Abstract
Formed in response to a World Health Organization recommendation, the Alpha One International Registry (AIR) is a multinational research program focused on alpha1‐antitrypsin (AAT) deficiency. Each of the nearly 20 participating countries maintains a National Registry of patients with AAT deficiency and contributes to an international database in Malmö, Sweden, that is designed to increase understanding of AAT deficiency as well as safeguard patient confidentiality. AIR members are engaged in active and wide‐ranging investigations to improve the diagnosis, monitoring and therapy of the disease. The AIR membership meets biennially to exchange views and research findings. The third biennial meeting was held in Barcelona, Spain, June 11–13, 2003. A wide range of AAT deficiency‐related topics were addressed, encompassing molecular and cellular pathophysiologic mechanisms, clinical epidemiology, diagnostic advances, current and investigational therapeutic approaches, and progress in registry development. Valuable cross‐fertilization of concepts and scientific observations was apparent between AAT deficiency research and other fields of biomedicine. The proceedings of the meeting are summarized in this report.
Introduction
Affecting one in 1,600 to 5,000 individuals, alpha1‐antitrypsin (AAT) deficiency is one of the most common hereditary disorders in Caucasians. AAT deficiency is characterized by low serum levels of AAT, a predisposition to early‐onset emphysema, and less commonly, liver disease, including neonatal jaundice and adult cirrhosis and hepatoma. In order to overcome the significant challenges that exist when investigating uncommon disorders, in recent years a concerted effort has been made to bring together all relevant information on research into the underlying pathophysiology and the clinical management of AAT deficiency, as illustrated by the establishment of a substantial number of registries for the disorder. Although much remains to be elucidated, progress continues to be made. Moreover, it has become clear that advances in AAT deficiency research can contribute to the understanding of other diseases and that approaches developed for other medical areas are now being applied to AAT deficiency.
The Alpha One International Registry (AIR), formed in 1998, is a multinational research project with the involvement of nearly 20 countries. AIR has committed to organizing biennial scientific meetings to update the diverse areas of research and clinical development relating to AAT deficiency Citation[[1]]. The 3rd AIR meeting was held in Barcelona, Spain in June 2003, involving 25 speakers and covering a wide‐ranging array of issues, from basic research to clinical findings. In a review of recent developments with regard to clinical management of AAT deficiency, Dr. Robert A. Sandhaus concluded that very little has changed in the management of lung and liver disease of AAT deficiency in the past two years. However, progress is steadily being made in unraveling the pathophysiology of both lung and liver and identifying risk factors for disease in AAT deficiency, and a number of new approaches to therapy are on the horizon. The presentations are summarized in this report.
The Laurell Lecture
In recognition of the 40th anniversary of the 1963 seminal work on AAT deficiency by Dr. Carl‐Bertil Laurell, Head of the Department of Clinical Chemistry in Malmö, Sweden Citation[[2]], the AIR Laurell Lecture was established, and Dr. Robin W. Carrell was chosen to present the first such lecture. Dr. Laurell's original observation has led to progress in medicine extending far beyond AAT deficiency. Dr. Carrell noted that an observation in a clinical laboratory in Sweden 40 years ago has opened up new fields in biology and medicine, and has done so long before the significance was recognized by science as a whole. Numerous avenues of research and clinical progress can be traced to Dr. Laurell's astute observation of the absence of the alpha1‐band among the electrophoretic abnormalities of lung patients. Dr. Carrell summarized Dr. Laurell's contribution with a quote by Goethe: “When the king finds gold, the subjects all begin to dig the soil.”
Not only is AAT the archetype of the serpin supergene family but it also has been the prototype for an entire category of disorders—the conformational diseases Citation[[3]]. The serine proteinase inhibitors, or serpins, are a superfamily of proteins that are found in organisms from plants to viruses to animals. In humans, the family includes in addition to AAT, proteins as diverse as α‐antichymotrypsin, C1 inhibitor, antithrombin, and plasminogen activator inhibitor‐1, which have key regulatory functions in the inflammatory, complement, coagulation, and fibrinolytic cascades Citation[[4]]. The serpins all possess an exposed mobile reactive center loop consisting of 14 amino acid residues that acts like a mouse trap for serine proteinases and enables the serpins to inhibit the serine proteases irreversibly (). The design of this reactive loop makes serpins very effective antiproteinases, but also leaves them susceptible to conformational transitions that can cause disease.
Figure 1. Tertiary structure of AAT.
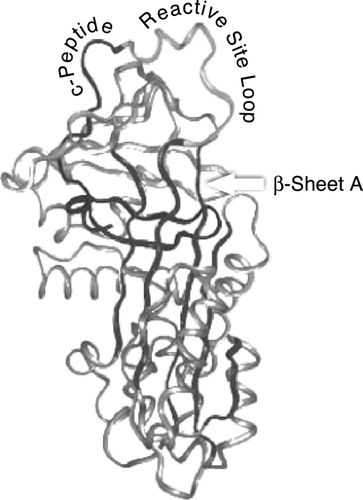
In individuals with the PiZ (proteinase inhibitor) phenotype, a single nucleotide mutation causes the AAT molecules to accumulate in the endoplasmic reticulum (ER) of hepatocytes rather than be secreted. In the ER, there is a rapid sequential interaction between the reactive center loop of one AAT molecule and the main β‐sheet of a second. The consequence in PiZ individuals is the formation of long polymers of AAT, with each molecule linked by its reactive loop to the sheet of the next Citation[[5]]. Characteristic inclusion bodies in the liver, which are readily recognized on periodic acid‐Schiff staining, develop over time. The accumulation of aberrant forms of individual proteins rather than a failure to produce the protein is the common theme in conformational diseases. Conformational diseases such as the common dementias, Alzheimer's disease, prion diseases, and the serpinopathies are all caused by structural rearrangements within a protein that transform it into a pathological species. Recently in a study of familial neurodegenerative disease, inclusion‐body formations containing neuroserpin, a brain‐specific serpin, were found sufficient to cause neurodegeneration Citation[[6]]. The onset and severity of the disease was found to be associated with the rate and magnitude of the neuronal protein aggregation.
Efforts are underway to develop small peptides that can selectively inhibit polymerization of the Z allele of the AAT protein Citation[[7]], and the early results have been promising. Dr. Carrell expressed confidence that in due course there will be a cure or treatment for this condition, but it could take a decade.
Lung Disease
The hallmark of AAT deficiency is the development of early‐onset severe lower zone emphysema in individuals with the PiZZ genotype. The unchecked action of neutrophil elastase on lung parenchyma leads to alveolar destruction.
AAT is the major defense in the lung against neutrophil elastase. Severe AAT deficiency is the only defined genetic risk factor for chronic obstructive pulmonary disease (COPD), and individuals with susceptible genotypes are at risk for severe, early‐onset COPD. Cigarette smoking markedly increases the risk and rate of development of emphysema in patients with severe AAT deficiency.
Dr. Robert A. Stockley reviewed the occurrence of exacerbations in AAT deficiency. In an attempt to establish a widely accepted definition, a COPD working group recently defined exacerbations as “a sustained worsening of the patient's condition, from the stable state and beyond normal day‐to‐day variations, that is acute in onset and necessitates a change in regular medication in a patient with underlying COPD” Citation[[8]]. The criteria for COPD exacerbation proposed by Anthonisen et al. Citation[[9]] are still widely accepted: breathlessness, sputum volume and sputum purulence. Increased numbers of exacerbations have been found to be related to a faster decline in lung function in individuals with AAT deficiency Citation[[10]]. Exacerbations have been associated with bacterial and viral infections, pollution, airway irritability/obstruction, and increased temperature, and there is a considerable challenge faced in delineating the contribution of each of these factors.
