Abstract
Tobacco smoke is the number one risk factor for chronic obstructive pulmonary disease (COPD) and contains a high concentration of oxidants. The lung has a high concentration of antioxidants and antioxidant enzymes; however, COPD patients show evidence of increased oxidative stress suggesting that endogenous antioxidants may be insufficient to prevent oxidative damage from cigarette smoke. The consequences of increased oxidative stress in the lung include increased transcription of inflammatory genes, increased protease activity, and increased mucus secretion. Oxidative stress is often associated with impaired skeletal muscle function and may be one of the causes of glucocorticoid resistance. While current pharmacologic approaches to the treatment of chronic obstructive pulmonary disease do not commonly include antioxidants, preclinical studies involving animal models suggest that antioxidant superoxide dismutase mimetics offer a potential new therapeutic approach to the prevention and treatment of chronic obstructive pulmonary disease.
Introduction to COPD
Chronic obstructive pulmonary disease (COPD) is the fourth leading cause of death in the world and is the fourth leading cause of chronic morbidity and mortality in the United States Citation[[1]]. COPD is a disease state that is characterized by airflow limitation that is not fully reversible and is associated with progressive decline in lung function as well as an abnormal inflammatory response. Clinically the disease is characterized by emphysema, which is the loss of lung parenchyma, and chronic bronchitis. The diagnosis of COPD is confirmed by spirometry. Typically the patients have an FEV1 of < 80% in combination with an FEV1/FVC ratio < 70%. The clinical signs of COPD include shortness of breath, as well as a cough with sputum production. The prevalence of COPD has been increasing over the past thirty years. Globally the prevalence rate in the mid 1990s in males was 9.3/1000 and in females was 7.3/1000 Citation[[2]]. The morbidity of COPD increases with age and is greater in men than women. Furthermore, it is predicted that in the coming decades COPD mortality will rise from its current position as the fourth leading cause of death in the world. It is estimated that COPD will be the fifth leading cause of disability‐adjusted life years lost worldwide Citation[[2]].
Overview of the Theories of Pathogenesis of COPD
The major risk factor for COPD is exposure to tobacco smoke. In the United States 80–90% of COPD patients are current or former tobacco smokers. The other well established risk factor for COPD is α1‐antitrypsin deficiency; however only an estimated 1–2% of COPD patients are α1‐antitrypsin deficient Citation[[3]]Citation[[4]]. The incidence of α1‐antitrypsin disease associated alleles in white populations is only 3–4%, thus α1‐antitrypsin only accounts for a small minority of the pathogenesis of COPD. Other risk factors for developing COPD include environmental tobacco smoke and heavy exposure to occupational dust or other irritants, and air pollution.
Only 10–20% of heavy smokers develop COPD, indicating that there must be other factors that determine whether long‐term exposures to noxious agents cause the disease. Although the link between tobacco smoke and COPD is well established, there is an incomplete understanding as to the molecular mechanisms by which tobacco smoke leads to COPD. Research on α1‐antitrypsin deficiency has shown that an imbalance in proteases and anti‐proteases can play a key role in the development of COPD. Additional hypotheses are that tobacco smoke and other unknown factors lead to increased death or apoptosis of lung cells and impaired regeneration of healthy lung tissue, eventually leading to emphysema. It has also been proposed that there is a predisposition towards inflammation from tobacco smoke and infections may underlie the enhanced progression of loss of lung function. Finally, cigarette smoke has trillions of oxidants in each puff Citation[[5]] and some believe that these oxidants are a major cause of COPD. The role of oxidative stress in the pathogenesis of COPD is the subject of this review.
Definitions of Oxidative and Nitrative Stress
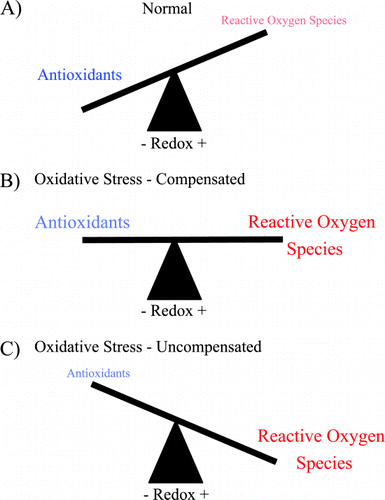
Airways are unique in both their exposure to high levels of environmental oxidants and their unusually high concentration of extracellular antioxidants. In the resting state, the balance between antioxidants and oxidants is sufficient to prevent the disruption of normal physiologic functions; however, either increases in oxidants or decreases in antioxidants can disrupt this balance. The imbalance between oxidants and antioxidants is termed oxidative stress (). There is increasing evidence that oxidative stress plays a role in the pathogenesis of COPD.
Figure 1. Oxidative stress is an imbalance between reactive oxygen species (ROS) and antioxidants. (A) Under normal conditions there are sufficient antioxidants in the lung to overcome endogenous ROS. (B) During pathologic states there is an increase in ROS that may be balanced by increase in antioxidants. (C) When either ROS production is excessive or antioxidants are inadequate, the balance is tipped toward oxidative stress and injury from ROS may occur. (Full color version available online.)
Both reactive oxygen (ROS) and reactive nitrogen species (RNS) contribute to the imbalance seen in COPD. Common ROS include superoxide, hydrogen peroxide, and hydroxyl radical. RNS include nitric oxide (NO) and its derivatives such as nitrogen dioxide and peroxynitrite. Other molecules that contribute to oxidative stress include protein radicals and lipid peroxide radicals. These molecules can cause nonspecific damage to cells and extracellular matrix, yet production of ROS and RNS in highly localized domains is also recognized as essential for normal physiologic function; for instance superoxide is a crucial component of phagocytic cell killing Citation[[6]] and NO mediates smooth muscle relaxation in both blood vessels Citation[[7]] and airways Citation[[8]].
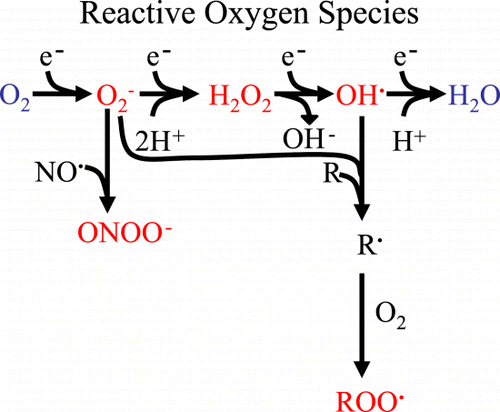
The features that make oxygen ideal for aerobic energy production (i.e. atmospheric abundance and a high affinity for electrons) are also its Achilles' heel. For instance, one electron addition to oxygen produces superoxide (O2−), a second electron yields hydrogen peroxide (H2O2), and a third electron leads to the formation of the hydroxyl radical (OH•). These three intermediates are called reactive oxygen species because they readily react with other molecules such as proteins, lipids, and DNA (). The hydroxyl radical (OH•) is the most reactive of all reactive oxygen species (extremely high rate constants > 109 M− 1s− 1 for reactions with most molecules) and typically reacts immediately with organic molecules at the site of production Citation[[9]]. When O2 gains a fourth electron, it has been fully reduced to water, and no longer readily reacts with other molecules. Other species of oxidants include: peroxyl (RO2•), alkoxyl (RO•), and hydroperoxyl (HO2•) free radicals, hypochlorous acid (HOCl), ozone (O3), singlet oxygen, and peroxynitrite (ONOO−). The term free radical denotes molecules with at least one unpaired electron. Oxygen (two unpaired electrons), superoxide, hydroxyl radical, and NO (each with one unpaired electron) are examples of free radicals. Hydrogen peroxide is not a free radical because all of its electrons are paired.
Figure 2. Derivation of reactive oxygen species. Sequential electron (e−) addition to oxygen results in the formation of reactive oxygen species (red): superoxide (O2‐), hydrogen peroxide (H202) and hydroxyl radical (OH). Superoxide can combine with nitric oxide (NO) to form peroxynitrite (ONOO‐). Both O2− and OH− can initiate lipid peroxidation to form lipid peroxides (ROO) that can propagate free radical chain reactions. (Full color version available online.)
Exogenous Sources of Oxidants
Cigarette Smoke
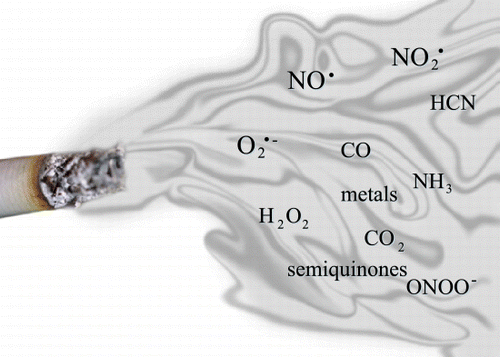
Cigarette smoke contains > 4700 chemicals including hydrogen peroxide, superoxide, hydroxyl radical and nitric oxide Citation[[5]]Citation[[9]]Citation[[10]] (). The constituents of cigarette smoke are often separated into a gas and particulate (tar) phase; both components contain abundant free radicals. The gas phase is less stable, but has been estimated to contain over 1015 free radicals per puff. The tar phase is more stable and has been estimated to contain over 1017 free radicals per gram Citation[[9]]. Quinones, semiquinones, hyrdroquinones, and metals are prominent components of smoke that may propagate reactions that produce high levels of ROS and RNS. Iron may also catalyze the production of free radicals through Fenton or Haber–Weiss reactions. The oxidants in cigarette smoke are postulated to injure lungs via several mechanisms including: 1) direct damage to lipids, nucleic acids, and proteins (see subsequent section), 2) depletion of crucial antioxidants such as glutathione, 3) initiation of redox‐cycling reactions, 4) promotion of carcinogenesis, 5) enhancement of the respiratory burst in macrophages and neutrophils, 6) inactivation of protease inhibitors such as α1 antitrypsin inhibitor, 7) a decrease in the binding affinity and translocation of steroid receptors, and 8) impairment of skeletal muscle dysfunction Citation[[9]].
Figure 3. Oxidants in cigarette smoke. Nitric oxide (NO), nitrogen dioxides (NO2), carbon monoxide (CO), ammonia (NH3), cyanide (HCN), hydrogen peroxide (H2O2), superoxide (O2−), and peroxynitrite (ONOO‐). (Full color version available online.)
Air Pollution
In December of 1952 in London there was a weeklong episode of severe air pollution associated with excess mortality. The majority of the excess deaths were due to exacerbations of bronchitis and the relative risk of death during this period was 9.3 Citation[[11]]. This event encouraged epidemiologists to document an association between the prevalence of COPD with geographic areas of high pollution. Air pollution contains a wide variety of chemicals that vary from one microenvironment to the next. Many of these particles contain reactive species such as nitrogen dioxide or ozone. The particulate matter in air pollution contains a large number of soluble metals including transition metals that are capable of redox cycling through the Fenton or Haber–Weiss reactions Citation[[12]]. Other components in air pollution that cause oxidative stress include polycyclic aromatic hydrocarbons and endotoxin.
