Abstract
Approximately 85% of all cases of chronic obstructive pulmonary disease are attributed to cigarette smoking. The only other established risk factor for the development of chronic obstructive pulmonary disease that is of comparable importance is the deficiency of alpha1‐antitrypsin, a rare genetic defect that is present in less than 1% of all cases. Other risk factors are not well characterized in the literature. This article describes one patient without a significant smoking history and a normal alpha1‐antitrypsin level that developed severe early‐onset emphysema and gives a brief discussion about other genetic and environmental risk factors for the development of emphysema.
Introduction
Cigarette smoking has been firmly established as the most important risk factor for the development of chronic obstructive pulmonary disease (COPD). It has been associated with a two‐ to threefold increase in the rate of decline in the forced expiratory volume in one second (FEV1) in smokers compared to nonsmokers Citation[[1]], and according to 1984 statistics and prior studies in the United States smoking accounts for 80% to 90% of the risk of developing COPD Citation[[2]]Citation[[3]]. Patients with COPD usually have at least a 20‐pack year history of smoking before symptoms develop, and patients are generally in their 6th decade of life at the time of presentation. The only other risk factor of comparable importance is the deficiency of α1‐protease inhibitor, also known as α1‐antitrypsin (A1AT). A1AT is a serum protein that is capable of inhibiting several types of proteolytic enzymes, particularly neutrophil elastase. It is coded by a single gene on chromosome 14, and it plays an important role in inflammatory states. Patients who have homozygous A1AT deficiency develop premature chronic airflow obstruction and panacinar emphysema. However, A1AT deficiency accounts for less than 1% of all cases of COPD Citation[[4]].
Approximately 15% of COPD cases cannot be attributed to cigarette smoking. Although other risk factors are mentioned in the American Thoracic Society's 1995 statement on the standards for the diagnosis and care of patients with COPD, the data on these risk factors are sparse and the characterization of the significance of these risk factors is poor Citation[[3]]. More recently, Silverman et al. described patients with severe, early‐onset COPD Citation[[5]]; however, these patients were current or ex‐smokers with disease out of proportion to their tobacco use.
To our knowledge, there is no literature on severe, early‐onset with emphysema without a significant smoking history and with normal A1AT levels. The case described in this article has severe, early‐onset emphysema based on pulmonary function testing and chest radiography. There is no history of tobacco use, and the serum A1AT level is within normal limits. The underlying cause or causes for the development of airflow obstruction in this patient are not known.
Case Presentation
A 32‐year‐old African American woman referred herself to a pulmonologist for a second opinion. She was diagnosed with asthma approximately 3 years prior to presentation, when following a course of beta blocker therapy for hypertension, she developed shortness of breath, chest congestion, and noticeable wheezing. Her beta blocker was discontinued, she was started on regularly scheduled inhaled triamcinolone, montelukast, and inhaled albuterol via a metered dose inhaler as needed, and her symptoms improved. She described dyspnea on exertion with walking two city blocks, which had progressively increased over the last 3 years. She relied on inhaled albuterol, using it three or four times daily. She noted daily cough, occasionally productive of white or yellow sputum. She denied wheezing, exertional chest discomfort, nausea, vomiting, choking, paroxysmal nocturnal dyspnea, orthopnea, or lower extremity edema. She also described rhinorrhea and nasal congestion, occurring predominantly in the spring and summer, but had never received skin testing for allergens. She was told that as a child she may have had asthma. She stated that she had a normal chest radiograph 3 years prior and that she had never had pulmonary function testing.
Her past medical history also included hypertension for which she took hydrochlorothiazide/lisinopril. She noted that she was born prematurely, but did not know further details. She denied tobacco or drug use. She drank alcohol at social occasions only. She was married and lived at home. She was an obstetrician/gynecologist, and she denied any occupational exposures that could be associated with lung disease. There were no pets at home, and she denied any changes to her living environment in the last 5 years. Her family history was significant for asthma in two maternal aunts, two maternal uncles, and a daughter. There was no family history of COPD. Her review of systems was otherwise negative.
