2% overall, to a juvenile cirrhosis. This tragic but relatively uncommon consequence of Z homozygosity overshadowed the much more frequent occurrence of a slowly progressive portal fibrosis, which in some 20% of homozygotes results in cirrhosis in later adult life Citation[19]Citation[29]. A key contribution to fitting all these findings together, within the framework of the molecular pathology of the alpha‐1 syndrome, was made by Jeppsson Citation[30] who showed that the hepatocyte inclusions Citation[31], which characterised the liver disease, were solely formed of the intact variant Z protein.
The original observation of the association of the plasma deficiency of α1‐antitrypsin with emphysema provided an immediate stimulus to research into mechanisms of obstructive lung disease. Although this was initiated in Sweden Citation[32] much of the activity was in the US, and in particular from groups with specialist respiratory interests including those of Lieberman Citation[33], Janoff Citation[34], Pierce Citation[35], Senior Citation[36] Snider Citation[37] and Crystal Citation[38]Citation[39]. Their work established the role of antitrypsin as an inhibitor of the elastase released by activated leukocytes. From this came the concept of the protease‐antiprotease balance as an essential requirement for respiratory health, with perturbations of this balance resulting in the progressive loss of lung elasticity. The findings opened insights into the contribution of proteolytic overload to the much more common occurrence of emphysema due to tobacco smoking. Biochemical and cellular studies Citation[40]Citation[41]Citation[42]Citation[43]Citation[44] studies showed the vulnerability of α1‐antitrypsin to oxidative inactivation (though disappointingly there has been little recent follow‐up of these findings of two decades ago). The predictable additive effect of smoking in conjunction with a plasma deficiency antitrypsin, fitted with the earlier findings of Larsson Citation[19] and others Citation[45] showing a catastrophic acceleration of the age of onset of emphysema in Z homozygotes who were also current or ex‐smokers.
The S and Z Variants
A theme of this review is the way an original seminal finding is disseminated and inspires the work of others. This dissemination occurs through multiple interactions that are appropriately illustrated at this stage if the author tells the story of his own involvement. This dates from my doctoral studies, on the molecular pathology of haemoglobin, in the Biochemistry Department of the University of Cambridge. With the completion of this work in 1967, came an invitation to speak at a British Council course for young investigators from Europe. After the talk I was approached by one of the participants in the course, Magne Fagerhöl, who told of the findings in Malmö and of the perplexing complexities of the variants of α1‐antitrypsin as revealed by the Pi system. He suggested that the technology and structural approach taken with the abnormalities of haemoglobin could also be used to determine the molecular basis of the Pi variants. The idea had an immediate attraction to me, as I was just about to return to a medical post in New Zealand where the population was predominantly of British descent and hence where there were relatively few instances of haemoglobinopathies. Moreover, this population predictably had the typical Northern European occurrence of the variants of α1‐antitrypsin, with 4% being MZ heterozygotes and 7% MS heterozygotes Citation[46]. So, having set up the technology for protein isolation and peptide mapping in Christchurch New Zealand, the search commenced to identify the molecular basis of this variation.
Initial studies focused on the arcane multiple banding of the Z protein, in the belief that the blocking of the intracellular processing of the Z protein resulted from aberrant glycosylation. It soon became apparent however, that the underlying abnormalities of the antitrypsin variants, like those of the abnormal haemoglobins, were due to single amino acid substitutions. During a visit to Laurell in 1972, I told him of our plans to analyse the S variant of α1‐antitrypsin by mapping its tryptic peptides. Malmö, by this time, had in place a system for the large‐scale isolation of the Z variant, which they planned to similarly peptide‐map. By 1975, when Laurell visited New Zealand, the peptide mapping of the S variant had been completed Citation[47] and we were able to show him that its abnormality was due to the substitution of a glutamic acid by a valine. Soon after this the abnormality of Z antitrypsin was solved by Jeppsson Citation[48], coincidental with the independent success of Yoshida and colleagues Citation[49] in the US. Both groups identified the replacement of a glutamic acid, different from that in the S variant, by a lysine.
The Reactive Site and the Pittsburgh Variant
An unexpected bonus from the identification of the S and Z variants came with the sequencing of the peptides adjacent to the mutations Citation[50]. As we now know, the S mutation affects the glutamate at residue 264 and the Z mutation the glutamate at 342. So further sequencing, by Owen and Brennan from the Christchurch group, soon bridged the two mutations to give a substantial fragment of the overall sequence of α1‐antitrypsin Citation[51]Citation[52]Citation[53]. The bonus was the finding that this extended sequence also contained a peptide fragment Citation[54] that had previously been recognised by James Travis from Athens Ga, as the putative reactive centre of the molecule Citation[40]Citation[55]. This intuitive deduction by Travis was based on the close homology of the sequence of this fragment with that of the reactive centres in the smaller and totally unrelated protease inhibitors of beans and other plants (). But at this stage a controversy arose. The large polypeptide sequence from α1‐antitrypsin, determined in New Zealand Citation[56] and independently in Japan Citation[57], was shown by Boswell Citation[58] to be homologous to that of the carboxy‐terminus of another plasma protease inhibitor, antithrombin. Moreover, the alignment of the two sequences clearly identified homologous reactive centres in both molecules. The controversial conclusion from this, that the reactive centre of antitrypsin was near the carboxy‐terminus of the molecule, was directly contrary to the findings of numbers of other groups, principally in the USA, who sited it at the amino‐terminus. The reason for this geographical polarisation of results later became clear. The US groups could afford the newly introduced automated peptide‐sequenators, as opposed to the tedious manual sequencing techniques used elsewhere. But the sequenators brought with them an unrecognised trap, as their preparative procedures can result in the cyclisation and hence blockage of amino‐terminal glutamates as present in α1‐antitrypsin. As a consequence only a single sequence appeared following cleavage of the active centre of antitrypsin, implying an amino‐terminal placement of the active site. The sequenator results, all from first‐class laboratories, were so consistent in supporting this apparent amino‐terminal placement that the controversy persisted Citation[59]Citation[60] well after other results unambiguously showed the opposite to be true. The way the debate was finally settled provides a clear example of how a clinical finding can break through scientific deadlocks to open unexpected new understandings.