Air pollution causes up to 30% of increase in COPD symptoms and case control studies have suggested that ozone has one of the strongest correlations Citation[[13]]Citation[[14]]. Each increase of 50 µg/m3 in air pollutants results in a 3–20% increase in the number of emergency room admissions due to chronic obstructive pulmonary disease Citation[[11]].
Endogenous Sources of Oxidants in Lungs
Although the lung is unique in its exposure to high concentrations of environmental oxidants, a large fraction of oxidants found in the lung are the by‐products of normal metabolism. Mitochondria are the largest source of free radicals because electrons leak from the electron transport chain onto oxygen to form superoxide. It is estimated that 1–3% of O2 reduced in mitochondria may form superoxide Citation[[9]]. A significant cytosolic source of superoxide is the enzyme xanthine dehydrogenase (a.k.a. xanthine oxidase) Citation[[15]]. Membrane oxidases that produce substantial quantities of superoxide include cytochrome P450 the b5 families of enzymes in the endoplasmic reticulum Citation[[16]] and the NADPH oxidases. Degenerate cloning of membrane oxidases has recently revealed a new family of membrane oxidases (Mox) that are found on advential smooth muscle cells of coronary arteries Citation[[17]] and aorta Citation[[18]]. Hydrogen peroxide is formed during the dismutation of superoxide, but also by glycolate oxidase in peroxisomes. Peroxisomes also contain hydrogen peroxide producing enzymes such as D‐amino acid oxidase, urate oxidase, L‐alpha‐hyroxyacid oxidase, and fatty acyl‐CoA oxidase Citation[[19]]. Soluble enzymes such as xanthine oxidase, aldehyde oxidase, dihydrorotate dehydrogenase, flavoprotein dehydrogenase and tryptophan dioxygenase can generate ROS Citation[[19]]. Hydroxyl radicals classically form in the presence of metals and hydrogen peroxide (Fenton reaction); however, decomposition from other molecules such as peroxynitrite may play a small role in hydroxyl radical formation Citation[[20]].
Reactive nitrogen species are primarily derived from nitric oxide. The sources of NO in the lung are three NO synthases (NOS): 1) neuronal NOS, found in the nerve plexus of the trachea Citation[[21]] (NOS1), 2) inducible NOS, found in respiratory epithelium and activated macrophages Citation[[22]] (NOS2), and 3) endothelial NOS, found in respiratory epithelium and vascular endothelium (NOS3) Citation[[23]]. In the resting state nitric oxide is considered a signaling molecule. When nitric oxide is produced in high concentrations, such as with inducible NOS, it can react with oxygen or superoxide to form the highly reactive compounds nitrogen dioxide and peroxynitrite. Only inducible NOS is highly upregulated by such cytokines as tumor necrosis factor–α (TNF‐α) and interleukin‐1ß Citation[[24]]. Thus, location determines the primary functions of nitric oxide in the lung: in pulmonary vessels nitric oxide is a vasodilator; in airway muscle nitric oxide functions as a bronchodilator; and in airway epithelium nitric oxide modulates the immune response.
Antioxidant Defenses in Lungs
Antioxidants can be classified as either enzymatic or non‐enzymatic and are the primary defense against ROS/RNS. Antioxidant enzymes include the families of superoxide dismutase (SOD), catalase, glutathione peroxidase, glutathione S‐transferase, and thioredoxin. Each family has isoenzymes that are distinguished primarily by their distribution. For instance, the three mammalian superoxide dismutases are cytosolic (SOD1) Citation[[25]], mitochondrial (SOD2) Citation[[26]], and extracellular (SOD3) Citation[[27]]. There are also multiple thioredoxin (Trx) and thioredoxin‐like (Txl‐1) proteins in the lung Citation[[28]]Citation[[29]]Citation[[30]].
The non‐enzymatic category of antioxidant defenses includes low molecular weight compounds such as glutathione, ascorbate, urate, alpha‐tocopherol, bilirubin, and lipoic acid. Concentrations of these antioxidants vary depending of both subcellular and anatomic location (). For instance, glutathione is one hundred fold more concentrated in airway epithelial lining fluid compared to plasma Citation[[31]]. Other high molecular weight molecules that might be considered antioxidants include proteins that have oxidizable thiol groups such as albumin or proteins that bind free metals such as transferrin. Albumin and transferrin are found in high concentration in serum, but are in much lower concentration in airway lining fluid Citation[[32]]. Thus, both the lung parenchyma and airways have several distinct antioxidant enzyme systems.
Table 1. Low Molecular Weight Antioxidants in Normal Subjects
Antioxidant Enzyme Families
SOD Family
One of the major enzymatic antioxidant families is the superoxide dismutase family, which catalyzes the dismutation of superoxide radicals into hydrogen peroxide and oxygen. The reaction is pseudo first‐order and almost diffusion limited (Michaelis‐Menten constant > 109 M− 1s− 1) Citation[[33]]. Three SOD isoenzymes have been identified in mammals. The major intracellular SOD is a 32 kDa copper and zinc (Cu/Zn SOD or SOD1) containing homodimer present throughout the cytoplasm and nucleus Citation[[34]]. Cu/Zn mutants are important in the pathogenesis of amyotrophic lateral sclerosis Citation[[35]]. The mitochondrial SOD (Mn‐SOD or SOD2) is a manganese‐containing 93‐kDa homotetramer that is synthesized in the cytoplasm and translocated to the inner matrix of mitochondria Citation[[26]]Citation[[36]]. Lack of Mn‐SOD may cause cardiomyopathies and neurodegenerative diseases Citation[[37]]Citation[[38]]Citation[[39]]. The last mammalian SOD to be discovered is primarily extracellular (EC‐SOD or SOD3) Citation[[27]].
CuZn SOD
CuZnSOD is the most abundant superoxide dismutase in most tissues. In the lung, CuZnSOD is highly expressed in bronchial epithelium, alveolar epithelium, mesenchymal cells, fibroblasts, and arterioles in capillary and endothelial cells Citation[[40]]Citation[[41]]. In the bronchial epithelium, CuZnSOD is highly expressed in ciliated epithelial cells. CuZnSOD is located primarily in the cytosol but is also present at substantial concentration in the nucleus. It is highly expressed in some organelles such as lysosomes Citation[[40]]. Studies with mice overexpressing transgenic CuZnSOD show that these mice are resistant to allergen induced lung toxicity and pulmonary oxygen toxicity Citation[[42]]Citation[[43]].
Manganese Superoxide Dismutase
MnSOD mRNA is prominently expressed in cells in airway walls, the septal tips of alveolar ducts and in arteriolar walls located adjacent to airways Citation[[44]]Citation[[45]]. In the lung, MnSOD is exclusively localized to mitochondria Citation[[40]]. Mice that do not express MnSOD die shortly after birth Citation[[37]]Citation[[38]]; however, the lungs are only minimally affected. The organs most affected by MnSOD genetic deficiency are the central nervous system, liver, and cardiovascular system Citation[[37]]Citation[[38]]. Mice that are heterozygous for the MnSOD deletion do not reveal sensitization to hyperoxia or pertussis toxin induced lung injury Citation[[46]]Citation[[47]]; however, decreased lung glutathione concentrations in these animals suggest that there is increased oxidative damage in the airways Citation[[48]]. Mice that overexpress transgenic MnSOD in the lungs are resistant to hyperoxia Citation[[49]] yet are not protected from excessive irradiation Citation[[50]]. Thus the role of MnSOD in modulating lung injury needs further investigation.
EC‐SOD
EC‐SOD is the primary extracellular SOD enzyme and is highly expressed in many organs. The uterus, pancreas, and lung have the highest EC‐SOD activity levels among solid organs; EC‐SOD activity in the human lung is approximately eight times that of the liver, six times that of the brain, two to three times that in heart, and one to two times that of the kidney Citation[[51]]. Within the lung, EC‐SOD protein is particularly abundant in blood vessels and airways (). EC‐SOD mRNA is abundant in airway epithelium and vascular endothelium and is expressed at levels four times of that in heart and 15 times that in liver Citation[[52]]. The protein is mainly associated with the connective tissue matrix around vessels and airways in the lung, but is also found in close proximity to airway and vascular smooth muscle cells Citation[[53]]Citation[[54]]. Electron microscopic immunocytochemistry reveals that EC‐SOD is seen in areas rich in type I collagen and associated with extracellular matrix around airway and vascular smooth muscle cells. The prominent expression of EC‐SOD around airways and airway smooth muscle suggests that it may play a role in allergic airway diseases such as asthma. Unstressed mice that are deficient in EC‐SOD have a normal phenotype but their blood vessels have impaired vasodilation to nitric oxide Citation[[55]].
Figure 4. Pulmonary distribution of extracellular superoxide dismutase. Histochemical localization of the superoxide dismutases in human lung using rabbit anti‐human extracellular superoxide dismutase extracellular superoxide dismutase (ECSOD) antibodies. ECSOD labeling of a small airway (A) and small pulmonary vessel (B). Strong labeling is seen in the airway and vessel walls (arrows). (A) Reprinted by permission from Ref. Citation[[206]] and (B) reprinted by permission from Ref. Citation[[207]]. (Full color version available online.)
![Figure 4. Pulmonary distribution of extracellular superoxide dismutase. Histochemical localization of the superoxide dismutases in human lung using rabbit anti‐human extracellular superoxide dismutase extracellular superoxide dismutase (ECSOD) antibodies. ECSOD labeling of a small airway (A) and small pulmonary vessel (B). Strong labeling is seen in the airway and vessel walls (arrows). (A) Reprinted by permission from Ref. Citation[[206]] and (B) reprinted by permission from Ref. Citation[[207]]. (Full color version available online.)](/cms/asset/ffaec02e-0308-4c60-b3c6-d721728c3c4a/icop_a_27031_uf0004_b.gif)
Although the role of EC‐SOD in airway diseases is not clear, two studies indirectly suggest that EC‐SOD activity might play a role in COPD. First, neutrophils secrete increased extracellular superoxide when stimulated with ultra‐fine carbon black particles Citation[[56]]. Second, EC‐SOD mimetics reduce airway inflammation and epithelial cell hyperplasia in animal models Citation[[57]]. Although the role of EC‐SOD has not been specifically investigated in COPD, EC‐SOD attenuates lung injury and inflammation in other pulmonary diseases. For instance, mice that overexpress EC‐SOD in the lung are partially protected from hyperoxia‐induced injury Citation[[58]]Citation[[59]], hemorrhagic shock Citation[[60]], influenza Citation[[61]], bleomycin Citation[[62]], and oil fly ash Citation[[63]]. These studies suggest that under normal conditions the constitutive levels of EC‐SOD may be sufficient to attenuate the effects of superoxide in extracellular spaces, but not during pathologic states.
Another mechanism by which EC‐SOD might influence airway physiology is through modulation of nitric oxide. Nitric oxide is a potent signaling molecule that may cause non‐adrenergic mediated bronchodilation Citation[[64]]Citation[[65]]Citation[[66]]. Superoxide inactivates nitric oxide by rapidly reacting with it to form peroxynitrite. A recent report implicates EC‐SOD as a modulator of nitric oxide mediated signaling in blood pressure control Citation[[55]]. The relative abundance of EC‐SOD in both bronchi and blood vessels raises the possibility the EC‐SOD might also modulate pulmonary blood flow or neurally mediated bronchodilation; however, the significance of EC‐SOD in modulating nitric oxide signaling pathways in airways remains to be shown.