A classic study by Hill et al. Citation[[11]] demonstrated that individuals with AAT deficiency have increased elastase activity at both the start and resolution of the exacerbation when compared with other COPD patients. Moreover, this study showed that both at the start and resolution of an exacerbation, the lung airways in AAT deficiency individuals are more neutrophilic, as evidenced by increased myeloperoxidase, sputum leukotriene B4 (LTB4) and sputum interleukin‐8 (IL‐8) levels. Increased exacerbation frequency has also been reported in those AAT deficiency individuals with chronic sputum expectoration Citation[[12]]. Exacerbations last approximately twice as long in AAT deficiency patients as in other COPD patients who have an equivalent degree of lung function impairment. Exacerbations do not occur in 40–50% of AAT deficiency individuals each year, but those AAT deficiency individuals who do experience exacerbations have 2.4 on average. Although this predictor is not clear‐cut, lower forced expiratory volume in 1 sec (FEV1) appears to be associated with increased risk of exacerbations. One study has indicated that patients on augmentation therapy have fewer exacerbations (i.e., the number of infections per year) compared with those not on augmentation therapy Citation[[13]].
High resolution computerized tomography (CT) scanning is the most sensitive non‐invasive method to diagnose emphysema. According to Dr. Saher Shaker, quantitative CT has shown good correlation with the pathological extent of emphysema, lung function tests, especially diffusion capacity, and to a lesser extent measures of airway obstruction like FEV1 Citation[[14]]. Quantitative CT is reproducible and more sensitive than lung function testing in monitoring the progression of emphysema and also correlates with the patient's health status. However, a major confounder in the use of quantitative CT is the depth of inspiration. The lung density almost doubles from full inspiration to full expiration. CT lung density should be adjusted for the level of inspiration, and from 10 to 75 percentile density, double logarithmic transformation and a coefficient of − 1.3 should be applied.
Another indicator of lung dysfunction is expiratory flow limitation (EFL), which is the maximal expiratory flow achieved during tidal breathing and signifies intrathoracic airflow obstruction. EFL promotes dynamic pulmonary hyperinflation and can adversely affect hemodynamics as well as contribute to dyspnea Citation[[15]]. Dr. Luciano Corda reported that EFL was observed frequently in AAT deficiency patients and that they had a significantly greater probability of exhibiting EFL when supine (70%) than seated (30%).
Role of Phenotype
The gene locus specifying the AAT protein is polymorphic, and more than 70 variants have now been identified, according to Dr. Sabina Janciauskiene. However, the M, S and Z alleles are most prevalent. The PiM allele, the predominant allele, results in normal AAT levels, as shown in Citation[[16]]. The PiS allele accounts for 2–3% of the alleles and is associated with mildly reduced AAT levels, whereas the PiZ allele accounts for only 1% of alleles but is associated with severely reduced AAT levels (16% of normal) and the characteristic emphysema of AAT deficiency.
Figure 2. Serum AAT levels in various Pi phenotypes. Based on the data of Brantly and Wittes Citation[[16]].
![Figure 2. Serum AAT levels in various Pi phenotypes. Based on the data of Brantly and Wittes Citation[[16]].](/cms/asset/6051cba4-a883-4ecf-80aa-ebb6ca1dac74/icop_a_11176901_uf0002_b.gif)
The risk of developing COPD in heterozygotes is still not clearly delineated and has been a subject of controversy. Dr. Morten Dahl reviewed the risk of lung disease in the heterozygote PiMZ phenotype. A longitudinal study spanning up to 18 years among 9,187 participants in the population‐based Copenhagen City Heart Study found that the 451 PiMZ heterozygotes had 31% lower levels of AAT than PiMM genotypes. Their rate of FEV1 decrease was 19% greater than the average values in persons with the PiMM genotype, and they had a 30% increase in risk for airway obstruction, a 50% increase in risk for COPD and a 50% increase in risk of hospitalization or death from COPD Citation[[17]]Citation[[18]]. Based on an as yet unpublished meta‐analysis of 17 studies, the odds ratio of developing COPD is 2.3 in PiMZ individuals and 13 in PiZZ individuals. In the meta‐analysis it was observed that the type of study design affected the outcome, with the odds ratio of COPD in PiMZ individuals being 1.3 in population‐based studies and 3.1 in case‐control studies.
While the risk of lung dysfunction is much lower in PiMZ than PiZZ individuals, the PiMZ genotype is present in the population in a much higher frequency than the PiZZ genotype. Consequently, the actual number of PiMZ patients with COPD could be similar to that of PiZZ patients with COPD. Evidence suggests that other genetic and environmental factors contribute to the development of COPD in PiMZ individuals and that in certain subsets PiMZ may be associated with a relatively large risk for developing COPD. On the other hand, data from the Copenhagen City Heart Study suggest that the PiMZ phenotype may be associated with reduced risk of ischemic cerebrovascular and ischemic heart disease, and in that regard the PiMZ phenotype could prove beneficial.
The prevalence of the PiSZ genotype in most European countries is 1 in 1000, according to Dr. Niels Seersholm. The genotype is more prevalent in Spain compared with other European countries Citation[[19]]. The prevalence in the USA is estimated to be 1 in 1270. However, few individuals of this genotype are in the various registries, and therefore the natural history of lung disease has not been extensively studied. In the Copenhagen City Heart Study Citation[[18]], airway obstruction was increased in PiSZ individuals (40%) compared with PiMM (15%). A Swedish study of 94 PiSZ individuals revealed that only a small fraction of persons with the PiSZ phenotype are at increased risk of developing pulmonary emphysema, and such individuals are affected at an older age than persons with the PiZ phenotype Citation[[20]]. A cohort study of 50 nonsmokers demonstrated that PiSZ may confer little or no added risk of lung disease Citation[[21]]. PiSZ smokers may carry a small additional risk. Life expectancy in PiSZ individuals appears to be normal.
Lung Transplants
As reported by Dr. Sandhaus, a recent analysis of the United Network for Organ Sharing (UNOS) lung transplant database showed that, with regard to mortality at 3, 5 and 10 years, AAT deficiency individuals receiving double lung transplants had better outcomes than any other group, including the cystic fibrosis patients. However, analysis of results from the National Emphysema Treatment Trial (NETT) indicated that patients with emphysema and lower lobe predominance, as is characteristic of AAT deficiency patients, did not benefit from lung reduction surgery Citation[[22]]. The NETT report did not stratify results according to disease etiology such as AAT deficiency, however.
Pathophysiology of Lung Disease
The pathogenesis of emphysema is still incompletely understood, and it is increasingly clear that complex, interacting pathways are involved in its initiation and progression and the failure of repair processes Citation[[23]]. While the protease/antiprotease hypothesis is still “alive and well”, animal models are elucidating how other cellular components and biochemical pathways can also contribute to the lung damage seen in emphysema, according to Dr. Robert M. Senior. Emphysema can be modeled in many ways, and mice have become the predominant species for these studies. Three categories of experimental design are currently being pursued: 1) inducing emphysema in normal mice by challenge with exogenous agents; 2) studying airspace enlargement in naturally occurring genetic mutant mouse strains, such as blotchy and pallid strains; and 3) creating targeted mutagenesis (“knockout”) or transgenic mice. Transgenic and gene‐related mouse models of emphysema are dominating research currently. Among the advantages are that many genes have been cloned, techniques for genetic manipulation of mice are well established, and large numbers of mice can be generated quickly. However, it is still not clear how accurately these models reflect human biology and pathology. There are now many examples linking proteases and emphysema, and different proteases appear to be involved in different models. In addition to neutrophil elastase, other potential enzymes implicated are macrophage elastase, interstitial collagenase, and cysteine proteases.