On physical examination she was a small, thin, well‐developed woman. She was afebrile, her blood pressure was 110/70 mmHg, her heart rate was 70 beats per minute, and her respiratory rate was 28 breaths per minute. Head, ears, eyes, nose, and throat examination revealed mild nasal mucosal erythema and rhinitis with clear posterior nasal drainage in the oropharynx. She did not have neck vein distention. Heart sounds were normal and there were no murmurs. Breath sounds were decreased, she was hyperresonant to percussion, and there was normal thoracoabdominal excursion bilaterally. Her anterior–posterior chest diameter was increased. No wheezing was appreciated with forced expiration. Abdominal exam was normal. There was no digital clubbing, cyanosis, or peripheral edema. Skin examination revealed no rashes.
Spirometry revealed an FEV1 of 0.67 L (22% predicted), a forced vital capacity (FVC) of 3.04 L (79% predicted), with a ratio of 22%. Administration of a short‐acting bronchodilator led to an increase in FVC by 0.49 L (16% increase). Measurement of lung volumes via body plethysmography revealed a total lung capacity (TLC) of 6.58 L (124% predicted) and a residual volume (RV) of 3.54 L (212% predicted). RV/TLC ratio was 54%. Diffusing capacity of carbon monoxide (DLCO) was 7.6 mL/mmHg/min (37% predicted). Within 3 years after initial presentation, her FEV1 decreased to 0.34 L, and her FVC decreased to 1.39 L.
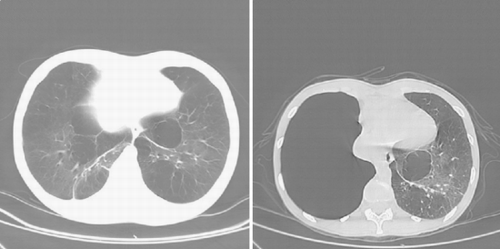
PA and lateral chest radiograph revealed hyperinflated lung fields and flattening of the diaphragm. CT of the thorax revealed emphysematous changes and bullous lung disease ( and ). Her A1AT level was 1.57 mg/mL (normal range 1.0–1.8 mg/mL). Her alpha2‐macroglobulin level was 2.49 mg/mL (within normal limits), and her alpha1‐antichymotrypsin level was 0.27 mg/mL (within normal limits). Genotyping of microsomal epoxide hydrolase revealed an FS genotype. Her serum HIV test was nonreactive by ELISA. She was evaluated for lung transplantation. Her follow‐up chest radiograph and CT scan 2 years later revealed marked progression of emphysematous changes and the development of a large bulla in the right lung with leftward mediastinal shift ( and ).
Figure 1. PA chest radiograph at presentation (left) and then 2 years later (right). Both radiographs demonstrate typical changes associated with emphysema, including hyperinflation, flattening of the diaphragm, and a paucity of pulmonary vascular markings peripherally. Note the rapid development of large bullous disease on the right.
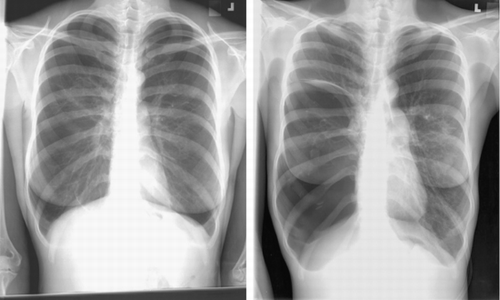
Figure 2. CT scan of the thorax, at presentation (left) and 2 years later (right), again demonstrating the rapid development of a large bulla in the right hemithorax with shift of the heart to the left.