Figure 3. Antitrypsin Pittsburgh. Alignment of the sequences (above), and comparison with the known reactive centers of the smaller protease inhibitors (below) revealed the homologous reactive centers of antithrombin and α1‐antitrypsin. This deductive siting was confirmed by the identification by Owen Citation[62] of the mutation in the Pittsburgh variant of α1‐antitrypsin, where the mutation of the reactive center methionine to arginine converted the antitrypsin to an inhibitor of thrombin. The use of agarose gel introduced by Laurell Citation[16] greatly increased the discrimination of plasma protein electrophoresis, as illustrated on left with the separation of the Pittsburgh (C) from normal (B) α1‐antitrypsin. Note also the presence of proalbumin (A) and the variation in all these components in quiescent (April 1980) versus the acute phase (September 1981) states (reproduced with permission from Ref. Citation[62] Copyright Massachusetts Medical Society). (View this art in color at www.dekker.com.)
![Figure 3. Antitrypsin Pittsburgh. Alignment of the sequences (above), and comparison with the known reactive centers of the smaller protease inhibitors (below) revealed the homologous reactive centers of antithrombin and α1‐antitrypsin. This deductive siting was confirmed by the identification by Owen Citation[62] of the mutation in the Pittsburgh variant of α1‐antitrypsin, where the mutation of the reactive center methionine to arginine converted the antitrypsin to an inhibitor of thrombin. The use of agarose gel introduced by Laurell Citation[16] greatly increased the discrimination of plasma protein electrophoresis, as illustrated on left with the separation of the Pittsburgh (C) from normal (B) α1‐antitrypsin. Note also the presence of proalbumin (A) and the variation in all these components in quiescent (April 1980) versus the acute phase (September 1981) states (reproduced with permission from Ref. Citation[62] Copyright Massachusetts Medical Society). (View this art in color at www.dekker.com.)](/cms/asset/04544362-17cc-4e59-aaf8-e03220867393/icop_a_11176889_uf0003_b.gif)
Dr Jessica Lewis, a haematologist in Pittsburgh Pa, reported in 1978 a case of a severe bleeding disorder, due to a variant antitrypsin that acted as an inhibitor of the thrombin‐fibrinogen interaction Citation[61]. Soon after this, the recognition of the homology and nature of the reactive sites of antitrypsin and antithrombin suggested a likely explanation, that was confirmed Citation[62] by the analysis of the variant Pittsburgh antitrypsin (). The peptide map of the Pittsburgh variant showed the replacement of the reactive centre methionine by an arginine, resulting in a change of function of the antitrypsin, from being an inhibitor of elastase to that of a highly active inhibitor of coagulation proteases. The finding unequivocally confirmed the carboxy‐terminal position of the active site and showed how the specificity of this family of inhibitors was critically dependent on the amino acid at its reactive centre. This natural experiment in protein engineering provided the first example of the way in which a single mutation can completely change the function of a protein. Furthermore, the independence of this new inhibitor of coagulation from heparin control also settled another debate, in thrombosis research, by showing that heparin acts directly on antithrombin by activating an inherent inhibitory activity. Yet another finding from this remarkable case opened a new field in biochemistry. The investigation by Brennan Citation[63]Citation[64] of the unexpected presence of proalbumin in the plasma of the Pittsburgh patient led to the first (and still largely unacknowledged) recognition of the nature and identity of the propeptide‐cleaving enzyme that has a key function in metabolism and endocrinology.
Structure and Defect in Processing
By 1978 a close collaboration had been established between the Malmö and Christchurch groups with the aim of characterising the full sequence and the nature of the heterogeneity of α1‐antitrypsin. The protein sequence was completed in 1982, to give definitive placements of the reactive site, the S and Z mutations and the points of attachment of the three oligosaccharide sidechains of the molecule Citation[65]. The multiple electrophoretic banding of α1‐antitrypsin was shown to be primarily due to the presence of different glycoforms of the protein, each with variations in the antennary structure Citation[66] of their oligosaccharides Citation[67]. Further contributions to the heterogeneity due to polymorphisms and post‐translational modifications were apparent on comparison of the protein and cDNA sequences of α1‐antitrypsin Citation[68]. The Sweden:New Zealand collaboration continued further, to establish the cause of the deficiency of the Z variant. Bathurst, Stenflo and others Citation[69]Citation[70]Citation[71] using mRNA isolated from normal (MM) and variant (ZZ) livers, showed both messages had equivalent rates of translation and of subsequent glycosylation in cell‐free systems. Injection of the mRNAs into a surrogate cell, the toad oöcyte, by Foreman and by Errington Citation[72]Citation[73], confirmed that there was equivalent initial synthesis of the two proteins. But whereas all of the M protein was secreted, most of the Z protein was blocked within the oöcyte at the final stage of processing. The conclusion by 1986 from these studies was that the mutation in Z antitrypsin probably “caused a minor perturbation of structure that affects the solubility of the incompletely processed protein Citation[74]Citation[75].”
It was clear that any further understanding of the molecular pathology of the alpha‐1 syndrome required a detailed knowledge of the three‐dimensional structure and function of α1‐antitrypsin. Huber and colleagues achieved the breakthrough in Munich in 1984 by solving the crystallographic structure of α1‐antitrypsin Citation[76], using material provided by Laurell. This first structure was initially considered a disappointment, as the antitrypsin was not in the active inhibitory form but had been cleaved at its reactive centre. In fact this unintentional cleavage provided a stimulus to research in the field far beyond that expected from just the structure itself. The cleaved form, with its change from a five‐stranded to a six‐stranded A‐sheet () provided an enigma and a challenge to structural biology and biochemistry as a whole. No protein had been known to undergo such a remarkable change in its folding. Subsequent completion of the structure of intact α1‐antitrypsin Citation[77] confirmed that cleavage of the reactive centre results in its displacement to the other end of the molecule, through a distance (huge in molecular terms) of 70Å. The reason for this extraordinary conformational change became apparent with the solving of the structure of the antitrypsin‐trypsin inhibitory complex by Huntington in 2000 Citation[78] (). The solving of this structure completed what is now a video sequence of structures showing how this unique mobile mechanism gives the serpin family of protease inhibitors the selective advantage that makes them the predominant inhibitors in higher organisms. The forced displacement drastically distorts the structure of the protease and results in its irreversible inactivation. Thus α1‐antitrypsin is a suicidal protein that protects the elastic tissue of the lung by efficiently and irreversibly inhibiting any elastases or other serine proteases, which diffuse away from the immediate periphery of inflammatory leukocytes.