Thioredoxin System
The thioredoxin system consists of multiple species including thioredoxins (e.g. Trx‐1 and Trx‐2), thioredoxin like proteins (e.g. Tlx‐1, Tlx‐2, Sptrx‐1, and Sptrx‐2) and thioredoxin reductases (e.g. TrxR‐1 and TrxR‐2) Citation[[29]]Citation[[67]]Citation[[68]]. These enzymes are important in protein disulfide reduction, but may have additional antioxidant functions. Thioredoxin is a 13‐kDa protein containing two redox‐active cysteines at its active site (Trp‐Cys‐Gly‐Pro‐Cys), which are highly conserved across species. Mammalian TrxR are selenoezymes that reduce oxidized thioredoxin and other protein disulfides. Thioredoxin reduces the viscosity of sputum in Cystic Fibrosis patients Citation[[69]], but its role in patients with COPD is unknown. In humans there are multiple thioredoxin genes including Trx‐1, which encode for the classical thioredoxin and Trx‐2, an isoenzyme that is located in the mitochondria. These proteins have been shown to be present in bronchial epithelium and alveolar macrophages Citation[[28]]Citation[[29]]. Spermatid‐specific like thioredoxins are not expressed in the lung Citation[[70]]. There have been a few studies of thioredoxin in non‐malignant lung disease. These show increased staining for both Trx and TrxR in interstitial lung diseases, such as usual interstitial pneumonia (UIP) and desquamative interstitial pneumonia (DIP), particularly in areas of metaplastic epithelium and in inflammatory cells Citation[[28]]. Exogenous thioredoxin has been shown to reduce ischemia reperfusion in the lung Citation[[71]]Citation[[72]]Citation[[73]]. Transgenic mice that overexpress human thioredoxin were more resistant to influenza virus infection than control mice Citation[[74]].
Glutathione System
Glutathione is the most abundant intracellular thiol‐based antioxidant. It has an important function in maintaining the redox status within cells in addition to detoxifying compounds via conjugation reactions through glutathione S transferases. The enzymes that are important in the metabolism of glutathione include glutathione peroxidases (GPx) and the glutathione regenerating enzyme, glutathione reductase (GR). The glutathione system has an important role intracellularly, however, glutathione is in very high concentrations in lung epithelial lining fluid as discussed above. Patients with COPD exacerbations have lower levels of glutathione peroxidase activity in red blood cells Citation[[75]].
The mechanisms by which glutathione (GSH) could attenuate COPD exacerbations are unknown, but should be interpreted within the context of its distribution in the lung. For instance, bronchoalveolar lavage fluid (BALF) contains a 100‐fold concentration of glutathione compared to blood Citation[[31]]Citation[[76]]. GSH is also highly concentrated in intracellular spaces Citation[[9]]. Although GSH has been shown to be elevated in the BALF of chronic smokers Citation[[77]], cigarette smoke acutely lowers intracellular glutathione Citation[[78]]Citation[[79]].
Evidence of Oxidative Stress in COPD
There is now overwhelming evidence implicating an association between oxidative stress and COPD; however, most of this evidence is indirect. The two indirect methods that are most often used to detect ROS are spin trapping and footprinting (also called fingerprinting). Spin trapping is a technique by which a radical reacts with a more stable molecule that can be readily measured in biologic systems. Spin trapping has been used to demonstrate increased ROS in the bronchoalveolar lavage fluid of patients with COPD Citation[[80]]. Footprinting involves the principle that ROS react with lipids, proteins, or nucleic acids to form specific markers of oxidative stress. Examples of footprint reactions include ROS attack on proteins to form carbonyls, peroxynitrite reacting with tyrosine to form nitrotyrosine, ROS reacting with lipids to liberate ethane and isoprostanes or DNA to form base pair adducts such as 8‐oxo‐2‐deoxyguanosine. Another commonly measured footprint is the oxidation of glutathione to oxidized glutathione. Footprinting has been used extensively to study local and systemic oxidative stress in COPD ().
Table 2. Free Radicals and Their Footprints in COPD
Many studies have demonstrated increased footprints of oxidative stress both locally (lung) and systemically (blood and urine) in COPD patients. Evidence for local oxidative stress has been found in exhaled breath condensates, sputum, and lavage fluid of both smokers and patients with COPD. Some, but not all of these studies have found a difference between smokers and patients with COPD. For instance, lipid peroxidation products such as aldehydes are increased in the exhaled breath condensates of smokers and patients with COPD, but only malondialdehyde is increased in patients with COPD compared to smokers without COPD Citation[[81]]. The presence of systemic oxidative stress in COPD is suggested by the elevation of plasma lipid peroxidation products such as malondialdehyde during COPD exacerbations Citation[[75]]Citation[[82]]Citation[[83]]. Urinary levels of 8‐isoprostanes are also increased in patients with COPD Citation[[84]]. Exhaled ethane is increased in COPD patients Citation[[85]]. Similarly, patients with COPD have increased exhaled breath condensate 8‐isoprostanes compared to healthy smokers and non‐smokers Citation[[86]]. The exhaled breath condensate pH has also been shown to correlate with 8‐isoprostanes and hydrogen peroxide in patients with COPD Citation[[87]] and the 8‐isoprostane levels increase with COPD exacerbations, but return to normal after 2–4 weeks Citation[[88]]. Lung tissue of patients with COPD has been shown to have increased lipid peroxidation product 4‐Hydroxy‐2‐nonenal Citation[[89]]. Long term N‐acetylcysteine does not appear to affect exhaled breath condensate lipid‐peroxidation as measured by TBARS Citation[[90]].
The EBC concentration of nitrates in COPD patients is no different from controls Citation[[91]]; however, the sputum of patients with COPD has increased nitrates and nitrites (NOx) Citation[[92]]. Nitrotyrosine levels are increased in the sputum of patients with COPD compared to healthy subjects and those with asthma Citation[[93]]. Four weeks of inhaled steroids reduce nitrotyrosine staining in sputum Citation[[94]]. Since exhaled nitric oxide is not elevated in patients with stable COPD Citation[[95]]Citation[[96]], further work is necessary to determine the specific role of NOx in COPD. It is likely that gaseous NO is consumed by formation of peroxynitrite in the presence of oxidative stress.
There is direct evidence that stable ROS are elevated in patients with COPD. For instance, patients with COPD have increased exhaled hydrogen peroxide Citation[[90]]Citation[[97]]Citation[[98]] and nitric oxide Citation[[66]]Citation[[85]]Citation[[99]]Citation[[100]]. Cigarette smoking has also been associated with increased superoxide production in airway leukocytes in both acute and chronic smokers Citation[[101]]. A causal link between air pollution and COPD exacerbations is suggested by the finding that neutrophils from patients with COPD produce more superoxide when exposed to ultrafine carbon black Citation[[56]]. Direct evidence implicating other ROS has been difficult to obtain because the evanescent nature of ROS make them difficult to measure in vivo. For instance, the hydroxyl radical reacts with molecules in its immediate vicinity, thus making it difficult to measure in vivo. Electron spin resonance is the only method that directly measures free radicals, but this method is not sensitive enough to detect free radicals in most in vivo conditions.
The Role of Oxidative Stress in the Pathogenesis of COPD
Gene Transcription
Transcription of genes requires multiple steps beginning at the level of chromatin. The basic unit of chromatin is the nucleosome, which consists of approximately 146 base pairs of DNA wrapped around multimeric subunits of nucleosomes. Genomic regions that are tightly wrapped around histones have suppressed gene transcription since the transcriptional complex does not have access to the DNA. One of the key events in unraveling the DNA from the histone complex is the acetylation of lysine residues on histone protein number 4 Citation[[102]]. The amount of acetylation is controlled by histone acetyl transferases (HAT), which promote histone acetylation, and histone deacetylases (HDAC), which promote histone deacetylation. Although the specific role of oxidative stress in modifying HAT and HDAC activity is unknown, transcription factors such as NF‐κB AP‐1, cyclic APM response element binding protein (CREB) and other transcription factors are sensitive to the redox state and modulate HAT activity (Citation[[19]]Citation[[103]]). Oxidative stress activates HAT activity in an epithelial line and leads to inflammatory gene transcription Citation[[104]]. There is evidence that HDAC2 activity is reduced by cigarette smoking and oxidative stress Citation[[105]] and that this may be due to nitration of HDAC2 by peroxynitrite formation Citation[[106]]. This reduction in HDAC2 activity appears to account for corticosteroid resistance in COPD patients Citation[[107]]. These pathways are also activated in the transcript of other pro‐inflammatory genes such as interleukin 1 and tumor necrosis factor α. The mechanism by which ROS modulate this activity is unknown, but the most likely mechanism is through the oxidation and reduction of disulfide bridges or thiol‐residues. For instance, when Rac‐GTPases are activated, they produce reactive oxygen species that down regulate Rho activity through oxidation of a key thiol residue on a low molecular weight tyrosine phosphatase that turns off Rho Citation[[108]].
The mechanisms by which tobacco smoke modulates transcription of genes in patients with chronic obstructive pulmonary disease are unknown. Signaling pathways that have been shown to be sensitive to tobacco smoke include protein kinase C (PKC) whose activity increases 2–3 fold when stimulated with 5% cigarette smoke extract Citation[[109]]. Cigarette smoke also modulates NF‐κB Citation[[110]] and leads to a prolonged increase in c‐fos transcripts Citation[[111]]. NF‐κB is increased in alveolar macrophages and airway epithelial cells of COPD patients Citation[[112]], particularly during exacerbations Citation[[113]]. A recent gene expression profiling study of rat respiratory tract epithelium which extensively characterized the effects of cigarette smoke on gene expression Citation[[114]] revealed that cigarette smoke causes a rapid induction of oxidative stress response genes and drug metabolizing enzymes such as hemeoxygenase 1 and NADPH quinonone‐oxido‐reductase. However, after three weeks of exposure, the increased expression of these genes was markedly reduced.
Latent Adenoviral Infections
An intriguing hypothesis for the pathogenesis of COPD is that latent adenoviral infections contribute to the disease through the pro‐inflammatory effects of the adenoviral E1A gene Citation[[115]]Citation[[116]]. The link between adenovirus infection and oxidative stress in the pathogenesis of COPD remains to be proven; however, adenovirus E1A has been shown to block the induction of ferritin, which sensitizes cells to cytotoxicity from hydrogen peroxide Citation[[117]]. Additionally, human alveolar epithelial cells that have been treated with E1A show increased production of mRNA for the cytokine interleukin 8 after treatment with hydrogen peroxide Citation[[118]].