Discussion
COPD has recently been defined by an expert panel as: “A disease state characterized by airflow limitation that is not fully reversible. Airflow limitation is usually both progressive and associated with an abnormal inflammatory response of the lungs to noxious particles or gases. Symptoms, functional abnormalities, and complications of COPD can all be explained on the basis on this underlying inflammation and the resulting pathology Citation[[6]].” Emphysema is a pathological term defined as the abnormal permanent enlargement of airspaces distal to the terminal bronchioles, accompanied by destruction of their walls and without obvious fibrosis Citation[[7]]. COPD is a clinical diagnosis based on history and physical exam with supporting evidence from pulmonary function testing and chest radiography. Computed tomography is a tool commonly used for patients with COPD to provide better definition of the lung parenchyma. Its role in the diagnosis of COPD is a supportive one, but it can be helpful if chest radiography is not diagnostic or during assessment for lung volume reduction surgery, bullectomy, or transplantation.
Only about 15% of active caucasian smokers and 5% of active Asian smokers develop COPD Citation[[8]], which suggests that other factors play a role. In addition, several studies have reported higher rates of airflow obstruction in first degree relatives of patients with COPD compared to controls Citation[[9]]Citation[[10]]Citation[[11]]Citation[[12]]. These observations emphasize that other genetic and environmental factors that are not well described must contribute to the development of chronic airflow obstruction.
Another possible genetic link with COPD is microsomal epoxide hydrolase, an enzyme involved with the metabolism of foreign compounds that enter the lung, which serves as protection against oxidative stress. Smith et al. found that more patients with COPD, compared to those without COPD, were homozygous for the forms of microsomal epoxide hydrolase that confer slow activity (SS) compared to those who were homozygous or heterozygous for fast activity (FF or FS) Citation[[13]]. Another area of research focuses on matrix metalloproteinases, a family of enzymes that degrade extracellular matrix proteins. A recent study found higher levels of MMP‐2 and MMP‐9 activity in the sputum of patients with asthma and COPD compared to those without pulmonary disease Citation[[14]]. Other possible genetic influences include the alpha1‐antichymotrypsin gene, the alpha2‐macroglobulin gene, the heme oxygenase‐1 gene, the tumor necrosis factor‐alpha gene, and the glutathione S‐transferase P1 gene Citation[[15]]Citation[[16]]Citation[[17]]Citation[[18]]Citation[[19]]Citation[[20]]Citation[[21]]. A complete discussion of the genetic determinants of COPD is beyond the scope of this paper.
Recent research has also focused on early life events, particularly childhood asthma and airway hyperresponsiveness, that may reduce maximal lung growth and increase risk for development of chronic airflow obstruction Citation[[22]]Citation[[23]]Citation[[24]]. For example, maternal smoking predisposes children to low birth weight and increased childhood respiratory infections. It has also been associated with an increased incidence of asthma in children with lower birth weights Citation[[25]]. As a strong correlation between childhood respiratory infections and the development of COPD exists Citation[[26]]Citation[[27]], there may be a link between maternal smoking and the development of chronic airflow obstruction.
Airway hyperresponsiveness has also been implicated as a risk factor for developing chronic airflow obstruction. In 1961, Orie and colleagues proposed that an “asthmatic constitution,” characterized by a predisposition to atopic disease, airway hyperresponsiveness (AHR), and eosinophilia, predisposed patients to COPD Citation[[28]]. This proposal was called the “Dutch hypothesis.” Consistent with this hypothesis are the facts that smoking can worsen airway hyperresponsiveness in asthmatics, and asthma by itself can progress to severe obstructive lung disease. However, many American investigators feel that the pathogenesis of COPD and asthma to be quite different Citation[[29]]. Although AHR is closely related to COPD and has been shown in numerous studies to be an independent risk factor for an accelerated decline in FEV1 Citation[[30]]Citation[[31]]Citation[[32]], there are multiple studies that show pathologic differences between emphysema and asthma, whether from analysis of histopathology, BAL fluid characteristics, cellular infiltrates, or radiographic patterns Citation[[33]]Citation[[34]]Citation[[35]]Citation[[36]]. A thorough discussion of this topic is beyond the scope of this paper, but the relationship between these two diseases remains unclear.