Figure 4. Structure of α1‐antitrypsin. (a) The first crystallographic structure of α1‐antitrypsin on the right Citation[76] showed how the cleaved reactive center loop (yellow) is incorporated as a middle strand in the main β‐sheet of the molecule (red). The intact molecule on the left shows the exposed centre. The vulnerable hinge of the loop, where the Z mutation occurs, is arrowed, and the equally vulnerable sliding shutter region is shown circled. (b) The reason for this extraordinary change in conformation is apparent from the structure of the complex of trypsin and α1‐antitrypsin. The cleaved serpin is seen to violently displace the protease (in purple) causing its loss of structure and consequent inactivation Citation[78]. (View this art in color at www.dekker.com.)
![Figure 4. Structure of α1‐antitrypsin. (a) The first crystallographic structure of α1‐antitrypsin on the right Citation[76] showed how the cleaved reactive center loop (yellow) is incorporated as a middle strand in the main β‐sheet of the molecule (red). The intact molecule on the left shows the exposed centre. The vulnerable hinge of the loop, where the Z mutation occurs, is arrowed, and the equally vulnerable sliding shutter region is shown circled. (b) The reason for this extraordinary change in conformation is apparent from the structure of the complex of trypsin and α1‐antitrypsin. The cleaved serpin is seen to violently displace the protease (in purple) causing its loss of structure and consequent inactivation Citation[78]. (View this art in color at www.dekker.com.)](/cms/asset/6f12f83d-c630-4ac5-9d9d-0cbe9afe709f/icop_a_11176889_uf0004_b.gif)
The Serpins
The birth of the serpins came from the initial alignment of the carboxy‐terminal sequence of α1‐antitrypsin Citation[79] with that of antithrombin Citation[80] (). But it was the recognition of a third member, the egg white protein ovalbumin that established the homologies as a new family of serine protease inhibitors Citation[81]. This family, which we now know as the serpins, was initially called the α1‐antitrypsin‐antithrombin III‐ovalbumin superfamily of serine proteinase inhibitors. The renaming Citation[4] with the acronym, the serpins, was not only a matter of convenience but was made with the deliberate intention of encouraging the study of the family as a whole. This reflected my much earlier experience with haemoglobin and its abnormalities. It is generally believed that the pioneer studies on the molecular function and pathology of haemoglobin were based on Perutz's crystal structure of haemoglobin; but this assumption is only partly correct. In reality, much of the early work, and almost all the studies of the molecular pathology of haemoglobin, were based on Kendrew's higher resolution structure of myoglobin. But the real insights in these early studies came from the alignment of the two globins on the basis of their shared three‐dimensional structure Citation[82], which yielded information well beyond that obtained from the study of the individual proteins. Thus the serpins were named on the precept of the globins, with the expectation of the dividends in knowledge that would come, as with the globins, from their homologous alignments. These expectations of twenty years ago have been richly rewarded. Based on α1‐antitrypsin as the archetype of the family, we now know that the serpins, diverse though they are in their functions, all share the same overall structure and have the same molecular mechanism of action Citation[5]Citation[8]Citation[83]Citation[84]Citation[85]Citation[86]. Even more significantly they share the same molecular pathology Citation[87]. The same mutations that in α1‐antitrypsin predispose to cirrhosis, are found in antithrombin to result in thrombosis, in C1‐inhibitor to result in angioedema and in neuroserpin in dementia Citation[7]Citation[11]. A notable example is the common Z variant of α1‐antitrypsin due to the mutation of a glutamate to a lysine, at the hinge‐point of the reactive site loop (,). The pathological significance of this particular mutation has been highlighted by its independent occurrence in two other serpins: in heparin cofactor II in man Citation[88], where it results in a loss of thrombin inhibitory activity, and in the fruit‐fly Drosophila where it causes a necrotic syndrome Citation[12].
Polymerisation and Deficiency
The study of the serpins as a family soon highlighted their conformational flexibility Citation[8]Citation[89]. This was evident not only from the folding rearrangements in the first structure of α1‐antitrypsin () but also from the identification of mutations in antithrombin and C1‐inhibitor that resulted in a loss of inhibitory activity Citation[87]. These dysfunctional mutations almost all occurred in the mobile hinges of the molecule. Of special interest, was a group of mutations affecting the region critical to the shutter‐like opening and closure of the main β‐pleated sheet of the molecule (). Mutations in this shutter region Citation[90]Citation[91] resulted in a plasma deficiency and liver aggregation of α1‐antitrypsin, identical to that associated with the common Z variant. These changes differ from those of most genetic deficiency diseases, which usually result from a complete deficiency of an underlying protein, due to a failure of its synthesis or initial folding. As opposed to this, the Z and shutter mutations in α1‐antitrypsin result in only a partial deficiency, with 15% of the Z protein being secreted into the plasma. With such conformationally unstable variants however, the bulk of the newly synthesised antitrypsin aggregates in the endoplasmic reticulum of the hepatocyte, predisposing to eventual cirrhosis. It is this added‐on, or gain‐of‐function disadvantage, as opposed to a simple deficiency, that typifies what we now know as the conformational diseases.
By 1990 then, there were several outstanding questions. What is the cause of the partial deficiency? Why does the antitrypsin aggregate at this final stage of intracellular processing? What is the unique links between mutations affecting the mobile regions of the molecule and the subsequent development of cirrhosis? The first answers to these questions came from an unexpected source ‐ the white of an egg. It was the completion by Stein of the crystal structure of the egg‐white serpin, ovalbumin, which revealed the hinge and sliding movements involved in the conformational change that typifies the family Citation[92]Citation[93]Citation[94]. With this understanding and from accompanying studies of antithrombin, it became apparent that serpins could undergo spontaneous and pathological changes in their conformation—but what was the precise change that caused the intracellular accumulation of α1‐antitrypsin? The solving, or more descriptively the untangling, of this problem came from the work of a young trainee respiratory physician, David Lomas, who joined the Cambridge group as a research fellow in 1990. He showed Citation[95] that the Z antitrypsin readily formed polymers, due to the insertion of the reactive loop of one molecule into the prematurely opened β‐sheet of the next (). Electron microscopy not only demonstrated the formation of necklace‐like polymers when Z α1‐antitrypsin was incubated in vitro, but also confirmed that inclusions extracted from the liver were formed of entanglements of these polymers.