Inflammation
Although asthma and COPD are both associated with increased airway inflammation, the type of inflammation is substantially different. The sputum of asthma patients contains a high percentage of eosinophils Citation[[100]] whereas the sputum in COPD patients contains predominantly neutrophils Citation[[119]]. Cigarette smokers have similar increases of neutrophils in their airways Citation[[120]] and there is an inverse correlation between neutrophilic inflammation and FEV1 Citation[[121]]. One mechanism by which cigarette smoke might increase lung neutrophils is by decreasing neutrophil deformability. Since the average pulmonary capillary diameter is only 5 µM and the average neutrophil diameter is 7 µM, the average neutrophil must deform to pass through the lungs. Cigarette smoking has been shown to acutely increase neutrophil deformability in the lungs in vitro Citation[[122]] and increase neutrophil lung‐transit time in vivo Citation[[123]]. The loss of deformability can be reduced by the antioxidant glutathione Citation[[122]]. Additional studies have shown that oxidants also play a role in recruiting neutrophils to the lung by causing the up regulation of neutrophil adhesion molecules Citation[[124]]. Thus, oxidants in cigarette smoke may increase lung inflammation by trapping neutrophils in the lungs both through decreased deformability and through increased adhesion to epithelium.
The recruitment of inflammatory cells to the lung in COPD patients may set up a positive‐feedback loop that perpetuates lung injury through other mechanisms, including the regulation of cytokine secretion. For instance, patients with COPD demonstrate increased levels of interleukin (IL)‐6, IL‐1 ß, TNF‐α and IL‐8 in sputum Citation[[125]] and respiratory epithelial cells of smokers and patients with COPD have been shown to secrete increased TGF‐ß Citation[[126]]. The link between cigarette smoke and cytokine secretion may be activation of NF‐κB, which is up regulated by oxidants Citation[[127]]. ROS, particularly hydrogen peroxide, have also been shown to regulate signal transduction in inflammatory cells via oxidation of key cysteine residues in phosphatases Citation[[108]].
Intracellular Adhesion Molecules
Cigarette smoke extract induces increased expression of intercellular adhesion molecule (ICAM)‐1, which is tightly controlled by NF‐κB, on airway epithelial monolayers compared with unstimulated cells in vitro Citation[[128]]. This enhanced ICAM‐1 expression is associated with a greater capacity of airway epithelial cells to bind mononuclear cells. ROS such as superoxide have recently been shown to be important in the induction of intracellular adhesion molecules such as E‐and P‐selectins Citation[[129]]Citation[[130]]Citation[[131]]. Recent evidence also implicates endothelial nitric oxide synthase (eNOS) in the induction of these adhesion molecules Citation[[132]]. In COPD patients the plasma levels of L‐selectin, E‐selectin and P‐selectin are not increased above controls, Citation[[133]]. Thus the relevance of oxidative stress in upregulating adhesion molecules remains to be proven in COPD patients.
Protease Activity
Neutrophil elastase and other lung proteases are capable of causing emphysema. Reduced activity of the α1‐protease inhibitors is strongly associated with COPD and is the only well‐established genetic risk factor. There are several links between protease activity and oxidative stress. First, ROS oxidize a key methionine in the α1‐antitrypsin leading to its inactivation Citation[[134]]. The oxidation of plasma proteins is almost exclusively due to the oxidant and pro‐oxidant species found in the tar phase of cigarette smoke Citation[[135]]. Second, induction of human neutrophil elastase is redox sensitive Citation[[136]]. Third, activation of lung epithelial cell secreted proteases leads to increased release of ROS Citation[[137]]. Additional investigations are needed to fully determine how ROS modulate protease activity and lead to degradation of lung tissue.
Mucus Secretion
Oxidants from cigarette smoke or inflammatory cells may be associated with chronic bronchitis and COPD exacerbations by impairing mucociliary clearance. Reactive oxygen species have been reported to stimulate mucin secretion Citation[[138]]. The superoxide‐mediated increase in mucus from respiratory epithelial cells is mediated by prostaglandins and can be decreased by inhibitors of arachidonic acid metabolism Citation[[139]]. Superoxide and hydrogen peroxide have also been shown to decrease beating of respiratory epithelium cilia, even as early as 15 minutes after exposure Citation[[140]]. A large, randomized trial is underway to determine whether the antioxidant N‐acetylcysteine can reduce exacerbations of chronic bronchitis Citation[[141]].
Apoptosis
Recently reports have suggested that apoptosis may also play a role in the pathogenesis of emphysema. In patients with COPD, increased numbers of apoptotic cells have been demonstrated in pulmonary endothelium Citation[[142]] and in alveolar walls Citation[[143]]Citation[[144]]. Airway lymphocytes Citation[[145]] and stimulated peripheral blood lymphocytes Citation[[146]] from patients with COPD have also shown increased apoptosis. The pathogenesis of increased apoptosis of lung tissue remains under active investigation, but is hypothesized to be due to decreased production of growth factors such as vascular endothelial growth factor (VEGF) Citation[[147]]Citation[[148]]. This hypothesis is supported by the finding that emphysematous lungs have decreased VEGF ligand Citation[[142]] and the observation that inhibition of the VEGF receptor will cause emphysema in rats Citation[[149]]. A role for oxidative stress is suggested because a SOD mimetic is capable of not only blocking the oxidative stress associated with VEGF blockade, but also is able to prevent the resultant emphysema () Citation[[150]].
Figure 5. Treatment with an SOD mimetic (M40419) attenuates anti‐VEGF‐(SU5416) mediated emphysema. (a) SU5416‐treated lungs show increased size of alveolar structures. (b) SU5416 + M40419‐treatment protects against alveolar loss and these animals show normal alveolar size and architecture (bar = 100 µm). Adapted with permission from Ref. Citation[[150]]. (Full color version available online.)
![Figure 5. Treatment with an SOD mimetic (M40419) attenuates anti‐VEGF‐(SU5416) mediated emphysema. (a) SU5416‐treated lungs show increased size of alveolar structures. (b) SU5416 + M40419‐treatment protects against alveolar loss and these animals show normal alveolar size and architecture (bar = 100 µm). Adapted with permission from Ref. Citation[[150]]. (Full color version available online.)](/cms/asset/c7f88757-a772-4914-ab04-b3095ab76835/icop_a_27031_uf0005_b.gif)
Oxidative Stress and Nitric Oxide Signaling in the Lung
Although there is strong evidence of a role for nitric oxide in asthma, data regarding the benefit of nitric oxide in COPD is less certain. Increases in exhaled nitric oxide correlate with sputum neutrophilia Citation[[151]] but the effect of COPD on exhaled NO is not yet clear. Some investigators have found that exhaled nitric oxide is higher in COPD patients during exacerbations compared to healthy controls Citation[[95]]Citation[[152]], but there is no difference when COPD patients are at baseline Citation[[153]]. Other investigators have found no increases in exhaled nitric oxide in patients with COPD Citation[[154]].
The role of nitric oxide in the lung is complicated because there are thee distinct sources of NOS, as previously discussed. Both nNOS and eNOS are constitutively active; however, it is primarily iNOS that is induced in asthma and in COPD exacerbations and is responsible for increased levels of exhaled nitric oxide during these diseases. Although inhaled nitric oxide reduces pulmonary vascular resistance in patients with COPD Citation[[155]] and decreases airway resistance in asthmatics, there is no improvement in oxygen saturation or airway resistance in patients with COPD Citation[[66]]Citation[[156]]Citation[[157]]. Thus a lack of significant bronchorelaxation associated with nitric oxide in COPD suggests that signaling pathways in bronchial smooth muscle are impaired. Concomitant increases in the production of reactive oxygen species during inflammation may be the source of this impairment. Additionally increases in exhaled NO may correlate with increases in tissue peroxynitrite rather than with increases in tissue NO if there is oxidative stress in the tissue with enhanced superoxide production.
The impairment of airway nitric oxide signaling in COPD patients is likely due to nitric oxide's reaction with reactive oxygen species. For instance, nitric oxide rapidly reacts with superoxide to form peroxynitrite Citation[[158]]. Peroxynitrite formation increases during inflammation and is toxic to microbes; however, peroxynitrite can also cause airway hyperresponsiveness Citation[[159]]. Presumably, peroxynitrite formation is also associated with a decrease in the amount of nitric oxide available for bronchodilation of smooth muscle.
One would predict peroxynitrite formation to be favored at sites of inflammation and infection, but one would also predict that in the normal lung peroxynitrite production would be inhibited at sites where nitric oxide mediated signaling is important. Two sites at which nitric oxide signaling is essential are respiratory smooth muscle and the vasculature. Indeed, EC‐SOD is abundant in both airway smooth muscle and pulmonary vasculature and may be a mechanism of preserving both vasoregulation and smooth muscle bronchodilation in response to nitric oxide. The specific role of EC‐SOD in COPD remains to be proven, but should be given consideration for a substantial role in both inhibiting airway inflammation and in preserving the bronchorelaxant effects of nitric oxide during oxidative stress.
Respiratory Muscle Dysfunction
There is significant skeletal muscle dysfunction in some patients with severe COPD Citation[[160]]. This dysfunction is especially prominent in the diaphragm Citation[[161]], but also in peripheral muscles Citation[[162]]. Several studies have demonstrated that COPD patients have evidence of increased oxidative stress, both peripherally and locally, particularly after exercise Citation[[10]]Citation[[162]]Citation[[163]]. Biopsies of skeletal muscles from patients with COPD have shown increased apoptosis Citation[[164]]. The contribution of oxidative stress to apoptosis in skeletal muscles is unknown. Other possible mechanisms by which oxidative stress may lead to decreased muscle function are unclear, but may involve alterations in electrolyte movement, mitochondrial respiration, or contractile proteins.
The relationship between skeletal muscle strength and oxidative stress is complicated. Oxidative stress has also been shown to increase skeletal muscle transcript levels for the antioxidant enzymes glutathione peroxidase, catalase, CuZnSOD and MnSOD Citation[[165]]. In healthy individuals, a training protocol leads to skeletal muscle adaptation to oxidative stress, presumably through the induction of these enzymes; however, patients with COPD have reduced capacity to adapt to oxidative stress with exercise training Citation[[166]].
Glucocorticoid Resistance
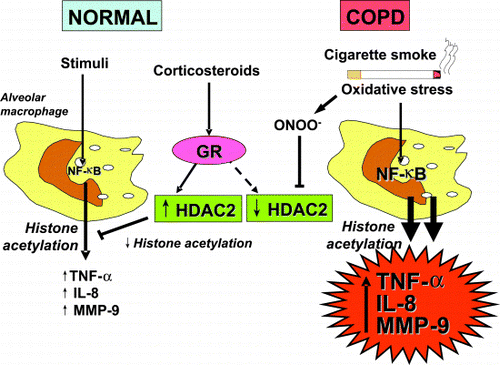
The chronic inflammation of COPD is resistant to the anti‐inflammatory effects of corticosteroids and this may be linked to oxidative stress. Steroid resistance occurs at the level of specific cells including alveolar macrophages, which show reduced responsiveness to corticosteroids in normal smokers and complete resistance in COPD patients Citation[[167]]. By contrast, patients with asthma are highly sensitive to the effects of low doses of corticosteroids. Some of the mechanisms by which corticosteroids switch off inflammatory genes in asthma have recently been elucidated Citation[[168]]. A major mechanism for switching off inflammatory genes is the reversal of histone acetylation activated by transcription factors by histone deacetylases (). Corticosteroids induce the translocation of glucocorticoid receptors from the cytoplasm to the nucleus and these bind to activate inflammatory gene transcriptional complexes. This leads to specific recruitment of HDAC2, which reverses histone acetylation and switches off the activated inflammatory genes Citation[[169]]. HDAC2 activity is markedly reduced in alveolar macrophages obtained from smokers and even further reduced in cells from COPD patients Citation[[105]]. The reduction in HDAC activity is significantly related to the reduced responsiveness to corticosteroids in these cells. The mechanism for reduction in HDAC activity is likely oxidative stress. Both oxidative stress (hydrogen peroxide) and cigarette smoke extract reduce HDAC activity in vitro. The mechanism may involve formation of peroxynitrite, since peroxynitrite mimics these effects and nitrates HDAC2, presumably impairing its catalytic activity and then its destruction Citation[[106]]. The same mechanisms may account for the resistance to corticosteroids seen in asthmatic patients who smoke Citation[[107]].