Occupational exposures may also play a role in the development of chronic airflow obstruction. In developing countries, exposure to smoke and chemical fumes, for example, during the production of fuels, may be an important risk factor in developing countries. Interestingly, farming, which increases exposure to organic antigens, increases risk of developing chronic bronchitis by two‐ to threefold Citation[[37]]. Other agents include silica, cadmium, and cotton dust Citation[[38]]Citation[[39]]Citation[[40]]Citation[[41]]; however, the roles of these agents in the development of COPD are unknown.
There have been reports that infection with HIV contributes to the development of chronic airflow obstruction. The underlying mechanism is still unknown. The Pulmonary Complications in HIV Infection Study Group performed pulmonary function tests on 1149 HIV‐infected patients in a prospective multicenter observational cohort study Citation[[42]]. They found that prior episodes of Pneumocystis carinii pneumonia or bacterial pneumonia led to permanent declines in FEV1, FVC, FEV1/FVC, and DLCO. Diaz et al. performed pulmonary function tests, bronchoalveolar lavage, and high‐resolution computed tomography on 114 young HIV‐positive patients without a history of opportunistic infection and compared the results to HIV‐negative controls Citation[[43]]. 15% (17 of 114) of HIV‐positive patients were diagnosed with emphysema compared to 2% (1 of 44) of the control group. The HIV‐positive patients with a smoking history of 12‐pack years or greater was 37% (14 of 38) compared to 0% (0 of 14) in the control group. Because a higher percentage of cytotoxic lymphocytes were found in the lavage fluid of the HIV‐positive group, their conclusion was that cytotoxic lymphocytes play an important role in accelerating the decline in lung function caused by smoking.
The patient presented in this report had seemingly unexplained severe, early‐onset emphysema. However, upon closer investigation, not all environmental or genetic risk factors were explored. For example, the patient was diagnosed with childhood asthma by history. Neither the severity of disease nor the adequacy of treatment is known in this case. We also cannot fully assess the patient’s exposure to air pollution or organic antigens. The patient denied occupational exposures, but she may not have known of any significant exposures in the distant past.
Some additional unanswered questions in this patient’s history include the following: were there any perinatal complications (eg neonatal respiratory distress syndrome, prolonged intubation, etc.)? Did her mother have any complications with pregnancy? Did her mother smoke during her pregnancy? Were corticosteroids administered prior to delivery? Was there any history of congenital abnormalities in the family? Was the strong family history of asthma truly asthma and not COPD? If the answers to any of these questions were positive, then these factors could have influenced her development of chronic airflow obstruction.
One must also consider other causes of obstructive airways disease, especially in patients like this one, a young nonsmoker. Other causes of chronic airflow obstruction include bronchiectasis, asthma, cystic fibrosis, allergic bronchopulmonary aspergillosis, chronic eosinophilic pneumonia, diseases of the small airways (eg., bronchiolitis obliterans, diffuse panbronchiolitis, respiratory broncihiolitis), and congenital pulmonary abnormalities (eg., Swyer–James–Macleod syndrome), to name a few. Other nonparenchymal respiratory diseases can present with wheezing and dyspnea, such as disorders of the upper airway, intrinsic pathology of the major airways (eg., tumor, postinflammatory stricture, amyloid, granulomatous processes, foreign body), and extrinsic compression of the airways (eg mediastinal masses). Many of these diagnoses were considered, but there was no evidence of these disease processes during the evaluation of this patient.