Figure 5. Polymerisation and inclusion bodies. Upper: Z antitrypsin readily undergoes loop‐sheet linkage Citation[95] to give bead‐like polymers (left) which become entangled (right) to form intracellular inclusions (electron micrographs × 220 000). Below ( × 20 000). Inclusions formed of entangled polymers of conformationally unstable serpins are seen with antitrypsin in the rough endoplasmic of reticulum of hepatocytes (on left), and with homologous mutations in neuroserpin in neurons, on right Citation[10].
![Figure 5. Polymerisation and inclusion bodies. Upper: Z antitrypsin readily undergoes loop‐sheet linkage Citation[95] to give bead‐like polymers (left) which become entangled (right) to form intracellular inclusions (electron micrographs × 220 000). Below ( × 20 000). Inclusions formed of entangled polymers of conformationally unstable serpins are seen with antitrypsin in the rough endoplasmic of reticulum of hepatocytes (on left), and with homologous mutations in neuroserpin in neurons, on right Citation[10].](/cms/asset/accfff0b-fb4b-4afc-a3f7-1b144cec7b7a/icop_a_11176889_uf0005_b.gif)
Strong supporting evidence for the central role of polymerisation came from an antitrypsin variant found in Japan Citation[91] that was also associated with inclusion body formation. This Siiyama variant had a replacement of a critical amino acid (Ser 53) in the shutter region of the molecule. Isolation of the Siiyama antitrypsin from the plasma confirmed the prediction that it would readily, and in fact spontaneously, undergo polymerisation Citation[96]. Foreman, using the toad oöcyte as a surrogate for the hepatocyte, elegantly confirmed the relationship between the molecular instability and consequent partial aggregation of the variant antitrypsins Citation[97]. The conclusion that the molecular pathology of Z α1‐antitrypsin was a direct consequence of its polymerisation was in keeping with the earlier observation of Cox Citation[98] of the spontaneous aggregation of plasma Z antitrypsin. However, the findings of Sifers Citation[99] suggested that the aggregation was primarily due to an impairment of intracellular degradation of the Z protein, with Perlmutter's results Citation[100] indicating an additional genetic contribution to this impaired turnover. So the central contribution of polymerisation to the disease syndrome remained a matter of debate until it was finally settled by a totally unexpected and landmark clinical case. And this arose because a general practitioner in upstate New York insisted on an autopsy on a patient with an atypical Alzheimer's disease.
Conformational Disease and Dementia
The misfolding Citation[99]Citation[101] and polymerisation Citation[95] of Z antitrypsin explained the deficiency of secretion into the plasma and hence the failure to effectively protect the lungs against proteolytic attack. But it was the consequences of the intracellular aggregation of the polymerised antitrypsin that opened wider concepts and led to the proposal of the new entity of the conformational diseases Citation[9]. These diseases each arise when an underlying protein undergoes a change in size or conformation with resultant self‐association and tissue deposition. Characteristically there is intracellular aggregation of the polymerised protein with, as in the alpha‐1 syndrome, a gradual and cumulative cell loss that results in a slow onset of disease. In particular, it was apparent that the cirrhosis of the Z homozygote provided a model for the progressive neurodegeneration leading to the common dementias such as Alzheimers and Parkinson's disease, as well as the prion encephalopathies Citation[102]Citation[103]Citation[104]. But not surprisingly there was a reluctance to accept that α1‐antitrypsin deficiency, which was commonly considered to be a rare genetic respiratory disorder, could act as a valid model for neurodegenerative diseases. The findings that broke through this conceptual deadlock came from the autopsy on the patient with the atypical Alzheimer‐like dementia.
The autopsy showed the presence of numerous inclusion bodies in the neurones of the brain. These inclusions had a striking similarity in appearance and histological staining to the inclusions seen in the liver in Z α1‐antitrypsin cirrhosis. The reason for this became apparent from investigations by Richard Davis and colleagues in Syracuse, who showed the inclusions were composed of a brain‐specific serpin‐neuroserpin Citation[105]. Furthermore, genetic analysis showed the presence in the propositus and in other affected family members, of a mutation in neuroserpin at the same site as the mutation (Ser 53) that is responsible for the highly polymerogenic Siiyama variant of α1‐antitrypsin. Lomas and colleagues showed that this late‐onset dementia shared the same detailed molecular pathology as the antitrypsin‐cirrhosis, by confirming the ready polymerisation of the mutant neuroserpin and the presence of entanglements of polymers in the neuronal inclusions Citation[10]. The central contribution of polymerisation to the molecular pathology has now been put beyond any doubt by the findings of further familial encephalopathies with neuroserpin inclusion bodies (FENIB) due to other mutations in neuroserpin Citation[106]. All of these mutations are identical to those independently known to cause liver inclusions with α1‐antitrypsin or episodic thrombosis with antithrombin Citation[11]Citation[107]. Thus the overall findings firmly establish the conformational basis of the alpha‐1 syndrome, with the central feature being the polymerisation of the conformationally unstable variants Citation[7]. The findings similarly support the deduction that the common encephalopathies and dementias arise from the conformational instability and aggregation of individual neuroproteins; with the onset and severity of the neurodegeneration being associated with the rate and magnitude of protein aggregation Citation[11]Citation[106].
A practical bonus from the realisation of the shared molecular pathology of the serpinopathies and the conformational dementias, is the prospect of shared approaches to therapy. Pharmaceutical companies are now making a huge research effort with the incentive of finding treatments for Alzheimer's and Parkinson's disease, which is also likely have relevance for the treatment of the serpinopathies. In return the serpins, and α1‐antitrypsin in particular, currently provide the only molecularly‐defined examples of conformational diseases on which to establish the design of structure based therapies.