Two recent papers have addressed the role of macrophage matrix metaloproteinase‐12 (MMP‐12)—also known as macrophage elastase—in inflammation and subsequent emphysema Citation[[24]]Citation[[25]]. Mice lacking α V beta 6 integrin have high levels of lung MMP‐12 and develop emphysema. Transforming growth factor (TGF‐b1) is activated by binding to alpha V β 6 integrin, and active TGF‐β down‐regulates the expression of MMP‐12 in macrophages. The hypothesis has been advanced that emphysema may occur in the α V β 6‐deficient mice due to overexpression of MMP‐12 following a failure to activate TGF‐β Citation[[24]]Citation[[26]]. In another mouse model of cigarette smoke‐induced emphysema, MMP‐12 was found to mediate smoke‐induced inflammation by releasing tissue necrosis factor‐alpha (TNF‐α) from macrophages, with subsequent endothelial activation, neutrophil influx, and proteolytic matrix breakdown caused by neutrophil‐derived proteases Citation[[25]]. Research in this field is very active with recent models revealing complex pathways. These models are opening new directions for the study of human emphysema.
Dr. Jean‐Michel Sallenave further elaborated on the role of cytokines in the pathogenesis of COPD Citation[[27]]. Exacerbations have been linked to increased expression of pro‐inflammatory cytokines, but the relative importance of these changes remains to be determined. Although there have been significant advances in understanding the pathophysiology of COPD, the role of inflammation in the pathogenesis of the condition is just beginning to be elucidated. Available evidence suggests that cytokines such as interleukin‐1 (IL‐1), TNF‐α, and interferon‐g (IFN‐γ) can initiate emphysema and that macrophages, neutrophils, and CD8 T cells also play a role. Cytokines such as IL‐8, the chemokine RANTES and interleukin‐13 derived from neutrophils, eosinophils and CD8 T cells, appear to be involved in COPD exacerbations. Even less explored is the role of cytokines and growth factors, such as vascular endothelial growth factor (VEGF), in lung hemostasis and repair. While there is not yet a clear picture of the varied roles of cytokines in emphysema, a growing number of models have demonstrated the proof of principle that cytokines do have a function in inducing emphysema, at least as assessed by animal models Citation[[25]]Citation[[28]]Citation[[29]].
Dr. Joan Albert Barberà reported on yet another cell type—the endothelial cell—that appears to play a role in the progression of COPD. Increased pulmonary artery pressure is a predictor of poor clinical outcome in COPD patients, and pulmonary vascular abnormalities start at early stages of the disease Citation[[30]]Citation[[31]]. Endothelial dysfunction can lead to pulmonary hypertension. Smooth muscle cell proliferation, as well as elastin and collagen deposition, in the thickened intimas of pulmonary arteries in moderate COPD patients and smokers has been reported Citation[[32]]. Cigarette smoke products may initiate vascular changes by a direct effect on endothelium and/or an inflammatory mechanism.
Liver Disease
PiZZ AAT deficiency is the most common genetic/metabolic cause of end stage liver disease in children, accounting for approximately 10% of pediatric liver transplantation in Europe/USA, according to Dr. Dino Hadzic. Severe liver disease has two peaks with decompensation either within the first 2 years of life or in puberty. The most common presentation is neonatal hepatitis, and liver deposits of AAT have been demonstrated as early as 17–20 weeks of gestation Citation[[33]]. Physiological perinatal prevalence of hydrophobic bile acids may add to prolonged neonatal jaundice, but the majority of children who will not need liver transplantation clear their jaundice by 6 months of age. Chronic liver disease will develop in 78% of PiZZ infants with neonatal hepatitis syndrome, and 28% will either require a transplant or die.
In contrast to PiZZ individuals, liver disease in PiSS and PiSZ individuals is much less well documented Citation[[34]]Citation[[35]]Citation[[36]]. The PiS allele is more common than PiZ in some parts of Europe, such as the Iberian peninsula where the prevalence may be as high as 28%. It has been reported that PiZZ patients with acute liver decompensation may exhibit a PiSZ‐like pattern on electrophoretic phenotyping, and this could lead to an erroneous exaggeration of the actual incidence of childhood liver disease associated with PiSZ Citation[[37]]. A study conducted at King's College Hospital in London included 9 PiSS and 20 PiSZ individuals. The PiSS individuals had a variety of liver problems, including biliary atresia, glycogen storage disease type III, urea cycle defect and steato‐hepatitis (Mauriac syndrome). However, after a mean follow‐up of 62 months in individuals with PiSS and 24 months in those with PiSZ, there was no clinical evidence of liver disease, except in relation to associated pathologies. Thus, possession of PiSS and PiSZ phenotype per se does not induce liver disease in childhood. Liver involvement in PiS individuals may be subclinical, and prospective community‐based studies in areas with high prevalence of the S allele are needed. Three studies that investigated the effect of rare Pi phenotypes on liver function have also recently been published Citation[[38]]Citation[[39]]Citation[[40]].
A number of presentations at the meeting underscored the need to explore further the pathophysiological processes taking place in the liver so that directed therapies can be developed. To date, there are no specific therapies for liver disease associated with AAT deficiency. The mutant PiZ protein has a single amino acid substitution that prevents it from folding properly. The AAT PiZ protein polymerizes and is retained in the ER rather than secreted into blood and body fluids. While the assumption has been that the retention of this aggregated PiZ glycoprotein elicits liver injury, the possibility exists that the accumulation of Z polymer per se does not cause liver disease, according to Dr. Richard Sifers. Dr. Jeffrey H. Teckman stressed that the mechanisms of liver injury, including the role of mutant AAT in triggering injury, are still poorly understood. For instance, the PiSaar mutation results in prolonged intracellular retention even though the proteins do not have polymeric properties Citation[[41]]. The mapping of the flux of AAT PiZ through the cell from synthesis, chaperone binding, polymerization, retention and degradation and even to the small amount that is secreted is key to understanding liver pathophysiology.
The glycoprotein ER‐associated degradation (GERAD) system that has been found to be a major component of the eukaryotic cell's “biosynthetic quality control” system Citation[[42]]. GERAD is one of the numerous checkpoints in eukaryotic cells that maintain fidelity of genetic information. On their passage through the cell, secretory proteins such as AAT are first transported by a molecular chaperone through the ER where they undergo conformational maturation. Recent studies have shown that an asparagine‐linked oligosaccharide of ATT serves a vital role in ER transport, processing and proper folding. The chaperone protein calnexin can only bind to ATT after the removal of two glucose molecules from the oligosaccharide. Binding of calnexin results in the shedding of another glucose and the folding of the AAT molecule. The PiZ molecule cannot be folded properly and therefore is marked for degradation. The oligosaccharide chain has also been found to play an essential role in the signal for degradation. The removal of one mannose plus a non‐native protein structure are both required for degradation.
Degradation of the misfolded or polymerized AAT molecules occurs primarily in the proteosome Citation[[43]]. However, nonproteosomal autophagy may also be involved. In autophagy, cytosol and intracellular organelles, such as ER, are first sequestered from the rest of the cytoplasm, allowing them to be degraded subsequently within lysosomes. The concentration of the PiZ protein may determine whether it is degraded by proteosomes or lysosomes. Mitochondrial autophagy may play a role in the mechanism of liver cell injury in AAT deficiency Citation[[44]], but mitochondrial injury is only one part of the story.