Cigarette smoking and A1AT deficiency are significant and well‐characterized risk factors for the development of COPD. The patient presented in this article most likely had a genetic predisposition combined with some poorly characterized environmental exposures that caused the rapid premature development of airflow obstruction. This case illustrates our need to better characterize other risk factors for developing COPD. Further research is needed to identify genetic defects as well as environmental exposures other than cigarette smoking that create or foster the protease/antiprotease imbalance that seems critical to the development of emphysema. This may lead to a better understanding of the pathogenesis of COPD, and eventually to better treatments.
| Abbreviations | ||
| A1AT: | = | Alpha 1‐Antitrypsin |
| AHR: | = | Airway Hyperresponsiveness |
| AP: | = | Anteroposterior |
| COPD: | = | Chronic Obstructive Pulmonary Disease |
| CT: | = | Computed Tomography |
| DLCO: | = | Diffusing Capacity of Carbon Monoxide |
| ELISA: | = | Enzyme‐Linked Immunosorbent Assay |
| FEV1: | = | Forced Expiratory Volume in 1 Second |
| FVC: | = | Forced Vital Capacity |
| HIV: | = | Human Immunodeficiency Virus |
| PA: | = | Posterior–Anterior |
| RV: | = | Residual Volume |
| TLC: | = | Total Lung Capacity |
References
- Xu X, Weiss S T, Rijcken B, Schouten J P. Smoking, changes in smoking habits, and rate of decline in FEV1: new insight into gender differences. Eur Respir J 1994; 7(6)1056–1061, [PUBMED], [INFOTRIEVE]
- US Surgeon General. The Health Consequences of Smoking: Chronic Obstructive Lung Disease. US Dept of Heath and Human Research, 84‐50205. 1984
- Tager I B, Speizer F E. Risk estimates for chronic bronchitis in smokers: a study of male–female differences. Am J Respir Crit Care Med 1976; 113: 619–625
- American Thoracic Society. Standards for the diagnosis and care of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1995; 152: S77–S121, [CSA]
- Silverman E K, Chapman H A, Drazen J M, Weiss S T, Rosner B, Campbell E J, O'Donnell W J, Reilly J J, Ginns L, Mentzer S, Wain J, Speizer F E. Genetic epidemiology of severe, early‐onset chronic obstructive pulmonary disease. Am J Resp Crit Care Med 1998; 157: 1770–1778, [PUBMED], [INFOTRIEVE], [CSA]
- Pauwels R A, Buist A S, Calverley P M, Jenkins C R, Hurd S S. GOLD Scientific Committee. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med 2001; 163: 1256–1276, [PUBMED], [INFOTRIEVE], [CSA]
- Piquette C A, Rennard S I, Snider G L. Chronic bronchitis and emphysema. Textbook of Respiratory Medicine3rd ed., J F Murray, J A Nadel. W.B. Saunders, Philadelphia, PA 2000; 1188–1245
- Barnes P J. Medical Progress: chronic obstructive pulmonary disease. N Engl J Med 2000; 343: 269–280, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Larson R K, Barman M L, Kueppers F, Fudenberg H H. Genetic and environmental determinants of chronic obstructive pulmonary disease. Ann Intern Med 1970; 72: 627–632, [PUBMED], [INFOTRIEVE]
- Kueppers F, Miller R D, Gordon H, Hepper N G, Offord K. Familial prevalence of chronic obstructive pulmonary disease in a matched pair study. Am J Med 1977; 63: 336–342, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Cohen B H, Diamond E L, Graves C G, Kreiss P, Levy D A, Menkes H A, Permutt S. A common familial component in lung cancer and chronic obstructive pulmonary disease. Lancet 1977; ii: 523–526, [CROSSREF]