Conclusion
This has been an account, from a personal view‐point, of how the seminal finding of Carl‐Bertil Laurell in 1962 opened completely new fields of understanding in biology and medicine. The finding of the deficiency of α1‐antitrypsin provided the handle and the motivation for research that has established α1‐antitrypsin as an archetypal protein, not only for the serpins but also for the elucidation of the conformational diseases in general. It is appropriate that this account should be published in a respiratory journal, as the key feature of this initial handle was the association of the deficiency with the development of emphysema. A theme too, in this account, has been the contribution of clinical medicine in providing the breakthroughs that have placed α1‐antitrypsin and the serpins at the forefront of biomedical research. A respiratory physician in Malmö decided to carry out plasma electrophoresis on all his patients, a haematologist in Pittsburgh followed‐up an unusual haemorraghic disorder, physicians in Japan further investigated unusual liver inclusions, and a practitioner and pathologists in up‐State New York pursued a case of atypical neurodegenerative disease. The bringing together of all these disparate findings is a consequence of the way Laurell provided, with α1‐antitrypsin, an immediate focus for the field and an inspiration for future research. He backed up his initial finding with the development of new technologies, such as rocket immunoelectroassay Citation[108] that facilitated this research. But most of all he motivated his younger colleagues by the precepts he set in the rigour and integrity of his science, together with the warmth of his personality and his enthusiasm for research.
Acknowledgments
Prof J‐O Jeppsson (Malmö) kindly provided details for Prof Laurell's biography and also the electrophoresis depiction in , and Prof David Lomas (Cambridge) read the draft of the paper. The author's research is supported by The Wellcome Trust and the Alpha‐1 Foundation.
References
- Laurell C‐B, Eriksson S. The electrophoretic α1‐globulin pattern of serum in α1‐antitrypsin deficiency. Scand J Clin Lab Invest 1963; 15: 132–140
- Schultze H E, Heide K, Haupt H. Alpha‐1‐antitrypsin aus humanserum. Klin Wochenschr 1962; 40: 427–429, [PUBMED], [INFOTRIEVE]
- Eriksson S, Laurell C‐B. A new abnormal serum globulin alpha‐1 antitrypsin. Acta Chem Scand 1963; 17: 150–153
- Carrell R, Travis J. α1—antitrypsin and the serpins: variation and countervariation. Trends Biochem Sci 1985; 10: 20–24, [CROSSREF]
- Silverman G A, Bird P I, Carrell R W, Church F C, Coughlin P B, Gettins P G, Irving J A, Lomas D A, Luke C J, Moyer R W, Pemberton P A, Remold‐O'Donnell E, Salvesen G S, Travis J, Whisstock J C. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J Biol Chem 2001; 276: 33293–33296, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Gettins P G. Serpin structure, mechanism and function. Chem Rev 2002; 102: 4751–4804, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Carrell R W, Lomas D A. Alpha1‐antitrypsin deficiency—a model for conformational diseases. N Engl J Med 2002; 346: 45–53, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Huber R, Carrell R W. Implications of the three‐dimensional structure of α1—antitrypsin for structure and function of serpins. Biochemistry 1989; 28: 8951–8966, [PUBMED], [INFOTRIEVE]
- Carrell R W, Lomas D A. Conformational diseases. Lancet 1997; 350: 134–138, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Davis R L, Shrimpton A E, Holohan P D, Bradshaw C, Feiglin D, Sonderegger P, Kinter J, Becker L M, Lacbawan F, Krasnewich D, Muenke M, Lawrence D A, Yerby M S, Shaw C‐M, Gooptu B, Finch J T, Carrell R W, Lomas D A. Familial dementia caused by polymerisation of mutant neuroserpin. Nature 1999; 401: 376–379, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Lomas D A, Carrell R W. Serpinopathies and the conformational dementias. Nat Rev, Genet 2002; 3: 759–768, [CROSSREF]
- Green C, Brown G, Dafforn T R, Reichhart J M, Morley T, Lomas D A, Gubb D. Drosophila necrotic mutations mirror disease‐associated variants of human serpins. Development 2003; 130: 1473–1478, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Laurell C‐B. Studies on the transportation and metabolism of iron in the body. Acta Physiol Scand 1947; 14: 1–129
- Holmberg C G, Laurell C‐B. Investigations in serum copper. II. Isolation of the copper containing protein, and a description of some of its properties. Acta Chem Scand 1948; 2: 550–556
- Laurell C‐B, Nyman M. Studies on the serum haptoglobin level in hemoglobinemia and its influence on renal excretion of hemoglobin. Blood 1957; 12: 493–506, [PUBMED], [INFOTRIEVE]
- Laurell C‐B, Lundh B. An electrophoretic method for estimation of some β‐globulins. Scan J Clin & Lab Invest 1962; 14: 490494
- Laurell C‐B, Laurell H, Waldenström J. Glycoproteins in serum from patients with myeloma, macroglobulinemia and related conditions. Am J Med 1957; 22: 24–36, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Eriksson S. Studies in α1‐antitryspin deficiency. Acta Med Scand 1965; suppl 432: 1–85
- Larsson C. Natural history and life expectancy in severe alpha1‐antitrypsin deficiency, PiZ. Acta Med Scand 1978; 204: 345–351, [PUBMED], [INFOTRIEVE]
- Sveger T. Liver disease in alpha1‐antitrypsin deficiency detected by screening of 200,000 infants. N Eng J Med 1976; 294: 1316–1321
- Fagerhol M K, Laurell C‐B. The polymorphism of 'prealbumins' and α1‐antitrypsin in human sera. Clin Chim Acta 1967; 16: 199–203, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Fagerhol M K, Cox D W. The Pi polymophism. Genetic, biochemical, and clinical aspects if human α1‐antitrypsin. Advances in Human Genetics. Plenum Press, New York 1981; 1–62; 371–372