A mouse model study has demonstrated an altered response to stress, such as fasting, in the AAT deficiency liver, including an incapacity for further increase in activated autophagic response, a developmental state‐specific rise in AAT‐containing globules, and higher mortality Citation[[45]]. The liver of AAT deficiency individuals could be very susceptible to perturbations and insults, and will suffer more injury than a normal liver for any given insult. Recent research in the laboratory of Dr. Teckman has suggested that many common medications may be injurious to the liver of AAT deficiency individuals.
Diagnosis and Rare Phenotypes of AAT Deficiency
Dr. Edward J. Campbell stressed that in all countries in which AAT deficiency is prevalent, the majority of individuals with the disease have not yet been detected. In Sweden and Denmark, approximately 15–20% of all deficient individuals have been detected. However, in the remainder of the world, that percentage is much lower. Of the more than 80,000 AAT deficiency individuals estimated to be in the USA, only 4,000 to 5,000 (5–6%) have been diagnosed.
World Health Organization recommendations were issued in 1997 that all patients with COPD, all adults and adolescents with asthma and all individuals with a family history of AAT deficiency, as well as neonates, children and adults with unexplained liver disease, should be screened, and that those with abnormal results on screening should undergo phenotyping Citation[[46]]. Three recommended categories of testing were immunoassay for serum levels of AAT, phenotyping based on isoelectrophoretic focusing of the AAT protein and genotyping using polymerase chain reaction (PCR) to analyze DNA. At least two independent tests are required to establish a diagnosis of AAT deficiency, and all approaches have their limitations.
The prevalence of genotypes other than PiZZ is approximately 6–10%, i.e., 17 of 182 (9.3%) US patients and 6 of 103 (5.8%) British patients. Diagnostic testing is complicated by the presence of Null alleles, which result in a total lack of circulating protein. Prior to the availability of DNA‐based testing, Null alleles could be identified with certainty only through familial patterns of inheritance. In PCR‐based genotyping, primers for both the PiZ mutation and the normal sequence allow “counting” of the PiZ alleles present. Individuals with an allele having normal sequence at residue 342, but only Z protein identified by phenotyping have a Null allele. Unlike the PiZ mutation which results from a single specific mutation, Null alleles can result from a number of mutations, including ones that result in T splicing abnormalities, deletion of AAT coding exons and premature stop codons. In an analysis of 25 AAT deficiency individuals with genotypes other than PiZZ and 29 individuals with suspected PiNull alleles, a total of 24 different deficient alleles, including 17 novel ones, were identified. The presence of Null alleles at relatively high frequency, and the variety of Null alleles represented, pose severe challenges for the use of genotyping alone for diagnosis of AAT deficiency.
In Northern Italy, a unique screening program has tracked rare AAT phenotypes and liver‐test abnormalities during infancy Citation[[38]]. The authors concluded that only a neonatal screening based on phenotyping can detect these rare carriers early in life.
The ATZ11 monoclonal antibody has been used for immunological detection of PiZ‐positive hepatocytes Citation[[47]] and detects a conformation‐dependent neoepitope on both polymerized AAT and native AAT‐elastase complex. Dr. Janciauskiene reported that this antibody detects AAT‐elastase complexes in the endothelium layer, and this finding requires further investigation. Moreover, AAT deficiency is related to high levels of circulating AAT‐polymers, and the detection of circulating and tissue‐bound AAT polymers might prove valuable for various clinical applications. For instance, ATZ11‐based enzyme‐linked immunosorbent plasma assays allow sensitive and specific detection of Z carriers Citation[[48]]Citation[[49]]. Tissue‐bound AAT‐polymers can serve as endothelial cell markers Citation[[50]].
With the sequencing of the human genome complete, researchers are now focusing on creating a map of human genetic variation based on common haplotype patterns. Dr. Sally Plummer reported on the construction of a complete single nucleotide polymorphism (SNP) map of the AAT gene. Using this approach, 16 polymorphisms have been identified in AAT to date. There appears to be one major haplotype with many other rare haplotypes. The goal is to identify informative haplotype tag SNPs that will allow a reduction in the number of loci that need to be included in future association studies.
Registries and Foundation Update
Dr. Charlie Strange reviewed findings and ongoing activities from two US‐based registries: the Alpha‐1 Research Registry Citation[[51]] and the National Heart Lung and Blood Institute (NHLBI) Registry Citation[[52]]Citation[[53]]. The Alpha‐1 Research Registry, which was initiated in 1997, has grown to 2280 individuals, and 274 new PiZZ individuals were enrolled in 2002. Several projects of the Alpha‐1 Research Registry are in progress such as the Family Linkage initiative, in which families are encouraged to register as a “linked” family, and the Alpha Coded Testing (ACT). In ACT, a mailed fingerstick test approach identified 709 PiMM individuals, 613 PiMZ, 97 PiMS, 53 PiZZ, 39 PiSZ and 8 PiSS.
The NHLBI‐sponsored Registry for AAT deficiency in North America enrolled 1,129 subjects from 1988 to 1992, and follow‐up continued through 1996. A recent analysis of data from the NHLBI Registry was performed to identify risk factors for decrease in FEV1. The study provided evidence that age, male sex, bronchial hyper‐responsiveness, baseline serum AAT concentrations and smoking were among the risk factors. Dust exposure and chronic bronchitis phenotype were new risk factors for decrease in FEV1 identified by the univariate analysis.
Dr. Claes‐Goran Löfdahl provided an update on AIR. This registry now consists of 1,296 individuals, primarily PiZZ phenotypes. The only other phenotype with substantial representation is PiSZ, comprising 7–8% of the registry. Ex‐smokers compose 60% of the registry, never smokers 31% and current smokers 9%. The predominant FEV1 (% predicted) in never smokers was 100%, whereas it was 20–40% in ex‐smokers and current smokers. In the ex‐smokers, the number of cigarette pack years was significantly correlated with decrease in lung function. However, even in never smokers, a decrease in lung function with increasing age can be seen, and by the age of 60, the mean value of FEV1 is in the lower range of normal. The decline in lung function from smoking was significantly less in PiSZ individuals compared with PiZZ individuals. When categorized according to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines, 35% of the PiZZ individuals were in Stage 0, i.e., at risk but with lung function still normal Citation[[54]]. The most rapid decline in lung function, as tracked by FEV1, was in those PiZZ individuals in GOLD Stage 3, i.e., severe COPD. The registry contains patients covering the spectrum of severity and may be well suited for interventional studies.
Dr. Tomas Sveger provided an update on a Swedish birth cohort with AAT deficiency that had been followed for almost 30 years Citation[[55]]. Neonatal screening for AAT deficiency undertaken in Sweden between 1972 and 1974 identified 129 infants with severe AAT deficiency (phenotype PiZ). This cohort has been followed prospectively, and participation is currently approximately 65% with an additional 15% still answering questionnaires Citation[[35]]Citation[[56]]Citation[[57]]. The participants are nearing 30 years of age and continue to be in excellent health. Their FEV1 values are within normal ranges and have not changed since about 18 years of age. None has clinical symptoms of liver disease, although a few have marginally increased liver enzymes occasionally. The question of whether emphysema and late onset liver disease can be prevented in those individuals who have neonatal detection of AAT deficiency and are given preventative advice remains unanswered.
The new Alpha‐1 Disease Management and Prevention (ADMAP) program from AlphaNet is about to be launched in the USA. This disease management program is a new approach, and grew out of the realization that the majority of those 2,700 AAT deficiency patients being treated in the USA are seen by a physicians who has only one AAT deficiency patient. Therefore, educational efforts directed at patients and families are probably the most effective way to increase knowledge about the disorder and bring the most current understanding of the disorder to the attention of healthcare professionals. The program is designed to foster collaborative self‐management, and will include nurse training and testing.