- Cohen B H. Chronic obstructive pulmonary disease: a challenge in genetic epidemiology. Am. J Epidemiol 1980; 112: 274–288, [PUBMED], [INFOTRIEVE]
- Smith C AD, Harrison D J. Association between polymorphism in gene for microsomal epoxide hydrolase and susceptibility to emphysema. Lancet 1997; 350: 630–633, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Cataldo D, Munaut C, Noel A, Frankenne F, Bartsch P, Foidart J M, Louis R. MMP‐2‐ and MMP‐9‐linked gelatinolytic activity in the sputum from patients with asthma and chronic obstructive pulmonary disease. Int Arch Allergy Immunol 2000; 123: 259–267, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Sanford A J, Chagani T, Weir T D, Connett J E, Anthonisen N R, Pare P D. Susceptibility genes for rapid decline of lung function in the Lung Health Study. Am J Respir Crit Care Med 2001; 163: 469–473, [CSA]
- Poller W, Meisen C, Olek K. DNA polymorphisms of the alpha 1‐antitrypsin gene region in patients with chronic obstructive pulmonary disease. Eur J Clin Investig 1990; 20: 1–7, [CSA]
- Ishii T, Matsuse T, Teramoto S, Matsui H, Hosoi T, Fukuchi Y, Ouchi Y. Association between alpha 1‐antichymotrypsin polymorphism and susceptibility to chronic obstructive pulmonary disease. Eur J Clin Investig 2000; 30: 543–548, [CROSSREF], [CSA]
- Poller W, Barth J, Voss B. Detection of an alternation of the α2‐macroglobulin gene in a patient with chronic lung disease and serum α2‐macroglobulin deficiency. Hum Genet 1989; 83: 93–96, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Yamada N, Yamaya M, Okinaga S, Nakayama K, Sekizawa K, Shibahara S, Sasaki H. Microsatellite polymorphism in the heme oxygenase‐1 gene promoter is associated with susceptibility to emphysema. Am J Hum Genet 2000; 55: 187–195, [CROSSREF]
- Huang S L, Su C H, Chang S C. Tumor necrosis factor–alpha gene polymorphism in chronic bronchitis. Am J Respir Crit Care Med 1997; 156: 1436–1439, [PUBMED], [INFOTRIEVE], [CSA]
- Ishii T, Matsuse T, Teramoto S, Matsui H, Miyao M, Hosoi T, Takahashi H, Fukuchi Y, Ouchi Y. Glutathione S‐transferase P1 (GSTP1) polymorphism in patients with chronic obstructive pulmonary disease. Thorax 1999; 54: 693–696, [PUBMED], [INFOTRIEVE]
- Weiss S T, Segal M R, Tager I B, Tosteson T, Redline S, Speizer F E. Effects of asthma on pulmonary function in children: a longitudinal population‐based study. Am Rev Respir Dis 1992; 145: 58–64, [PUBMED], [INFOTRIEVE]
- Gold DR, Wypij D, Wang X, Speizer F E, Pugh M, Ware J H, Ferris B G, Jr, Dockery D W. Gender‐ and race‐specific effects of asthma and wheeze on level and growth of lung function in children in six US cities. Am J Respir Crit Care Med 1994; 149: 1198–1208, [PUBMED], [INFOTRIEVE], [CSA]
- Wang X, Mensinga T T, Schouten J P, Rijcken B, Weiss S T. Determinants of maximaly attained level of pulmonary function. Am J Respir Crit Care Med 2004; 169: 941–949, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Shaheen S. The beginnings of chronic airflow obstruction. BMJ Bull 1970; 53: 58–70
- Shaheen S O, Barker D J, Shiell A W, Crocker F J, Wield G A, Holgate S T. The relationship between pneumonia in early childhood and impaired lung function in late adult life. Am J Respir Crit Care Med 1994; 149: 616–619, [PUBMED], [INFOTRIEVE], [CSA]
- Shaheen S O, Sterne J A, Florey C D. Birth weight, childhood lower respiratory tract infection, and adult lung function. Thorax 1998; 53: 549–553, [PUBMED], [INFOTRIEVE]