Figure 6. Stimulation of normal alveolar macrophages activates nuclear factor‐κB (NF‐κB) and other transcription factors to switch on histone acetyltransferase leading to histone acetylation and transcription of genes encoding inflammatory proteins, such as tumor necrosis factor‐α (TNF‐α), interleukin‐8 (IL‐8) and matrix metalloproteinase‐9 (MMP‐9). Corticosteroids reverse this by binding to glucocorticoid receptors (GR) and recruiting histone deacetylase‐2 (HDAC2). This reverses the histone acetylation induced by NF‐κB and switches off the activated inflammatory genes. In COPD patients cigarette smoke activates macrophages, as in normal subjects, but oxidative stress (perhaps acting through the formation of peroxynitrite) impairs the activity of HDAC2. This both amplifies the inflammatory response to NF‐κB activation, and reduces the anti‐inflammatory effect of corticosteroids as HDAC2 is now unable to reverse histone acetylation. (Full color version available online.)
The Role of Antioxidants in COPD
Endogenous and Dietary Antioxidants
There is a well‐established epidemiologic relationship between dietary intake of antioxidants and lung function Citation[[170]]. Diets rich in fresh fruit and vegetables that are rich in antioxidants are associated with improved lung function and have been proposed as treatment strategies for patients with COPD Citation[[171]]Citation[[172]]. It is unclear which specific dietary antioxidants are responsible for the decrease in lung function, but epidemiologic studies have indirectly suggested a role for vitamin E Citation[[173]]Citation[[174]], vitamin A Citation[[175]], and Vitamin C () Citation[[176]]; however, not all studies have reported a correlation between these dietary antioxidants and lung function Citation[[177]]. Direct evidence implicating specific dietary antioxidants is more complicated.
Figure 7. Low levels of plasma ascorbate increase the risk of COPD independently of smoking history. The relative risks of having COPD in light (white bar), medium (black bar) and heavy (cross hatched bar) smokers compared to never smokers are shown for each quintile of plasma ascorbate concentration. The risk of COPD was highest in the patients with the very low quintile of plasma concentration (3–37 µM), compared to low (28–49 µM), median (50–58 µM), high (59–68 µM), or very high quintiles (69–174 µM). Adapted from Ref. (176).
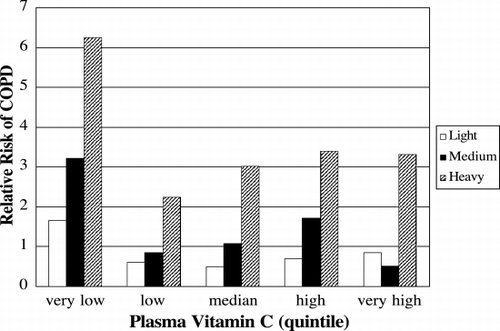
Direct measurement of antioxidants in both airways and plasma has suggested an association between COPD and low antioxidant levels. Alveolar macrophages obtained from bronchoalveolar lavage of COPD patients show decreased levels of glutathione Citation[[178]]; however, chronic supplementation with N‐acetylcysteine does not increase glutathione in bronchoalveolar lavage fluid or lung tissue in patients with COPD Citation[[179]]. Smoking is associated with increased concentrations of airway total antioxidant activity and specific antioxidants such as glutathione, particularly in chronic smokers Citation[[101]]. Airway glutathione concentrations are correlated with airflow obstruction in patients with COPD Citation[[180]]. In plasma, both smokers Citation[[181]] and patients with COPD exacerbations Citation[[82]] have decreased plasma total airway antioxidants. Some studies have failed to find a correlation between plasma total antioxidant activity and % predicted FEV1 Citation[[182]], thus additional investigation is necessary to elucidate the role of dietary antioxidants in COPD.
Enzymatic antioxidants may also play a role in COPD, however there are fewer investigations of these. Homozygosity of microsomal epoxide hydrolase, an enzyme that detoxifies epoxides, has been associated with COPD Citation[[183]]. Other studies have revealed a slightly increased risk of COPD with deletions in the glutathione‐S‐transferase allele Citation[[184]]. In smokers there is a GT dinucleotide repeat length polymorphism in the 5′‐flanking region of human hemoxygenase‐1 gene that is of increased prevalence in patients with emphysema Citation[[185]]. Decreased red blood cell catalase activity has been associated with COPD Citation[[186]]. Red blood cell glutathione peroxidase has been shown to increase during COPD exacerbations Citation[[75]]. To our knowledge, there are no reports of lung superoxide dismutase activity, thioredoxins, or other peroxidases in COPD.
Pharmacologic Antioxidants
N‐Acetylcysteine
N‐acetylcysteine (NAC) is hydrolyzed intracellularly to cysteine and stimulates glutathione synthesis. NAC reduces lung injury in many animal models of disease Citation[[187]]Citation[[188]]Citation[[189]]Citation[[190]]Citation[[191]]Citation[[192]]Citation[[193]]Citation[[194]]Citation[[195]] and is used to treat acetaminophen overdosage in humans. Several randominized‐controlled studies have shown that 600 mg NAC given daily will reduce exacerbations of COPD (). The mechanism by which NAC protects the lung are unknown, but are presumed to be through upregulation of the glutathione system.
Table 3. Randomized Controlled Trials Using N‐Acetylcysteine to Treat COPD Patients
Antioxidant Mimetics
Several classes of antioxidant mimetic have been created that share the properties of being relatively low in molecular weight and having high rate constants for enzymatic scavenging of free radical species (). A metal atom (commonly manganese) acts as the active center for electron transfer to or from free radical species. Mimetics of the macrocyclic class illustrated in are restricted to one electron transfer and are therefore more specific for scavenging O2− while the salen and porphyrin classes carry out both one and two electron transfers and are potentially effective in scavenging a broader range of reactive oxygen species. One of the macrocyclic classes of antioxidant mimetics has been shown to prevent the formation of emphysema in response to VEGF‐receptor blockade () Citation[[150]].
Figure 8. Three classes of superoxide dismutase mimetics. Side chains (R‐) can be substituted.

A manganoporphyrin antioxidant mimetic has been shown to significantly reduce inflammation and squamous metaplasia induced by tobacco smoke () Citation[[57]]. In this study animals were treated with the mimetic AEOL 10113 by intratracheal aerosolization once per week and then exposed to high doses of tobacco smoke (80 mg particulate/m3) for 6 hours per day for three successive days and then repeated for 8 weekly cycles. At the end of 8 weeks, animals exposed to tobacco smoke alone had 12% of their trachea showing squamous metaplasia while those receiving tobacco smoke exposure plus the antioxidant mimetic had only 2% airway squamous metaplasia. Both the number of inflammatory cells and mediators of inflammation expressed in bronchoalveolar lavage fluids were decreased by treatment with the mimetic Citation[[57]].
Figure 9. Effects of tobacco smoke on airways. Catalytic antioxidants attenuate squamous metaplasia from tobacco smoke. (A) Epithelial cell changes in airways from tobacco smoke‐exposed and filtered air (control) rats (epithelium is indicated between arrows). Lung sections were stained with hematoxylin and eosin for general morphology. Airway epithelium from filtered air control rats do not show the striking squamous cell metaplasia with prominent filamentous‐like cytoplasmic extensions (*) observed in the airways from rats exposed to tobacco smoke for 8 weeks. (B) Squamous metaplasia in rat lungs with or without a superoxide dismutase mimetic. The percent of airway surface expressing the type of squamous metaplasia illustrated in is shown. Data are presented as mean ± SE (n = 5−6). *p < 0.05 for comparison to the tobacco smoke alone group. Adapted from Ref. Citation[[57]]. (Full color version available online.)
![Figure 9. Effects of tobacco smoke on airways. Catalytic antioxidants attenuate squamous metaplasia from tobacco smoke. (A) Epithelial cell changes in airways from tobacco smoke‐exposed and filtered air (control) rats (epithelium is indicated between arrows). Lung sections were stained with hematoxylin and eosin for general morphology. Airway epithelium from filtered air control rats do not show the striking squamous cell metaplasia with prominent filamentous‐like cytoplasmic extensions (*) observed in the airways from rats exposed to tobacco smoke for 8 weeks. (B) Squamous metaplasia in rat lungs with or without a superoxide dismutase mimetic. The percent of airway surface expressing the type of squamous metaplasia illustrated in Fig. 9A is shown. Data are presented as mean ± SE (n = 5−6). *p < 0.05 for comparison to the tobacco smoke alone group. Adapted from Ref. Citation[[57]]. (Full color version available online.)](/cms/asset/4cb06350-f0ec-4302-a976-132774203098/icop_a_27031_uf0009_b.gif)
Other strategies for reducing oxidant stress such as the use of spin traps as free radical scavengers [e.g. nitrones Citation[[196]]] or iNOS inhibitors to prevent the formation of peroxynitrite could also be effective in preventing airway and lung inflammation. These alternative approaches have not yet been tested in rigorous models of emphysema.
Conclusions
Current efforts to ameliorate the symptoms of COPD focus on the use of bronchodilators to counteract the airway narrowing induced by cigarette smoking. There are no known medications that have been shown to substantially alter the natural history of COPD. Although there are substantial data to implicate oxidative stress in the pathogenesis of COPD, there have been few studies that have successfully used antioxidant strategies to modulate the progression of COPD. The need for strategies to supplement endogenous antioxidants or modulate oxidative stress is great. These approaches may represent one of the most promising avenues of research for disease modifying drugs for a disease that exacts a significant toll on quality of life and productivity.
Acknowledgments
This work was supported by National Institutes of Health HL‐04407 (R.P.B.), 5 P01 HL031992‐20 (J.D.C), and the Colorado Tobacco Research Program 3I‐005(R.P.B.).
Dr. Crapo is a founder and CEO of Aeolus Pharmaceuticals, which is developing antioxidant mimetics for human clinical trials.