Augmentation Therapy for Lung Disease
Dr. James K. Stoller reviewed seven salient studies on clinical efficacy of augmentation therapy Citation[[13]]Citation[[14]]Citation[[53]]Citation[[58]]Citation[[59]]Citation[[60]]Citation[[61]]. These studies ranged from observational studies to cohort studies (with either historical or concurrent controls) to one randomized controlled trial. The randomized trial of 56 ex‐smokers with AAT deficiency receiving monthly therapy found that augmentation conferred overall no significant benefit in slowing FEV1 decline, but the analysis of lung density measured by CT showed a trend to slower decline Citation[[14]].
The NHLBI Registry study was a nonrandomized comparison of 927 patients either receiving or not receiving AAT replacement. The rate of FEV1 decline was significantly less in those receiving augmentation therapy (66 ± 5 vs. 93 ± 11 mL/year; p = 0.03) only in the subgroup with FEV1 35–49% predicted Citation[[53]]. In this study subjects receiving augmentation therapy also had a significantly decreased mortality risk. In another large nonrandomized cohort study comparing 198 German patients receiving weekly augmentation infusions with 97 untreated Danish patients Citation[[58]], the mean annual decline in FEV1 was significantly less in treated patients only in the subgroup with FEV1 31–65% predicted (62 vs. 83 mL, p = 0.04). Both these studies are concordant in showing that augmentation therapy recipients experience a slower rate of decline in FEV1, especially in those individuals with a moderate degree of airflow obstruction.
In a longitudinal follow‐up of AAT deficiency patients before and during augmentation therapy the largest benefit was observed in those with better preserved lung function Citation[[60]]. This is in contrast to the findings of other observational studies which showed the largest benefits in moderate degrees of airflow obstruction. A novel Web‐based survey of individuals with the PiZ phenotype found that augmentation therapy appeared to be associated with a marked reduction in the frequency and severity of lung infections Citation[[13]]. Two or more infections per year occurred in 65% of respondents before augmentation therapy but in only 18% after therapy began, whereas 55% of those not receiving augmentation therapy experienced two or more infections per year. In a study on the effect of augmentation therapy on bronchial inflammation, short‐term therapy increased lung fluid AAT concentrations to protective levels and was associated with a fall in LTB4, which is thought to be central to the airway inflammation in AAT deficiency Citation[[61]]. In contrast to the benefit from augmentation therapy in the other studies, eight weeks of augmentation therapy in 12 AAT deficiency individuals did not appreciably reduce the rate of elastin degradation, as measured by the rate of urinary excretion of desmosine Citation[[59]]. Dr. Stoller concluded that more studies are needed and that the results of observational studies need to be interpreted with caution.
Prolastin® (Bayer AG, Leverkusen, Germany), which has been available since 1987 and is plasma‐derived, has been the only augmentation therapy available. Two new plasma‐derived augmentation products are currently being introduced in the USA: Aralast™ (Baxter BioScience, Westlake Village, California, USA) and Zemaira™ (Aventis Behring, Marburg, Germany). Also in development are augmentation products, both plasma‐derived and recombinant, that can be inhaled.
Dr. Jorge Jorquera reported that a new product developed by Instituto Grifols, S.A (Barcelona, Spain) and expected to be introduced in 2003, was stable for at least 15 days at both 5°C and 30°C when the contents of several vials of therapy were reconstituted in polypropylene bags equipped with a sterility filter. Because of sterility concerns, the recommended shelf‐life of currently available augmentation products is only hours. A system with an extended shelf‐life could prove of great utility in home care settings.
Potential New Therapeutic Approaches
Thus far the mainstay of therapy has been administration of exogenous AAT in order to augment circulating levels. A variety of additional approaches in early stages of development are also being pursued. Among these are restoring endogenous capacity for AAT synthesis and secretion through gene therapy. A potential cellular therapy involves the transplantation of immortalized hepatocytes. An alternative cellular therapy would entail use of embryonal stem cells to generate replacement hepatocytes. Other investigational approaches include synthetic elastase inhibitors that might reduce the need for AAT, hyaluronic acid that might prevent elastase‐mediated damage, retinoids that might reverse pathologic changes occurring due to AAT deficiency and chaperone molecules that might minimize mislocalization and misfolding of mutant AAT.
A Phase I AAT gene therapy clinical trial is approved and scheduled to start at the University of Florida College of Medicine, according to Dr. Terry Spencer. The trial will involve the intramuscular (IM) injection of the AAT gene using a recombinant adeno‐associated virus (rAAV) vector. This safety study will be an open‐label, single‐dose escalation study with 4 cohorts of 3 subjects each. rAAV‐2 is a non‐pathogenic parvovirus that is a common inhabitant of the upper respiratory tract. Gene therapy, which is a direct extension of protein replacement strategy, has the potential for single‐dose treatment and may be able to provide stable plasma levels.
Skeletal muscle has been shown in animal studies to be particularly efficient for expressing DNA encoded by the AAV vector Citation[[62]]. AAT secretion following IM gene therapy was sustained for up to a year and a half in a mouse model, and there is persistence of rAAV‐hAAT DNA in skeletal muscle for up to 18 months. Further studies are currently being done with another vector, AAV‐1, aimed at increasing the levels of AAT expression. Efficient transgene expression and increased AAT serum levels have also been successfully demonstrated in a non‐human primate model Citation[[63]]. This model was additionally used to demonstrate that the risks of immune reaction and germline transmission after intramuscular injection of rAAV‐bAAT are relatively low within the range of vector doses studied. Efficient, sustained expression of AAT transgene has been demonstrated in mice, rabbits and baboons Citation[[64]]. Other gene therapy approaches for AAT deficiency are also being explored Citation[[65]]Citation[[66]]Citation[[67]].
An approach in the preclinical investigation phase Citation[[68]] described by Dr. Mark Zern is the immortalization of hepatocytes through the transduction of hepatocytes with the catalytic component of human telomerase, the telomerase reverse transcriptase (TERT). Telomeres are located on the tip of chromosomes. Each time a cell divides the telomeres shorten, and eventually the cell dies. Telomerase lengthens telomeres, allowing the cell to divide indefinitely. The number of cells used in therapy could be tailored to the patient. Individuals with primary lung disease may merely require the transplantation of an adequate number of cells to provide the AAT protein. Those with liver and lung disease will require enough cells to provide for adequate liver function as well as enough AAT protein for their lungs. Immortalized hepatocytes could potentially be used for direct liver transplantation or in a bioartificial liver. In preclinical studies, human fetal hepatocytes transduced with TERT have been in continuous culture for one and a half years, compared to controls which went into senescence at approximately 100 days. The immortalized cells were not found to be tumorigenic in mouse studies.
Another approach proposed by Dr. Juan Domínguez‐Bendala is the use of embryonic stem cells to create replacement hepatocytes. This is a promising new area, but without relevant publications thus far. The development of this approach will be hindered unless a suitable animal model of AAT deficiency can be developed. In the first report of its kind, Dr. Zern described the preliminary success of his research group, under the leadership of Dr. Hitoshi Shirahasi, in achieving the differentiation of human embryonic stem cells along a hepatocyte lineage.