- Orie NG, Sluiter H J, Devries K. The host factor in bronchitis. Bronchitis, An International Symposium, N GM Orie, H J Sluiter. Assen, Netherlands 1961; 43–59
- Burrows B, Earle R H. Course and prognosis of chronic obstructive lung disease: a prospective study of 200 patients. N Engl J Med 1969; 280: 397–404, [PUBMED], [INFOTRIEVE]
- Rijken B, Scouten J P, Xu X, Rosner B, Weiss S T. Bronchial hyperresponsiveness to histamine is associated with accelerated decline in FEV1. Am J Respir Crit Care Med 1995; 151: 1377–1382, [CSA]
- Tracey M, Villar A, Dow L, Coggon D, Lampe F C, Holgate S T. The influence of increased bronchial responsiveness, atopy, serum IgE on decline in FEV1: a longitudinal study in the elderly. Am J Respir Crit Care Med 1995; 151: 656–662, [PUBMED], [INFOTRIEVE], [CSA]
- O'Connor G T, Sparrow D, Weiss S T. A prospective study of metacholine airway hyperresponsiveness as a predictor of pulmonary function decline: the Normative Aging Study. Am J Respir Crit Care Med 1995; 152: 87–92, [PUBMED], [INFOTRIEVE], [CSA]
- Postma D S, Kerstjens H AM. Characteristics of airway hyperresponsiveness in asthma and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 158: S187–S192, [PUBMED], [INFOTRIEVE], [CSA]
- Jeffrey P K. Differences and similarities between chronic obstructive pulmonary disease and asthma. Clin Exp Allergy 1999; 29(suppl 2)14–26, [CROSSREF]
- Saetta M, Turato G, Maestrelli P, Mapp C E, Fabbri L M. Cellular and structural bases of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 163: 1304–1309, [PUBMED], [INFOTRIEVE], [CSA]
- Fabbri L M, Romagnoli M, Corbetta L, Casoni G, Busljjetic K, Turato G, Ligabue G, Ciaccia A, Saetta M, Papi A. Differences in airway inflammation in patients with fixed airflow obstruction due to asthma or chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2003; 167: 418–424, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Melbostad E, Eduard W, Magnus P. Chronic bronchitis in farmers. Scand J Work, Environ & Health 1997; 23: 271–280, [CSA]
- Davison A G, Fayers P M, Taylor A J, Venables K M, Darbyshire J, Pickering C A, Chettle D R, Franklin D, Guthrie C J, Scott M C. Cadmium fume inhalation and emphysema. Lancet 1988; 1: 663–667, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Cowie R L, Mabena S K. Silicosis, chronic airflow limitation, and chronic bronchitis in South African gold miners. Am Rev Respir Dis 1991; 143: 80–84, [PUBMED], [INFOTRIEVE], [CSA]
- Christiani D C, Ye T T, Wegman D H, Eisen E A, Dai H L, Lu P L. Cotton dust exposure, across‐shift drop in FEV1, and five‐year change in lung function. Am J Respir Crit Care Med 1994; 150: 1250–1255, [PUBMED], [INFOTRIEVE], [CSA]
- Cowie R L. The influence of silicosis on deteriorating lung function in gold miners. Chest 1998; 113: 340–343, [PUBMED], [INFOTRIEVE]
- Morris A M, Huang L, Baccetti P, Turner J, Hopewell P C, Wallace J M, Kvale P A, Rosen M J. Permanent declines in pulmonary function following pneumonia in human immunodeficiency virus‐infected persons. The pulmonary complications of HIV infection study group. Am J Respir Crit Care Med 2000; 162: 612–616, [PUBMED], [INFOTRIEVE], [CSA]
- Diaz P T, King M A, Pacht E R, Wewers M D, Gadek J E, Nagaraja H N, Drake J, Clanton T L. Increased susceptibility to pulmonary emphysema among HIV‐seropositive smokers. Ann Intern Med 2000; 132: 369–372, [PUBMED], [INFOTRIEVE], [CSA]