References
- Pauwels R A, Buist A S, Calverley P M, Jenkins C R, Hurd S S. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med 2001; 163(5)1256–1276, [PUBMED], [INFOTRIEVE], [CSA]
- Murray C JL, Lopez A D. Harvard School of Public Health, World Health Organization, and World Bank. The Global Burden of Disease: A Comprehensive Assessment of Mortality and Disability from Diseases, Injuries, and Risk Factors in 1990 and Projected to 2020. Harvard University Press, Cambridge, MA 1996, 43 p
- Silverman E K, Palmer L J, Mosley J D, Barth M, Senter J M, Brown A, Drazen J M, Kwiatkowski D J, Chapman H A, Campbell E J, Province M A, Rao D C, Reilly J J, Ginns L C, Speizer F E, Weiss S T. Genomewide linkage analysis of quantitative spirometric phenotypes in severe early‐onset chronic obstructive pulmonary disease. Am J Hum Genet 2002; 70(5)1229–1239, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Silverman E K. Genetic epidemiology of COPD. Chest 2002; 121(3 Suppl)1S–6S, [PUBMED], [INFOTRIEVE]
- Pryor W A. K. Stone. Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann N Y Acad Sci 1993; 686: 12–27, discussion 27–8.[PUBMED], [INFOTRIEVE]
- Ottonello L, Dapino P, Pastorino G, Dallegri F, Sacchetti C. Neutrophil dysfunction and increased susceptibility to infection. Eur J Clin Invest 1995; 25(9)687–692, [PUBMED], [INFOTRIEVE], [CSA]
- Palmer R M, Ferrige A G, Moncada S. Nitric oxide release accounts for the biological activity of endothelium‐derived relaxing factor. Nature 1987; 327(6122)524–526, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Dupuy P M, Shore S A, Drazen J M, Frostell C, Hill W A, Zapol W M. Bronchodilator action of inhaled nitric oxide in guinea pigs. J Clin Invest 1992; 90(2)421–428, [PUBMED], [INFOTRIEVE], [CSA]
- Halliwell B, Gutteridge J MC. Free Radicals in Biology and Medicine3rd ed. Oxford Science Publications, Oxford University Press, OxfordEngland 1999; 936, xxxi, [19] of col. plates
- Langen R C, Korn S H, Wouters E F. ROS in the local and systemic pathogenesis of COPD. Free Radic Biol Med 2003; 35(3)226–235, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Sunyer J. Urban air pollution and chronic obstructive pulmonary disease: a review. Eur Respir J 2001; 17(5)1024–1033, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Kelly F J. Oxidative stress: its role in air pollution and adverse health effects. Occup Environ Med 2003; 60(8)612–616, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Karakatsani A, Andreadaki S, Katsouyanni K, Dimitroulis I, Trichopoulos D, Benetou V, Trichopoulou A. Air pollution in relation to manifestations of chronic pulmonary disease: a nested case‐control study in Athens, Greece. Eur J Epidemiol 2003; 18(1)45–53, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Desqueyroux H, Pujet J C, Prosper M, Le Moullec Y, Momas I. Effects of air pollution on adults with chronic obstructive pulmonary disease. Arch Environ Health 2002; 57(6)554–560, [PUBMED], [INFOTRIEVE], [CSA]
- Quinlan G J, Lamb N J, Tilley R, Evans T W, Gutteridge J M. Plasma hypoxanthine levels in ARDS: implications for oxidative stress, morbidity, and mortality. Am J Respir Crit Care Med 1997; 155(2)479–484, [PUBMED], [INFOTRIEVE], [CSA]
- Goeptar A R, Scheerens H, Vermeulen N P. Oxygen and xenobiotic reductase activities of cytochrome P450. Crit Rev Toxicol 1995; 25(1)25–65, [PUBMED], [INFOTRIEVE], [CSA]
- Azumi H, Inoue N, Takeshita S, Rikitake Y, Kawashima S, Hayashi Y, Itoh H, Yokoyama M. Expression of NADH/NADPH oxidase p22phox in human coronary arteries. Circulation 1999; 100(14)1494–1498, [PUBMED], [INFOTRIEVE]
- Marumo T, Schini‐Kerth V B, Brandes R P, Busse R. Glucocorticoids inhibit superoxide anion production and p22 phox mRNA expression in human aortic smooth muscle cells. Hypertension 1998; 32(6)1083–1088, [PUBMED], [INFOTRIEVE], [CSA]
- Thannickal V J, Fanburg B L. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol, 279(6)L1005–L1028, [CSA]
- Merenyi G, Lind J, Goldstein S, Czapski G. Peroxynitrous acid homolyzes into *OH and *NO2 radicals. Chem Res Toxicol 1998; 11(7)712–713, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Guembe L, Villaro A C. Histochemical demonstration of neuronal nitric oxide synthase during development of mouse respiratory tract. Am J Respir Cell Mol Biol 1999; 20(2)342–351, [PUBMED], [INFOTRIEVE], [CSA]
- Kobzik L, Bredt D S, Lowenstein C J, Drazen J, Gaston B, Sugarbaker D, Stamler J S. Nitric oxide synthase in human and rat lung: immunocytochemical and histochemical localization. Am J Respir Cell Mol Biol 1993; 9(4)371–377, [PUBMED], [INFOTRIEVE], [CSA]
- Forstermann U, Pollock J S, Schmidt H H, Heller M, Murad F. Calmodulin‐dependent endothelium‐derived relaxing factor/nitric oxide synthase activity is present in the particulate and cytosolic fractions of bovine aortic endothelial cells. Proc Natl Acad Sci U S A 1991; 88(5)1788–1792, [PUBMED], [INFOTRIEVE]
- Radomski M W, Vallance P, Whitley G, Foxwell N, Moncada S. Platelet adhesion to human vascular endothelium is modulated by constitutive and cytokine induced nitric oxide. Cardiovasc Res 1993; 27(7)1380–1382, [PUBMED], [INFOTRIEVE], [CSA]
- McCord J M, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem 1969; 244(22)6049–6055, [PUBMED], [INFOTRIEVE]
- Weisiger R A, Fridovich I. Mitochondrial superoxide simutase. Site of synthesis and intramitochondrial localization. J Biol Chem 1973; 248(13)4793–4796, [PUBMED], [INFOTRIEVE]
- Marklund S L. Human copper‐containing superoxide dismutase of high molecular weight. Proc Natl Acad Sci U S A 1982; 79(24)7634–7638, [PUBMED], [INFOTRIEVE]
- Tiitto L, Kaarteenaho‐Wiik R, Sormunen R, Holmgren A, Paakko P, Soini Y, Kinnula V L. Expression of the thioredoxin system in interstitial lung disease. J Pathol 2003; 201(3)363–370, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Sadek C M, Jimenez A, Damdimopoulos A E, Kieselbach T, Nord M, Gustafsson J A, Spyrou G, Davis E C, Oko R, van der Hoorn F A, Miranda‐Vizuete A. Characterization of human thioredoxin‐like 2. A novel microtubule‐binding thioredoxin expressed predominantly in the cilia of lung airway epithelium and spermatid manchette and axoneme. J Biol Chem 2003; 278(15)13133–13142, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Arner E S, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem 2000; 267(20)6102–6109, [PUBMED], [INFOTRIEVE], [CROSSREF]
- van der Vliet A, O'Neill C A, Cross C E, Koostra J M, Volz W G, Halliwell B, Louie S. Determination of low‐molecular‐mass antioxidant concentrations in human respiratory tract lining fluids. Am J Physiol 1999; 276(2 Pt 1)L289–L296, [PUBMED], [INFOTRIEVE]
- Reynolds H Y, Newball H H. Analysis of proteins and respiratory cells obtained from human lungs by bronchial lavage. J Lab Clin Med 1974; 84(4)559–573, [PUBMED], [INFOTRIEVE]
- Forman H J, Fridovich I. Superoxide dismutase: a comparison of rate constants. Arch Biochem Biophys 1973; 158(1)396–400, [PUBMED], [INFOTRIEVE]
- Crapo J, Oury T, Rabouille C, Slot J, Chang L. Copper, zinc superoxide dismutase is primarily a cytosolic protein in human cells. PNAS 1992; 89: 10405–10409, [PUBMED], [INFOTRIEVE]
- Rosen D R, Siddique T, Patterson D, Figlewicz D A, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan J P, Deng H X, Rahmani Z, Krizus A, McKenna‐Yasek D, Cayabyab A, Gaston S M, Berger R, Tanzi R, Halperin J, Herzfeldt B, Van den Bergh, Mulder D W, Smyth C, Laing N G, Soriano E, Pericak‐Vance M A, Halnes J, Rouleau G, Crusella J S, Horvitz H R, Brown R H. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993; 362(6415)59–62, [CROSSREF]
- Weisiger R A, Fridovich I. Superoxide dismutase. Organelle specificity. J Biol Chem 1973; 248(10)3582–3592, [PUBMED], [INFOTRIEVE]
- Li Y, Huang T T, Carlson E J, Melov S, Ursell P C, Olson J L, Noble L J, Yoshimura M P, Berger C, Chan P H, Wallace D C, Epstein C J. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet 1995; 11: 376–381, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Lebovitz R M, Zhang H, Vogel H, Cartwright J., Jr., Dionne L, Lu N, Huang S, Matzuk M M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase‐deficient mice. Proc Natl Acad Sci U S A 1996; 93(18)9782–9787, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Melov S, Schneider J A, Day B J, Hinerfeld D, Coskun P, Mirra S S, Crapo J D, Wallace D C. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet 1998; 18(2)159–163, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Chang L Y, Kang B H, Slot J W, Vincent R, Crapo J D. Immunocytochemical localization of the sites of superoxide dismutase induction by hyperoxia in rat lungs. Lab Invest 1995; 73(1)29–39, [PUBMED], [INFOTRIEVE], [CSA]
- Coursin D B, Cihla H P, Oberley T D, Oberley L W. Immunolocalization of antioxidant enzymes and isozymes of glutathione S‐transferase in normal rat lung. Am J Physiol 1992; 263(6 Pt 1)L679–L691, [PUBMED], [INFOTRIEVE]
- Larsen G L, White C W, Takeda K, Loader J E, Nguyen D D, Joetham A, Groner Y, Gelfand E W. Mice that overexpress Cu/Zn superoxide dismutase are resistant to allergen‐induced changes in airway control. Am J Physiol Lung Cell Mol Physiol 2000; 279(2)L350–L359, [PUBMED], [INFOTRIEVE], [CSA]
- White C W, Avraham K B, Shanley P F, Groner Y. Transgenic mice with expression of elevated levels of copper‐zinc superoxide dismutase in the lungs are resistant to pulmonary oxygen toxicity. J Clin Invest 1991; 87(6)2162–2168, [PUBMED], [INFOTRIEVE], [CSA]
- Clyde B L, Chang L Y, Auten R L, Ho Y S, Crapo J D. Distribution of manganese superoxide dismutase mRNA in normal and hyperoxic rat lung. Am J Respir Cell Mol Biol 1993; 8(5)530–537, [PUBMED], [INFOTRIEVE]
- Gilks C B, Price K, Wright J L, Churg A. Antioxidant gene expression in rat lung after exposure to cigarette smoke. Am J Pathol 1998; 152(1)269–278, [PUBMED], [INFOTRIEVE]