- Fagerhol M K. The Pi system. Genetic variants of α1‐antitrypsin. Ser Haematol 1968; 1: 153–161
- Cox D W. New variants of α1‐antitrypsin: comparison of Pi phenotypng techniques. Am J Hum Genet 1981; 33: 354–365, [PUBMED], [INFOTRIEVE]
- Eriksson S, Carlson J, Velez R. Risk of cirrhosis and primary liver cancer in alpha1‐antitrypsin deficiency. N Eng J Med 1986; 314: 736–739
- Sharp H L. Alpha‐1‐antitrypsin deficiency. Hosp Pract 1971; 6: 83–96
- Sharp H L, Bridges R A, Krivit W, Freier E F. Cirrhosis associated with alpha‐1‐antitrypsin deficiency: a previously unrecognised inherited disorder. J Lab Clin Med 1969; 73: 934–939, [PUBMED], [INFOTRIEVE]
- Sveger T. α1‐antitrypsin deficiency in early childhood. Pediatrics 1978; 62: 22–25, [PUBMED], [INFOTRIEVE]
- Berg N O, Eriksson S. Liver disease in adults with alpha‐1‐antitrypsin deficiency. N Engl J Med 1972; 287: 1264–1267, [PUBMED], [INFOTRIEVE]
- Jeppsson J‐O, Larsson C, Eriksson S. Characterization of α1‐antitrypsin in the inclusion bodies from the liver in α1‐antitrypsin deficiency. N Engl J Med 1975; 293: 576–579, [PUBMED], [INFOTRIEVE]
- Eriksson S, Larsson C. Purification and partial characterization of PAS‐positive inclusion bodies from the liver in alpha1‐antitrypsin deficiency. NEJM 1975; 292: 176–180, [PUBMED], [INFOTRIEVE]
- Balldin G, Ohlsson K, Olsson A S. Studies on the influence of Trasylol on the partition of trypsin between the human plasma protease inhibitors in vitro. Hoppe Seylers Z Physiol Chem 1978; 359: 691–697, [PUBMED], [INFOTRIEVE]
- Lieberman J, Gaidults L, Schleissner L A. Intermediate alpha1‐antitrypsin deficiency resulting from a null gene (M‐phenotype). Chest 1976; 70: 532–535, [PUBMED], [INFOTRIEVE]
- Janoff A, Carp H, Lee D K, Drew R T. Cigarette smoke inhalation decreases α1‐antitrypsin activity in rat lung. Science 1979; 206: 1313–1314, [PUBMED], [INFOTRIEVE]
- Pierce J A. Antitrypsin and emphysema. Perspective and prospects. JAMA 1988; 19: 2890–2894, [CROSSREF]
- Senior R M, Tegner H, Kuhn C, Ohlsson K, Starcher B C, Pierce J A. The induction of pulmonary emphysema with human leukocyte elastase. Am Rev Respir Dis 1977; 116: 469–475, [PUBMED], [INFOTRIEVE]
- Snider G L, Lucey E C, Christensen T G, Stone P J, Calore J D, Catanese A, Franzblau C. Emphysema and bronchial secretory cell metaplasia induced in hamsters by human neutrophil products. Am Rev Respir Dis 1984; 129: 155–160, [PUBMED], [INFOTRIEVE]
- Gadek J E, Fells G A, Crystal R G. Cigarette smoking induces functional antiprotease deficiency in the lower respiratory tract of humans. Science 1979; 206: 1315–1316, [PUBMED], [INFOTRIEVE]
- Brantly M, Nukiwa T, Crystal R G. Molecular basis of α1‐antitrypsin deficiency. Am J Med 1988; 84(suppl 6A)13–31, [PUBMED], [INFOTRIEVE]
- Johnson D, Travis J. Structural evidence for methionine at the reactive site of human α‐1‐proteinase inhibitor. J Biol Chem 1978; 253: 7142–7144, [PUBMED], [INFOTRIEVE]
- Johnson D, Travis J. The oxidative inactivation of human alpha‐1‐proteinase inhibitor. Further evidence for methionine at the reactive center. J Biol Chem 1979; 254: 4022–4026, [PUBMED], [INFOTRIEVE]
- Carp H, Janoff A. Possible mechanisms of emphysema in smokers. In vitro suppression of serum elastase‐inhibitory capacity by fresh cigarette smoke and its prevention by antioxidants. Am Rev Respir Dis 1978; 118: 617–621, [PUBMED], [INFOTRIEVE]
- Carp H, Janoff A. In vitro suppression of serum elastase‐inhibitory capacity by reactive oxygen species generated by phagocytosing polymorphonuclear leukocytes. J Clin Invest 1979; 63: 793–797, [PUBMED], [INFOTRIEVE]
- Janoff A, Chan S K, Abboud R T, Richter A, Fera T, Johal S. Is alpha 1‐protease inhibitor inactivated by smoking?. Science 1984; 224: 755–756, [PUBMED], [INFOTRIEVE]
- Janus E D, Carrell R W. α1‐antitrypsin deficiency in New Zealand. NZ J Med 1975; 81: 461–467
- Janus E D, Joyce P R, Sheat J M, Carrell R W. Alpha‐1‐antitrypsin variants in New Zealand. NZMJ 1975; 82: 289–291
- Owen M C, Carrell R W. Alpha‐1‐antitrypsin: molecular abnormality of S variant. Br Med J 1976; 1: 130–131
- Jeppsson J‐O. Amino acid substitution Glu Lys in α1‐antitrypsin PiZ. FEBS Lett 1976; 65: 195–197, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Yoshida A, Lieberman J, Gaidulis L, Ewing C. Molecular abnormality of human alpha1‐antitrypsin variant (Pi‐ZZ) associated with plasma activity deficiency. Proc Natl Acad Sci USA 1976; 73: 1324–1328, [PUBMED], [INFOTRIEVE]
- Jeppsson J‐O, Laurell C‐B, Fagerhol M. Properties of isolated human α1‐antitrypsins of Pi types M, S and Z. Eur J Biochem 1978; 83: 143–153, [PUBMED], [INFOTRIEVE]
- Owen M C, Carrell R W, Brennan S O. The abnormality of the S variant of human α1‐antitrypsin. Biochim Biophys Acta 1976; 453: 257–261, [PUBMED], [INFOTRIEVE]
- Owen M C, Carrell R W. α1‐antitrypsin: sequence of the Z variant tryptic peptide. FEBS Lett 1977; 79
- Owen M C, Lorier M, Carrell R W. α1‐antitrypsin: structural relationships of the substitutions of the S and Z variants. FEBS Lett 1978; 88: 234–236, [PUBMED], [INFOTRIEVE]
- Kress L F, Kurecki T, Chan S K, Laskowski M S. Characterization of the inactive fragment resulting from limited proteolysis of human alpha1‐proteinase inhibitor by Crotalus adamanteus proteinase II. J Biol Chem 1979; 254: 5317–5320, [PUBMED], [INFOTRIEVE]