Among other therapeutic approaches discussed by Dr. Sandhaus are synthetic elastase inhibitors, hyaluronic acid, retinoids and chaperone proteins. An endogenous polysaccharide in normal lung, hyaluronic acid is depleted in emphysema. In hamsters intratracheal administration of hyaluronic acid blocked the marked decrease in airspace enlargement after elastase instillment Citation[[69]]. The protective effect of hyaluronic acid may reflect its ability to bind to lung elastic fibers, thus preventing elastase‐mediated breakdown. Retinoids are important in regulating lung growth and development, and in a rat model of emphysema all‐trans‐retinoic acid reversed the loss of lung elastic recoil and the reductions in numbers of alveoli and volume‐corrected alveolar surface area Citation[[70]]. Protein chaperones might potentially reverse the cellular mislocalization or misfolding of mutant AAT. One chemical chaperone, 4‐phenylbutyric acid, increased secretion of functionally active AAT PiZ in a model cell culture system Citation[[71]]. The diverse and numerous presentations at the meeting brought into focus the extensive efforts being made on many fronts to improve the understanding, diagnosis and treatment of AAT deficiency.
References
- Luisetti M, Miravitlles M, Stockley R A. Alpha1‐antitrypsin deficiency: a report from the 2nd meeting of the Alpha One International Registry, Rapallo (Genoa, Italy), 2001. Eur Respir J 2002; 20(4)1050–1056, [PUBMED], [INFOTRIEVE]
- Laurell C B, Eriksson S. The electrophoretic a1‐globulin pattern of serum in a1‐antitrypsin deficiency. Scand J Clin Lab Invest 1963; 15: 133–140
- Carrell R W, Lomas D A. Conformational disease. Lancet 1997; 350(9071)134–138, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Lomas D A, Carrell R W. Serpinopathies and the conformational dementias. Nat Rev, Genet 2002; 3(10)759–768, [CROSSREF]
- Carrell R W, Lomas D A. Alpha1‐antitrypsin deficiency—a model for conformational diseases. N Engl J Med 2002; 346(1)45–53, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Davis R L, Shrimpton A E, Carrell R W, Lomas D A, Gerhard L, Baumann B, Lawrence D A, Yepes M, Kim T S, Ghetti B, Piccardo P, Takao M, Lacbawan F, Muenke M, Sifers R N, Bradshaw C B, Kent P F, Collins G H, Larocca D, Holohan P D. Association between conformational mutations in neuroserpin and onset and severity of dementia. Lancet 2002; 359(9325)2242–2247, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Mahadeva R, Dafforn T R, Carrell R W, Lomas D A. 6‐mer peptide selectively anneals to a pathogenic serpin conformation and blocks polymerization. Implications for the prevention of Z alpha(1)‐antitrypsin‐related cirrhosis. J Biol Chem 2002; 277(9)6771–6774, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Rodriguez‐Roisin R. Toward a consensus definition for COPD exacerbations. Chest 2000; 117(5 suppl 2)398S–401S, [CROSSREF]
- Anthonisen N R, Manfreda J, Warren C P, Hershfield E S, Harding G K, Nelson N A. Antibiotic therapy in exacerbations of chronic obstructive pulmonary disease. Ann Intern Med 1987; 106(2)196–204, [PUBMED], [INFOTRIEVE]
- Dowson L J, Guest P J, Stockley R A. Longitudinal changes in physiological, radiological, and health status measurements in alpha(1)‐antitrypsin deficiency and factors associated with decline. Am J Respir Crit Care Med 2001; 164(10 Pt. 1)1805–1809, [PUBMED], [INFOTRIEVE], [CSA]
- Hill A T, Campbell E J, Bayley D L, Hill S L, Stockley R A. Evidence for excessive bronchial inflammation during an acute exacerbation of chronic obstructive pulmonary disease in patients with alpha(1)‐antitrypsin deficiency (PiZ). Am J Respir Crit Care Med 1999; 160(6)1968–1975, [PUBMED], [INFOTRIEVE], [CSA]
- Dowson L J, Guest P J, Stockley R A. The relationship of chronic sputum expectoration to physiologic, radiologic, and health status characteristics in alpha(1)‐antitrypsin deficiency (PiZ). Chest 2002; 122(4)1247–1255, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Lieberman J. Augmentation therapy reduces frequency of lung infections in antitrypsin deficiency: a new hypothesis with supporting data. Chest 2000; 118(5)1480–1485, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Dirksen A, Dijkman J H, Madsen F, Stoel B, Hutchison D C, Ulrik C S, Skovgaard L T, Kok‐Jensen A, Rudolphus A, Seersholm N, Vrooman H A, Reiber J H, Hansen N C, Heckscher T, Viskum K, Stolk J. A randomized clinical trial of alpha(1)‐antitrypsin augmentation therapy. Am J Respir Crit Care Med 1999; 160(5 Pt. 1)1468–1472, [PUBMED], [INFOTRIEVE], [CSA]
- Koulouris N G. Negative expiratory pressure: a new tool. Monaldi Arch Chest Dis 2002; 57(1)69–75, [PUBMED], [INFOTRIEVE], [CSA]
- Brantly M L, Wittes J T, Vogelmeier C F, Hubbard R C, Fells G A, Crystal R G. Use of a highly purified alpha 1‐antitrypsin standard to establish ranges for the common normal and deficient alpha 1‐antitrypsin phenotypes. Chest 1991; 100(3)703–708, [PUBMED], [INFOTRIEVE]
- Dahl M, Nordestgaard B G, Lange P, Vestbo J, Tybjaerg‐Hansen A. Molecular diagnosis of intermediate and severe alpha(1)‐antitrypsin deficiency: MZ individuals with chronic obstructive pulmonary disease may have lower lung function than MM individuals. Clin Chem 2001; 47(1)56–62, [PUBMED], [INFOTRIEVE], [CSA]
- Dahl M, Tybjaerg‐Hansen A, Lange P, Vestbo J, Nordestgaard B G. Change in lung function and morbidity from chronic obstructive pulmonary disease in alpha1‐antitrypsin MZ heterozygotes: a longitudinal study of the general population. Ann Intern Med 2002; 136(4)270–279
- de Serres F J. Worldwide racial and ethnic distribution of alpha1‐antitrypsin deficiency: summary of an analysis of published genetic epidemiologic surveys. Chest 2002; 122(5)1818–1829, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Seersholm N, Kok‐Jensen A. Intermediate alpha 1‐antitrypsin deficiency PiSZ: a risk factor for pulmonary emphysema?. Respir Med 1998; 92(2)241–245, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Turino G M, Barker A F, Brantly M L, Cohen A B, Connelly R P, Crystal R G, Eden E, Schluchter M D, Stoller J K. Clinical features of individuals with PI*SZ phenotype of alpha 1‐antitrypsin deficiency. Alpha 1‐Antitrypsin Deficiency Registry Study Group. Am J Respir Crit Care Med 1996; 154(6 Pt. 1)1718–1725, [PUBMED], [INFOTRIEVE], [CSA]