- Jackson R M, Helton E S, Viera L, Ohman T. Survival, lung injury, and lung protein nitration in heterozygous MnSOD knockout mice in hyperoxia. Exp Lung Res 1999; 25(7)631–646, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Tsan M F, White J E, Caska B, Epstein C J, Lee C Y. Susceptibility of heterozygous MnSOD gene‐knockout mice to oxygen toxicity. Am J Respir Cell Mol Biol 1998; 19(1)114–120, [PUBMED], [INFOTRIEVE], [CSA]
- Williams M D, Van Remmen H, Conrad C C, Huang T T, Epstein C J, Richardson A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J Biol Chem 1998; 273(43)28510–28515, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Wispe J R, Warner B B, Clark J C, Dey C R, Neuman J, Glasser S W, Crapo J D, Chang L Y, Whitsett J A. Human Mn‐superoxide dismutase in pulmonary epithelial cells of transgenic mice confers protection from oxygen injury. J Biol Chem 1992; 267(33)23937–23941, [PUBMED], [INFOTRIEVE]
- Epperly M W, Travis E L, Whitsett J A, Raineri I, Epstein C J, Greenberger J S. Overexpression of manganese superoxide dismutase (MnSOD) in whole lung or alveolar type II cells of MnSOD transgenic mice does not provide intrinsic lung irradiation protection. Int J Cancer 2001; 96(1)11–21, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Marklund S L. Extracellular superoxide dismutase in human tissues and human cell lines. J Clin Invest 1984; 74(4)1398–1403, [PUBMED], [INFOTRIEVE]
- Su W Y, Folz R, Chen J S, Crapo J D, Chang L Y. Extracellular superoxide dismutase mRNA expressions in the human lung by in situ hybridization. Am J Respir Cell Mol Biol 1997; 16(2)162–170, [PUBMED], [INFOTRIEVE], [CSA]
- Oury T D, Day B J, Crapo J D. Extracellular superoxide dismutase in vessels and airways of humans and baboons. Free Radic Biol Med 1996; 20(7)957–965, [PUBMED], [INFOTRIEVE], [CSA]
- Oury T D, Chang L Y, Marklund S L, Day B J, Crapo J D. Immunocytochemical localization of extracellular superoxide dismutase in human lung. Lab Invest 1994; 70(6)889–898, [PUBMED], [INFOTRIEVE], [CSA]
- Jonsson L M, Rees D D, Edlund T, Marklund S L. Nitric oxide and blood pressure in mice lacking extracellular‐superoxide dismutase. Free Radic Res 2002; 36(7)755–758, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- van Beurden W J, Wielders P L, Scheepers P J, van Herwaarden C L, Dekhuijzen P N. Superoxide production by peripheral polymorphonuclear leukocytes in patients with COPD. Respir Med 2003; 97(4)401–406, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Smith K R, Uyeminami D L, Kodavanti U P, Crapo J D, Chang L Y, Pinkerton K E. Inhibition of tobacco smoke‐induced lung inflammation by a catalytic antioxidant. Free Radic Biol Med 2002; 33(8)1106–1114, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Carlsson L M, Jonsson J, Edlund T, Marklund S L. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc Natl Acad Sci U S A 1995; 92(14)6264–6268, [PUBMED], [INFOTRIEVE], [CSA]
- Folz R J, Abushamaa A M, Suliman H B. Extracellular superoxide dismutase in the airways of transgenic mice reduces inflammation and attenuates lung toxicity following hyperoxia. J Clin Invest 1999; 103(7)1055–1066, [PUBMED], [INFOTRIEVE], [CSA]
- Bowler R P, Arcaroli J, Crapo J D, Ross A, Slot J W, Abraham E. Extracellular superoxide dismutase attenuates lung injury after hemorrhage. Am J Respir Crit Care Med 2001; 164(2)290–294, [PUBMED], [INFOTRIEVE], [CSA]
- Suliman H B, Ryan L K, Bishop L, Folz R J. Prevention of influenza‐induced lung injury in mice overexpressing extracellular superoxide dismutase. Am J Physiol Lung Cell Mol Physiol 2001; 280(1)L69–L78, [PUBMED], [INFOTRIEVE], [CSA]
- Bowler R P, Nicks M, Warnick K, Crapo J D. Role of extracellular superoxide dismutase in bleomycin‐induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2002; 282(4)L719–L726, [PUBMED], [INFOTRIEVE], [CSA]
- Ghio A J, Suliman H B, Carter J D, Abushamaa A M, Folz R J. Overexpression of extracellular superoxide dismutase decreases lung injury after exposure to oil fly ash. Am J Physiol Lung Cell Mol Physiol 2002; 283(1)L211–L218, [PUBMED], [INFOTRIEVE], [CSA]
- Belvisi M G, Stretton C D, Yacoub M, Barnes P J. Nitric oxide is the endogenous neurotransmitter of bronchodilator nerves in humans. Eur J Pharmacol 1992; 210(2)221–222, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Di Maria G U, Spicuzza L, Mistretta A, Mazzarella G. Role of endogenous nitric oxide in asthma. Allergy 2000; 55(suppl 61)31–35, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Hogman M, Frostell C G, Hedenstrom H, Hedenstierna G. Inhalation of nitric oxide modulates adult human bronchial tone. Am Rev Respir Dis 1993; 148(6 Pt 1)1474–1478, [PUBMED], [INFOTRIEVE], [CSA]
- Gromer S, Urig S, Becker K. The thioredoxin system‐from science to clinic. Med Res Rev 2004; 24(1)40–89, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Nordberg J, Arner E S. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med 2001; 31(11)1287–1312, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Rancourt R C, Tai S, King M, Heltshe S L, Penvari C, Accurso F J, White C W. Thioredoxin liquefies and decreases the viscoelasticity of cystic fibrosis sputum. Am J Physiol Lung Cell Mol Physiol 2004; 286(5)L931–L938, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Sadek C M, Damdimopoulos A E, Pelto‐Huikko M, Gustafsson J A, Spyrou G, Mira‐Vizuete A. Sptrx‐2, a fusion protein composed of one thioredoxin and three tandemly repeated NDP‐kinase domains is expressed in human testis germ cells. Genes Cells 2001; 6(12)1077–1090, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Wada H, Hirata T, Decampos K N, Hitomi S, Slutsky A S. Effect of the combination of human thioredoxin and L‐cysteine on ischemia‐reperfusion injury in isolated rat lungs. Eur Surg Res 1995; 27(6)363–370, [PUBMED], [INFOTRIEVE], [CSA]
- Fukuse T, Hirata T, Yokomise H, Hasegawa S, Inui K, Mitsui A, Hirakawa T, Hitomi S, Yodoi J, Wada H. Attenuation of ischaemia reperfusion injury by human thioredoxin. Thorax 1995; 50(4)387–391, [PUBMED], [INFOTRIEVE]
- Okubo K, Kosaka S, Isowa N, Hirata T, Hitomi S, Yodoi J, Nakano M, Wada H. Amelioration of ischemia‐reperfusion injury by human thioredoxin in rabbit lung. J Thorac Cardiovasc Surg 1997; 113(1)1–9, [PUBMED], [INFOTRIEVE]
- Nakamura H, Tamura S, Watanabe I, Iwasaki T, Yodoi J. Enhanced resistancy of thioredoxin‐transgenic mice against influenza virus‐induced pneumonia. Immunol Lett 2002; 82(1–2)165–170, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Sahin U, Unlu M, Ozguner F, Sutcu R, Akkaya A, Delibas N. Lipid peroxidation and glutathione peroxidase activity in chronic obstructive pulmonary disease exacerbation: prognostic value of malondialdehyde. J Basic Clin Physiol Pharmacol 2001; 12(1)59–68, [PUBMED], [INFOTRIEVE], [CSA]
- Cantin A M, North S L, Hubbard R C, Crystal R G. Normal alveolar epithelial lining fluid contains high levels of glutathione. J Appl Physiol 1987; 63(1)152–157, [PUBMED], [INFOTRIEVE], [CSA]
- Linden M, Hakansson L, Ohlsson K, Sjodin K, Tegner H, Tunek A, Venge P. Glutathione in bronchoalveolar lavage fluid from smokers is related to humoral markers of inflammatory cell activity. Inflammation 1989; 13(6)651–658, [PUBMED], [INFOTRIEVE], [CSA]
- Li X Y, Donaldson K, Rahman I, MacNee W. An investigation of the role of glutathione in increased epithelial permeability induced by cigarette smoke in vivo and in vitro. Am J Respir Crit Care Med 1994; 149(6)1518–1525, [PUBMED], [INFOTRIEVE], [CSA]
- Rahman I, Li X Y, Donaldson K, Harrison D J, MacNee W. Glutathione homeostasis in alveolar epithelial cells in vitro and lung in vivo under oxidative stress. Am J Physiol 1995; 269(3 Pt 1)L285–L292, [PUBMED], [INFOTRIEVE]
- Pinamonti S, Leis M, Barbieri A, Leoni D, Muzzoli M, Sostero S, Chicca M C, Carrieri A, Ravenna F, Fabbri L M, Ciaccia A. Detection of xanthine oxidase activity products by EPR and HPLC in bronchoalveolar lavage fluid from patients with chronic obstructive pulmonary disease. Free Radic Biol Med 1998; 25(7)771–779, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Corradi M, Rubinstein I, Andreoli R, Manini P, Caglieri A, Poli D, Alinovi R, Mutti A. Aldehydes in exhaled breath condensate of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2003; 167(10)1380–1386, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Rahman I, Skwarska E, MacNee W. Attenuation of oxidant/antioxidant imbalance during treatment of exacerbations of chronic obstructive pulmonary disease. Thorax 1997; 52(6)565–568, [PUBMED], [INFOTRIEVE]
- Calikoglu M, Unlu A, Tamer L, Ercan B, Bugdayci R, Atik U. The levels of serum vitamin C, malonyldialdehyde and erythrocyte reduced glutathione in chronic obstructive pulmonary disease and in healthy smokers. Clin Chem Lab Med 2002; 40(10)1028–1031, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Pratico D, Basili S, Vieri M, Cordova C, Violi F, Fitzgerald G A. Chronic obstructive pulmonary disease is associated with an increase in urinary levels of isoprostane F2alpha‐III, an index of oxidant stress. Am J Respir Crit Care Med 1998; 158(6)1709–1714, [PUBMED], [INFOTRIEVE], [CSA]
- Paredi P, Kharitonov S A, Leak D, Ward S, Cramer D, Barnes P J. Exhaled ethane, a marker of lipid peroxidation, is elevated in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2000; 162(2 Pt 1)369–373, [PUBMED], [INFOTRIEVE], [CSA]
- Montuschi P, Collins J V, Ciabattoni G, Lazzeri N, Corradi M, Kharitonov S A, Barnes P J. Exhaled 8‐isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am J Respir Crit Care Med 2000; 162(3 Pt 1)1175–1177, [PUBMED], [INFOTRIEVE], [CSA]