- Travis J, Salvesen G S. Human plasma proteinase inhibitors. Ann Rev Biochem 1983; 52: 655–709, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Carrell R W, Owen M C, Brennan S O. α1‐antitrypsin: structural homology with antithrombin III. NZ J Med 1979; 90: 517
- Morii M, Odani S, Ikenaka T. Characterization of a peptide released during the reaction of human alpha 1‐antitrypsin and bovine alpha‐chymotrypsin. J Biochem (Tokyo) 1979; 86: 915–921
- Boswell D R, Owen M C, Brennan S O, Carrell R W, McLachlan A D. Homology of α1‐antitrypsin and antithrombin‐III: identification of active sites. Protides Biol Fluids 1980; 28: 129–132
- Martodam R R, Liener I E. The interaction of α1‐antitrypsin with trypsin, chymotrypsin and human leukocyte elastase as revealed by end group analysis. Biochim Biophys Acta 1981; 667: 328–340, [PUBMED], [INFOTRIEVE]
- Boswell R D, Jeppsson J‐O, Brennan S O, Carrell R W. The reactive site of α1‐antitrypsin is C‐terminal, not N‐terminal. Biochim Biophys Acta 1983; 744: 212–218, [PUBMED], [INFOTRIEVE]
- Lewis J H, Iammarino R M, Spero J A, Hasiba U. Antithrombin pittsburgh: an α1‐antitrypsin variant causing hemorrhagic disease. Blood 1978; 51: 129–137, [PUBMED], [INFOTRIEVE]
- Owen M C, Brennan S O, Lewis J H, Carrell R W. Mutation of antitrypsin to antithrombin. α1‐antitrypsin pittsburgh (358 Met to Arg), a fatal bleeding disorder. N Engl J Med 1983; 309: 694–698, [PUBMED], [INFOTRIEVE]
- Brennan S O, Owen M C, Boswell D R, Lewis J H, Carrell R W. Circulating proalbumin associated with a variant protease inhibitor. Biochim Biophys Acta 1984; 802: 24–28, [PUBMED], [INFOTRIEVE]
- Bathurst I C, Brennan S O, Carrell R W, Cousens L‐S, Brake A J, Barr P J. Yeast KEX2 protease has the properties of a human proalbumin converting enzyme. Science 1987; 235: 348–350, [PUBMED], [INFOTRIEVE]
- Carrell R W, Jeppsson J‐O, Laurell C‐B, Brennan S O, Owen M C, Vaughan L, Boswell D R. Structure and variation of human α1‐antitrypsin. Nature 1982; 298: 329–334, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Mega T, Lujan E, Yoshida A. Studies on the oligosaccharide chains of human α1‐protease inhibitor. I. Isolation of glycopeptides. J Biol Chem 1980; 255: 4053–4056, [PUBMED], [INFOTRIEVE]
- Vaughan L, Lorier M A, Carrell R W. α1‐antitrypsin microheterogeneity: isolation and physioogical significance of isoforms. Biochim Biophys Acta 1982; 701: 339–345, [PUBMED], [INFOTRIEVE]
- Kurachi K, Chandra T, Degen S JF, White T T, Marchioro T L, Woo S LC, Davie E W. Cloning and sequence of cDNA coding for alpha‐1‐antitrypsin. Proc Natl Acad Sci USA 1981; 78: 6826–6830, [PUBMED], [INFOTRIEVE]
- Errington D M, Bathurst I C, Janus E D, Carrell R W. In vitro synthesis of M and Z forms of human α1‐antitrypsin. FEBS Lett 1982; 148: 83–86, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Bathurst I C, Stenflo J, Errington D M, Carrell R W. Translation and processing of normal (PiMM) and abnormal (PiZZ) human α1‐antitrypsin. FEBS Lett 1983; 153: 270–274, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Riley J H, Bathurst I C, Edbrooke M R, Carrell R W, Craig R K. α1‐antitrypsin and serum albumin mRNA accumulation in normal, acute phase and ZZ human liver. FEBS Lett 1985; 189: 361–366, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Foreman R C, Judah J D, Colman A. Xenopus oocytes can synthesise but do not secrete the Z variant of human α1‐antitrypsin. FEBS Lett 1984; 168: 84–88, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Errington D M, Bathurst I C, Carrell R W. Human α1‐antitrypsin expression in Xenopus oocytes. Secretion of the normal (PiM) and abnormal (PiZ) forms. Eur J Biochem 1985; 153: 361–365, [PUBMED], [INFOTRIEVE]
- Carrell R W, Bathurst I C, Brennan S O. The molecular pathology of human α1‐antitrypsin. 49th Colloquium of Biochemical Society. Biochem Soc Symp, London 1984; 55–66
- Carrell R W. α1‐antitrypsin: molecular pathology, leukocytes and tissue damage. J Clin Invest 1986; 78: 1427–1431, [PUBMED], [INFOTRIEVE]
- Loebermann H, Tokuoka R, Deisenhofer J, Huber R. Human a1‐proteinase inhibitor. Crystal structure analysis of two crystal modifications, molecular model and preliminary analysis of the implications for function. J Mol Biol 1984; 177: 531–556, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Elliott P R, Lomas D A, Carrell R W, Abrahams J P. Inhibitory conformation of the reactive loop of α1‐antitrypsin. Nat Struct Biol 1996; 3: 676–681, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Huntington J A, Read R J, Carrell R W. Structure of a serpin‐protease complex shows inhibition by deformation. Nature 2000; 407: 923–926, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Carrell R, Owen M, Brennan S, Vaughan L. Carboxy terminal fragment of human α1‐antitrypsin from hydroxylamine cleavage: homology with antithrombin III. Biochem Biophys Res Commun 1979; 91: 1032–1037, [PUBMED], [INFOTRIEVE]
- Petersen T E, Dudek‐Wojciechowska G, Sottrup‐Jensen L, Magnusson S. Primary structure of antithrombin III (heparin cofactor): partial homology between alpha‐1‐antitrypsin and antithrombin III. The Physiological Inhibitors of Coagulation and Fibrinolysis, D Collen, B Wimas, M Verstraete. Elsevier‐North Holland, Amsterdam 1979; 43–54
- Hunt L T, Dayhoff M O. A surprising new protein superfamily containing ovalbumin, antithrombin III, and alpha1‐proteinase inhibitor. Biochem Biophys Res Commun 1980; 95: 864–871, [PUBMED], [INFOTRIEVE]
- Perutz M F, Kendrew J C, Watson H C. Structure and function of haemoglobin II. Some relations between polypeptide chain configuration and amino acid sequence. J Mol Biol 1965; 13: 669–678
- Carrell R W, Boswell D R. Serpins: the superfamily of plasma serine proteinase inhibitors. Proteinase Inhibitors, A Barrett, G Salvesen. Elsevier, Amsterdam 1986; 403–420