- Fishman A, Martinez F, Naunheim K, Piantadosi S, Wise R, Ries A, Weinmann G, Wood D E. A randomized trial comparing lung‐volume‐reduction surgery with medical therapy for severe emphysema. N Engl J Med 2003; 348(21)2059–2073, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Mahadeva R, Shapiro S D. Chronic obstructive pulmonary disease * 3: experimental animal models of pulmonary emphysema. Thorax 2002; 57(10)908–914, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Morris D G, Huang X, Kaminski N, Wang Y, Shapiro S D, Dolganov G, Glick A, Sheppard D. Loss of integrin alpha(v)beta6‐mediated TGF‐beta activation causes Mmp12‐dependent emphysema. Nature 2003; 422(6928)169–173, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Churg A, Wang R D, Tai H, Wang X, Xie C, Dai J, Shapiro S D, Wright J L. Macrophage metalloelastase mediates acute cigarette smoke‐induced inflammation via tumor necrosis factor‐alpha release. Am J Respir Crit Care Med 2003; 167(8)1083–1089, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Roberts A B. Medicine: smoke signals for lung disease. Nature 2003; 422(6928)130–131, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Reid P T, Sallenave J M. Cytokines in the pathogenesis of chronic obstructive pulmonary disease. Curr Pharm Des 2003; 9(1)25–38, [PUBMED], [INFOTRIEVE], [CSA]
- Fujita M, Shannon J M, Irvin C G, Fagan K A, Cool C, Augustin A, Mason R J. Overexpression of tumor necrosis factor‐alpha produces an increase in lung volumes and pulmonary hypertension. Am J Physiol, Lung Cell Mol Physiol 2001; 280(1)L39–L49, [CSA]
- Lucey E C, Keane J, Kuang P P, Snider G L, Goldstein R H. Severity of elastase‐induced emphysema is decreased in tumor necrosis factor‐alpha and interleukin‐1beta receptor‐deficient mice. Lab Invest 2002; 82(1)79–85, [PUBMED], [INFOTRIEVE], [CSA]
- Kessler R, Faller M, Fourgaut G, Mennecier B, Weitzenblum E. Predictive factors of hospitalization for acute exacerbation in a series of 64 patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999; 159(1)158–164, [PUBMED], [INFOTRIEVE], [CSA]
- Kessler R, Faller M, Weitzenblum E, Chaouat A, Aykut A, Ducolone A, Ehrhart M, Oswald‐Mammosser M. “Natural history” of pulmonary hypertension in a series of 131 patients with chronic obstructive lung disease. Am J Respir Crit Care Med 2001; 164(2)219–224, [PUBMED], [INFOTRIEVE], [CSA]
- Santos S, Peinado V I, Ramirez J, Melgosa T, Roca J, Rodriguez‐Roisin R, Barbera J A. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur Respir J 2002; 19(4)632–638, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Malone M, Mieli‐Vergani G, Mowat A P, Portmann B. The fetal liver in PiZZ alpha‐1‐antitrypsin deficiency: a report of five cases. Pediatr Pathol 1989; 9(6)623–631, [PUBMED], [INFOTRIEVE]
- Pittschieler K, Massi G. Liver involvement in infants with PiSZ phenotype of alpha 1‐antitrypsin deficiency. J Pediatr Gastroenterol Nutr 1992; 15(3)315–318, [PUBMED], [INFOTRIEVE]
- Sveger T, Eriksson S. The liver in adolescents with alpha 1‐antitrypsin deficiency. Hepatology 1995; 22(2)514–517, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Mahadeva R, Chang W S, Dafforn T R, Oakley D J, Foreman R C, Calvin J, Wight D G, Lomas D A. Heteropolymerization of S, I, and Z alpha1‐antitrypsin and liver cirrhosis. J Clin Invest 1999; 103(7)999–1006, [PUBMED], [INFOTRIEVE], [CSA]
- Whitehouse D B, Lovegrove J U, Mieli‐Vergani G, Mowat A P, Hopkinson D A. ‘SZ like’ alpha 1‐antitrypsin phenotypes in PI ZZ children with liver disease. Ann Hum Genet 1994; 58(Pt. 1)11–17, [PUBMED], [INFOTRIEVE]
- Pittschieler K. Rare alpha‐1‐antitrypsin phenotypes and liver‐test abnormalities during infancy. Acta Paediatr 2001; 90(4)406–408, [PUBMED], [INFOTRIEVE]
- Seixas S, Lopes A I, Rocha J, Silva L, Salgueiro C, Salazar‐de‐Sousa J, Batista A. Association between the defective Pro369Ser mutation and in vivo intrahepatic 1‐antitrypsin accumulation. J Med Genet 2001; 38(7)472–474, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Canva V, Piotte S, Aubert J P, Porchet N, Lecomte‐Houcke M, Huet G, Zenjari T, Roumilhac D, Pruvot F R, Degand P, Paris J C, Balduyck M. Heterozygous M3Mmalton alpha1‐antitrypsin deficiency associated with end‐stage liver disease: case report and review. Clin Chem 2001; 47(8)1490–1496, [PUBMED], [INFOTRIEVE], [CSA]
- Lin L, Schmidt B, Teckman J, Perlmutter D H. A naturally occurring nonpolymerogenic mutant of alpha 1‐antitrypsin characterized by prolonged retention in the endoplasmic reticulum. J Biol Chem 2001; 276(36)33893–33898, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Cabral C M, Liu Y, Moremen K W, Sifers R N. Organizational diversity among distinct glycoprotein endoplasmic reticulum‐associated degradation programs. Mol Biol Cell 2002; 13(8)2639–2650, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Teckman J H, Burrows J, Hidvegi T, Schmidt B, Hale P D, Perlmutter D H. The proteasome participates in degradation of mutant alpha 1‐antitrypsin Z in the endoplasmic reticulum of hepatoma‐derived hepatocytes. J Biol Chem 2001; 276(48)44865–44872, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Perlmutter D H. Liver injury in alpha1‐antitrypsin deficiency: an aggregated protein induces mitochondrial injury. J Clin Invest 2002; 110(11)1579–1583, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Teckman J H, An J K, Loethen S, Perlmutter D H. Fasting in alpha1‐antitrypsin deficient liver: constitutive activation of autophagy. Am J Physiol: Gastrointest Liver Physiol 2002; 283(5)G1156–G1165, [CSA]
- World Health Organization. Alpha 1‐antitrypsin deficiency: memorandum from a WHO meeting. Bull World Health Organ 1997; 75(5)397–415, [CSA]
- Fischer H P, Ortiz‐Pallardo M E, Ko Y, Esch C, Zhou H. Chronic liver disease in heterozygous alpha1‐antitrypsin deficiency PiZ. J Hepatol 2000; 33(6)883–892, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Carlson J, Eriksson S. Chronic ‘cryptogenic’ liver disease and malignant hepatoma in intermediate alpha 1‐antitrypsin deficiency identified by a Pi Z‐specific monoclonal antibody. Scand J Gastroenterol 1985; 20(7)835–842, [PUBMED], [INFOTRIEVE]
- Elzouki A N, Segelmark M, Wieslander J, Eriksson S. Strong link between the alpha 1‐antitrypsin PiZ allele and Wegener's granulomatosis. J Intern Med 1994; 236(5)543–548, [PUBMED], [INFOTRIEVE]
- Janciauskiene S, Dominaitiene R, Sternby N H, Piitulainen E, Eriksson S. Detection of circulating and endothelial cell polymers of Z and wild type alpha 1‐antitrypsin by a monoclonal antibody. J Biol Chem 2002; 277(29)26540–26546, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Stoller J K, Brantly M, Fleming L E, Bean J A, Walsh J. Formation and current results of a patient‐organized registry for alpha(1)‐antitrypsin deficiency. Chest 2000; 118(3)843–848, [PUBMED], [INFOTRIEVE]
- McElvaney N G, Stoller J K, Buist A S, Prakash U B, Brantly M L, Schluchter M D, Crystal R D. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of alpha 1‐antitrypsin deficiency. Alpha 1‐Antitrypsin Deficiency Registry Study Group. Chest 1997; 111(2)394–403, [PUBMED], [INFOTRIEVE]