- Potempa J, Korzus E, Travis J. The serpin superfamily of proteinase inhibitors: structure, function, and regulation. J Biol Chem 1994; 269: 15957–15960, [PUBMED], [INFOTRIEVE]
- Irving J A, Pike R N, Lesk A M, Whisstock J C. Phylogeny of the serpin superfamily: implications of patterns of amino acid conservation for structure and function. Genome Res 2000; 10: 1845–1864, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Huntington J, Carrell R. The serpins: nature's molecular mousetraps. Science Progress. 2001
- Stein P E, Carrell R W. What do dysfunctional serpins tell us about molecular mobility and disease?. Nat Struct Biol 1995; 2: 96–113, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Villa P, Aznar J, Vaya A, Espana F, Ferrando F, Mira Y, Estelles A. Hereditary homozygous heparin cofactor II deficiency and the risk of developing venous thrombosis. Thromb Haemost 1999; 82: 1011–1014, [PUBMED], [INFOTRIEVE]
- Carrell R W, Evans D L, Stein P E. Mobile reactive centre of serpins and the control of thrombosis. Nature 1991; 353: 576–578, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Roberts E A, Cox D W, Medline A, Wanless I R. Occurrence of alpha‐1‐antitrypsin deficiency in 155 patients with alcoholic liver disease. Am J Clin Pathol 1984; 82: 424–427, [PUBMED], [INFOTRIEVE]
- Seyama K, Nukiwa T, Takabe K, Takahashi H, Miyake K, Kira S. Siiyama (serine 53 (TCC) to phenylalanine 53 (TTC)). A new α1‐antitrypsin‐deficient variant with mutation on a predicted conserved residue of the serpin backbone. J Biol Chem 1991; 266: 12627–12632, [PUBMED], [INFOTRIEVE]
- Stein P E, Leslie A GW, Finch J T, Turnell W G, McLaughlin P J, Carrell R W. Crystal structure of ovalbumin as a model for the reactive centre of serpins. Nature 1990; 347: 99–102, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Stein P E, Leslie A GW, Finch J T, Carrell R W. Crystal structure of uncleaved ovalbumin at 1.95Å resolution. J Mol Biol 1991; 221: 941–959, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Stein P, Chothia C. Serpin tertiary structure transformation. J Mol Biol 1991; 221: 615–621, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Lomas D A, Evans D L, Finch J T, Carrell R W. The mechanism of Z α1‐antitrypsin accumulation in the liver. Nature 1992; 357: 605–607, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Lomas D A, Finch J T, Seyama K, Nukiwa T, Carrell R W. α1‐antitrypsin Siiyama (Ser53Phe); further evidence for intracellular loop‐sheet polymerisation. J Biol Chem 1993; 268: 15333–15335, [PUBMED], [INFOTRIEVE]
- Sidhar S K, Lomas D A, Carrell R W, Foreman R C. Mutations which impede loop/sheet polymerisation enhance the secretion of human α1‐antitrypsin deficiency variants. J Biol Chem 1995; 270: 8393–8396, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Cox D W, Billingsley G D, Callahan J W. Aggregation of plasma Z type α1‐antitrypsin suggests basic defect for the deficiency. FEBS Lett 1986; 205: 255–260, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Le A, Graham K S, Sifers R N. Intracellular degradation of the transport‐impaired human PiZ α1‐antitrypsin variant. Biochemical mapping of the degradative event among compartments in the secretory pathway. J Biol Chem 1990; 265: 14001–14007, [PUBMED], [INFOTRIEVE]
- Wu Y, Whitman I, Molmenti E, Moore K, Hippenmeyer P, Perlmutter D H. A lag in intracellular degradation of mutant α1‐antitrypsin correlates with liver disease phenotype in homozygous PiZZ α1‐antitrypsin deficiency. PNAS 1994; 91: 9014–9018, [PUBMED], [INFOTRIEVE]
- Yu M‐H, Lee K N, Kim J. The Z type variation of human α1‐antitrypsin causes a protein folding defect. Nat Struc Biol 1995; 2: 363–367, [CROSSREF]
- Carrell R W, Gooptu B. Conformational changes and disease—including serpins, prions and alzheimer's. Curr Opin Struct Biol 1998; 8: 799–809, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Tran P B, Miller R J. Aggregates in neurodegenerative disease: crowds and power?. Neurosci 1999; 22: 194–197
- Kopito R R, Ron D. Conformational Disease. Nat Cell Biol 2000; 2: E207–E209, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Davis R L, Holohan P D, Shrimpton A E, Tatum A H, Daucher J, Collins G H, Todd R, Bradshaw C, Kent P, Feiglin D, Rosenbaum A, Yerby M S, Shaw C M, Lacbawan F, Lawrence D A. Familial encephalopathy with neuroserpin inclusions bodies. Am J Pathol 1999; 155: 1901–1913, [PUBMED], [INFOTRIEVE]
- Davis R L, Shrimpton A E, Carrell R W, Lomas D A, Gerhard L, Baumann B, Lawrence D A, Yepes M, Kim T S, Ghetti B, Paccardo P, Takao M, Lacbawan F, Muenke M, Sifers R N, Bradshaw C B, Kent P F, Collins G H, Larocca D, Holohan P D. Association between conformational mutations in neuroserpin and onset and severity of dementia. Lancet 2002; 359: 2242–2247, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Zhou A, Stein P E, Huntington J A, Carrell R W. Serpin polymerization is prevented by a hydrogen bond network that is centered on His‐334 and stabilized by glycerol. J Biol Chem 2003; 278: 15116–15122, [PUBMED], [INFOTRIEVE]
- Laurell C‐B. Quantitative estimation of proteins by electrophoresis in agarose gel containing antibodies. Anal Biochem 1966; 15: 45–52, [PUBMED], [INFOTRIEVE]
- Vaughan L, Lorier M A, Carrell R W. α1‐antitrypsin microheterogeneity. Isolation and physiological significance. Biochim Biophys Acta 1982; 701: 339–345, [PUBMED], [INFOTRIEVE]
- Yoshida A, Wessels M. Origin of the multiple components of human alpha 1‐antitrypsin. Biochem Genet 1978; 16: 641–649, [PUBMED], [INFOTRIEVE], [CROSSREF]