- National Heart Lung and Blood Institute. Survival and FEV1 decline in individuals with severe deficiency of alpha1‐antitrypsin. The Alpha‐1‐Antitrypsin Deficiency Registry Study Group. Am J Respir Crit Care Med 1998; 158(1)49–59, [CSA]
- Gomez F P, Rodriguez‐Roisin R. Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines for chronic obstructive pulmonary disease. Curr Opin Pulm Med 2002; 8(2)81–86, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Sveger T. Liver disease in alpha1‐antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med 1976; 294(24)1316–1321, [PUBMED], [INFOTRIEVE]
- Sveger T, Ohlsson K, Piitulainen E. Adolescents with alpha1‐antitrypsin deficiency have high alpha2‐macroglobulin and low neutrophil lipocalin and elastase levels in plasma. Pediatr Res 1998; 44(6)939–941, [PUBMED], [INFOTRIEVE], [CSA]
- Piitulainen E, Sveger T. Respiratory symptoms and lung function in young adults with severe alpha(1)‐antitrypsin deficiency (PiZZ). Thorax 2002; 57(8)705–708, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Seersholm N, Wencker M, Banik N, Viskum K, Dirksen A, Kok‐Jensen A, Konietzko N. Does alpha1‐antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary alpha1‐antitrypsin deficiency? Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WATL) Alpha1‐AT Study Group. Eur Respir J 1997; 10(10)2260–2263, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Gottlieb D J, Luisetti M, Stone P J, Allegra L, Cantey‐Kiser J M, Grassi C, Snider G L. Short‐term supplementation therapy does not affect elastin degradation in severe alpha(1)‐antitrypsin deficiency. The American–Italian AATD Study Group. Am J Respir Crit Care Med 2000; 162(6)2069–2072, [PUBMED], [INFOTRIEVE], [CSA]
- Wencker M, Fuhrmann B, Banik N, Konietzko N. Longitudinal follow‐up of patients with alpha(1)‐protease inhibitor deficiency before and during therapy with IV alpha(1)‐protease inhibitor. Chest 2001; 119(3)737–744, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Stockley R A, Bayley D L, Unsal I, Dowson L J. The effect of augmentation therapy on bronchial inflammation in alpha1‐antitrypsin deficiency. Am J Respir Crit Care Med 2002; 165(11)1494–1498, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Song S, Morgan M, Ellis T, Poirier A, Chesnut K, Wang J, Brantly M, Muzyczka N, Byrne B J, Atkinson M, Flotte T R. Sustained secretion of human alpha‐1‐antitrypsin from murine muscle transduced with adeno‐associated virus vectors. Proc Natl Acad Sci U S A 1998; 95(24)14384–14388, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Song S, Scott‐Jorgensen M, Wang J, Poirier A, Crawford J, Campbell‐Thompson M, Flotte T R. Intramuscular administration of recombinant adeno‐associated virus 2 alpha‐1 antitrypsin (rAAV‐SERPINA1) vectors in a nonhuman primate model: safety and immunologic aspects. Molec Ther 2002; 6(3)329–335, [CROSSREF], [CSA]
- Flotte T R. Recombinant adeno‐associated virus gene therapy for cystic fibrosis and alpha(1)‐antitrypsin deficiency. Chest 2002; 121(3 Suppl.)98S–102S, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Stoll S M, Sclimenti C R, Baba E J, Meuse L, Kay M A, Calos M P. Epstein‐Barr virus/human vector provides high‐level, long‐term expression of alpha1‐antitrypsin in mice. Molec Ther 2001; 4(2)122–129, [CROSSREF], [CSA]
- Metz R, DiCola M, Kurihara T, Bailey A, Frank B, Roecklein B, Blaese M. Mode of action of RNA/DNA oligonucleotides: progress in the development of gene repair as a therapy for alpha(1)‐antitrypsin deficiency. Chest 2002; 121(3 suppl)91S–97S, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Dasi F, Benet M, Crespo J, Crespo A, Alino S F. Asialofetuin liposome‐mediated human alpha1‐antitrypsin gene transfer in vivo results in stationary long‐term gene expression. J Mol Med 2001; 79(4)205–212, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Wege H, Le H T, Chui M S, Liu L, Wu J, Giri R, Malhi H, Sappal B S, Kumaran V, Gupta S, Zern M A. Telomerase reconstitution immortalizes human fetal hepatocytes without disrupting their differentiation potential. Gastroenterology 2003; 124(2)432–444, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Cantor J O, Cerreta J M, Keller S, Turino G M. Modulation of airspace enlargement in elastase‐induced emphysema by intratracheal instillment of hyaluronidase and hyaluronic acid. Exp Lung Res 1995; 21(3)423–436, [PUBMED], [INFOTRIEVE], [CSA]
- Massaro G D, Massaro D. Retinoic acid treatment abrogates elastase‐induced pulmonary emphysema in rats. Nat Med 1997; 3(6)675–677, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Burrows J A, Willis L K, Perlmutter D H. Chemical chaperones mediate increased secretion of mutant alpha 1‐antitrypsin (alpha 1‐AT) Z: a potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1‐AT deficiency. Proc Natl Acad Sci U S A 2000; 97(4)1796–1801, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
Appendix
Meeting Chairman
Robert A. Stockley (UK, Chairman of AIR)
Scientific Coordinators
Marc Miravitlles (Spain)
Niels Seersholm (Denmark)
Scientific Committee
Robert A. Stockley (UK)
Marc Miravitlles (Spain)
Maurizio Luisetti (Italy)
Bruce C. Trapnell (USA)
Niels Seersholm (Denmark)
David A. Lomas (UK)
Dino Hadzic (UK)
Invited Speakers and Chairs
Gill Ainsle (Cape Town, South Africa)
Bruno Balbi (Pavia, Italy)
Joan A. Barberà (Barcelona, Spain)
Mark L. Brantly (Gainesville, Florida, USA)
Edward Campbell (Salt Lake City, Utah, USA)
Robin Carrell (Cambridge, UK)
Morten Dahl (Copenhagen, Denmark)
Asger Dirksen (Copenhagen, Denmark)
Juan Domínguez‐Bendala (Miami, Florida, USA)
Sarah Everett (Boston, Massachusetts, USA)
Shane Fitch (Cádiz, Spain)
Dino Hadzic (London, UK)
John Humphries (Research Triangle Park, North Carolina, USA)
Sabina Janciauskiene (Copenhagen, Denmark)
Claes‐Goran Löfdhal (Lund, Sweden)
David A. Lomas (Cambridge, UK)
Maurizio Luisetti (Pavia, Italy)
Marc Miravitlles (Barcelona, Spain)
Eeva Piitulainen (Malmö, Sweden)
Sally Plummer (Nottingham, UK)
Erich Russi (Zurich, Switzerland)
Jean‐Michel Sallenave (Edinburgh, Scotland)
Sandy Sandhaus (Denver, Colorado, USA)
Niels Seersholm (Copenhagen, Denmark)
Bob Senior (St. Louis, Missouri, USA)
Saher Shaker (Copenhagen, Denmark)
Richard Sifers (Houston, Texas, USA)
Edwin K. Silverman (Boston, Massachusetts, USA)
Gordon L. Snider (Boston, Massachusetts, USA)
Terry Spencer (Gainesville, Florida, USA)
Robert A. Stockley (Birmingham, UK)
Jan Stolk (Leiden, Netherlands)
James K. Stoller (Cleveland, Ohio, USA)
Charlie Strange (Charleston, South Carolina, USA)
Tomas Sveger (Malmö, Sweden)
Jeffrey H. Teckman (St. Louis, Missouri, USA)
Bruce C. Trapnell (Cincinnati, Ohio, USA)
Gerard M. Turino (New York, New York, USA)
Claus Vogelmeier (Marburg, Germany)