- Kostikas K, Papatheodorou G, Ganas K, Psathakis K, Panagou P, Loukides S. pH in expired breath condensate of patients with inflammatory airway diseases. Am J Respir Crit Care Med 2002; 165(10)1364–1370, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Biernacki W A, Kharitonov S A, Barnes P J. Increased leukotriene B4 and 8‐isoprostane in exhaled breath condensate of patients with exacerbations of COPD. Thorax 2003; 58(4)294–298, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Rahman I, van Schadewijk A A, Crowther A J, Hiemstra P S, Stolk J, MacNee W, De Boer W I. 4‐Hydroxy‐2‐nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002; 166(4)490–495, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Kasielski M, Nowak D. Long‐term administration of N‐acetylcysteine decreases hydrogen peroxide exhalation in subjects with chronic obstructive pulmonary disease. Respir Med 2001; 95(6)448–456, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Corradi M, Pesci A, Casana R, Alinovi R, Goldoni M, Vettori M V, Cuomo A. Nitrate in exhaled breath condensate of patients with different airway diseases. Nitric Oxide 2003; 8(1)26–30, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Kanazawa H, Shiraishi S, Hirata K, Yoshikawa J. Imbalance between levels of nitrogen oxides and peroxynitrite inhibitory activity in chronic obstructive pulmonary disease. Thorax 2003; 58(2)106–109, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Ichinose M, Sugiura H, Yamagata S, Koarai A, Shirato K. Increase in reactive nitrogen species production in chronic obstructive pulmonary disease airways. Am J Respir Crit Care Med 2000; 162(2 Pt 1)701–706, [PUBMED], [INFOTRIEVE], [CSA]
- Sugiura H, Ichinose M, Yamagata S, Koarai A, Shirato K, Hattori T. Correlation between change in pulmonary function and suppression of reactive nitrogen species production following steroid treatment in COPD. Thorax 2003; 58(4)299–305, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Maziak W, Loukides S, Culpitt S, Sullivan P, Kharitonov S A, Barnes P J. Exhaled nitric oxide in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 157(3 Pt 1)998–1002, [PUBMED], [INFOTRIEVE], [CSA]
- Kharitonov S A, Barnes P J. Biomarkers of some pulmonary diseases in exhaled breath. Biomarkers 2002; 7(1)1–32, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Dekhuijzen P N, Aben K K, Dekker I, Aarts L P, Wielders P L, van Herwaarden C L, Bast A. Increased exhalation of hydrogen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1996; 154(3 Pt 1)813–816, [PUBMED], [INFOTRIEVE], [CSA]
- vanBeurden W J, Dekhuijzen P N, Harff G A, Smeenk F W. Variability of exhaled hydrogen peroxide in stable COPD patients and matched healthy controls. Respiration 2002; 69(3)211–216, [CROSSREF], [CSA]
- Ansarin K, Chatkin J M, Ferreira I M, Gutierrez C A, Zamel N, Chapman K R. Exhaled nitric oxide in chronic obstructive pulmonary disease: relationship to pulmonary function. Eur Respir J 2001; 17(5)934–938, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Fabbri L M, Romagnoli M, Corbetta L, Casoni G, Busljetic K, Turato G, Ligabue G, Ciaccia A, Saetta M, Papi A. Differences in airway inflammation in patients with fixed airflow obstruction due to asthma or chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2003; 167(3)418–424, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Morrison D, Rahman I, Lannan S, MacNee W. Epithelial permeability, inflammation, and oxidant stress in the air spaces of smokers. Am J Respir Crit Care Med 1999; 159(2)473–479, [PUBMED], [INFOTRIEVE], [CSA]
- Imhof A, Wolffe A P. Transcription: gene control by targeted histone acetylation. Curr Biol 1998; 8(12)R422–R424, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Rahman I. Oxidative stress, chromatin remodeling and gene transcription in inflammation and chronic lung diseases. J Biochem Mol Biol 2003; 36(1)95–109, [PUBMED], [INFOTRIEVE]
- Tomita K, Barnes P J, Adcock I M. The effect of oxidative stress on histone acetylation and IL‐8 release. Biochem Biophys Res Commun 2003; 301(2)572–577, [PUBMED], [INFOTRIEVE], [CSA]
- Ito K, Lim S, Caramori G, Chung K F, Barnes P J, Adcock I M. Cigarette smoking reduces histone deacetylase 2 expression, enhances cytokine expression, and inhibits glucocorticoid actions in alveolar macrophages. Faseb J 2001; 15(6)1110–1112, [PUBMED], [INFOTRIEVE], [CSA]
- Ito K, Hanazawa T, Tomita K, Barnes P J, Adcock I M. Oxidative stress reduces histone deacetylase 2 activity and enhances IL‐8 gene expression: role of tyrosine nitration. Biochem Biophys Res Commun 2004; 315(1)240–245, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Barnes P J, Ito K, Adcock I M. Corticosteroid resistance in chronic obstructive pulmonary disease: inactivation of histone deacetylase. Lancet 2004; 363(9410)731–733, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Nimnual A S, Taylor L J, Bar‐Sagi D. Redox‐dependent downregulation of Rho by Rac. Nat Cell Biol 2003; 5(3)236–241, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Wyatt T A, Heires A J, Sanderson S D, Floreani A A. Protein kinase C activation is required for cigarette smoke‐enhanced C5a‐mediated release of interleukin‐8 in human bronchial epithelial cells. Am J Respir Cell Mol Biol 1999; 21(2)283–288, [PUBMED], [INFOTRIEVE], [CSA]
- Gebel S, Muller T. The activity of NF‐kappaB in Swiss 3T3 cells exposed to aqueous extracts of cigarette smoke is dependent on thioredoxin. Toxicol Sci 2001; 59(1)75–81, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Muller T. Expression of c‐fos in quiescent Swiss 3T3 cells exposed to aqueous cigarette smoke fractions. Cancer Res 1995; 55(9)1927–1932, [PUBMED], [INFOTRIEVE]
- Di Stefano A, Caramori G, Oates T, Capelli A, Lusuardi M, Gnemmi I, Ioli F, Chung K F, Donner C F, Barnes P J, Adcock I M. Increased expression of nuclear factor‐kappaB in bronchial biopsies from smokers and patients with COPD. Eur Respir J 2002; 20(3)556–563, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Caramori G, Romagnoli M, Casolari P, Bellettato C, Casoni G, Boschetto P, Fan Chung K, Barnes P J, Adcock I M, Ciaccia A, Fabbri L M, Papi A. Nuclear localisation of p65 in sputum macrophages but not in sputum neutrophils during COPD exacerbations. Thorax 2003; 58(4)348–351, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Gebel S, Gerstmayer B, Bosio A, Haussmann H J, Van Miert E, Muller T. Gene expression profiling in respiratory tissues from rats exposed to mainstream cigarette smoke. Carcinogenesis 2004; 25(2)169–178, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Hogg J C. Role of latent viral infections in chronic obstructive pulmonary disease and asthma. Am J Respir Crit Care Med 2001; 164(10 Pt 02)S71–S75, [PUBMED], [INFOTRIEVE], [CSA]
- Ogawa E, Elliott W M, Hughes F, Eichholtz T J, Hogg J C, Hayashi S. Latent adenoviral infection induces production of growth factors relevant to airway remodeling in COPD. Am J Physiol Lung Cell Mol Physiol 2004; 286(1)L189–L197, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Orino K, Tsuji Y, Torti F M, Torti S V. Adenovirus E1A blocks oxidant‐dependent ferritin induction and sensitizes cells to pro‐oxidant cytotoxicity. FEBS Lett 1999; 461(3)334–338, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Gilmour P S, Rahman I, Hayashi S, Hogg J C, Donaldson K, MacNee W. Adenoviral E1A primes alveolar epithelial cells to PM(10)‐induced transcription of interleukin‐8. Am J Physiol Lung Cell Mol Physiol 2001; 281(3)L598–L606, [PUBMED], [INFOTRIEVE], [CSA]
- Stockley R A. Neutrophils and the pathogenesis of COPD. Chest. 2002; 121(5 suppl)151S–155S, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Hunninghake G W, Crystal R G. Cigarette smoking and lung destruction. Accumulation of neutrophils in the lungs of cigarette smokers. Am Rev Respir Dis 1983; 128(5)833–838, [PUBMED], [INFOTRIEVE], [CSA]
- Di Stefano A, Capelli A, Lusuardi M, Balbo P, Vecchio C, Maestrelli P, Mapp C E, Fabbri L M, Donner C F, Saetta M. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med 1998; 158(4)1277–1285, [PUBMED], [INFOTRIEVE], [CSA]
- Drost E M, Selby C, Bridgeman M M, MacNee W. Decreased leukocyte deformability after acute cigarette smoking in humans. Am Rev Respir Dis 1993; 148(5)1277–1283, [PUBMED], [INFOTRIEVE], [CSA]
- MacNee W, Wiggs B, Belzberg A S, Hogg J C. The effect of cigarette smoking on neutrophil kinetics in human lungs. N Engl J Med. 1989; 321(14)924–928, [PUBMED], [INFOTRIEVE], [CSA]
- Lehr H A, Kress E, Menger M D, Friedl H P, Hubner C, Arfors K E, Messmer K. Cigarette smoke elicits leukocyte adhesion to endothelium in hamsters: inhibition by CuZn‐SOD. Free Radic Biol Med 1993; 14(6)573–581, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Chung K F. Cytokines in chronic obstructive pulmonary disease. Eur Respir J Suppl 2001; 34: 50s–59s, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Takizawa H, Tanaka M, Takami K, Ohtoshi T, Ito K, Satoh M, Okada Y, Yamasawa F, Nakahara K, Umeda A. Increased expression of transforming growth factor‐beta1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD). Am J Respir Crit Care Med 2001; 163(6)1476–1483, [PUBMED], [INFOTRIEVE], [CSA]
- Janssen‐Heininger Y M, Poynter M E, Baeuerle P A. Recent advances towards understanding redox mechanisms in the activation of nuclear factor kappaB. Free Radic Biol Med 2000; 28(9)1317–1327, [CROSSREF], [CSA]
- Floreani A A, Wyatt T A, Stoner J, Sanderson S D, Thompson E G, Allen‐Gipson D, Heires A J. Smoke and C5a induce airway epithelial intercellular adhesion molecule‐1 and cell adhesion. Am J Respir Cell Mol Biol 2003; 29(4)472–482, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Folch E, Salas A, Panes J, Gelpi E, Rosello‐Catafau J, Anderson D C, Navarro S, Pique J M, Fernandez‐Cruz L, Closa D. Role of P‐selectin and ICAM‐1 in pancreatitis‐induced lung inflammation in rats: significance of oxidative stress. Ann Surg 1999; 230(6)792–798, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Akgur F M, Brown M F, Zibari G B, McDonald J C, Epstein C J, Ross C R, Granger D N. Role of superoxide in hemorrhagic shock‐induced P‐selectin expression. Am J Physiol Heart Circ Physiol 2000; 279(2)H791–H797, [PUBMED], [INFOTRIEVE], [CSA]
- Russell J, Epstein C J, Grisham M B, Alexander J S, Yeh K Y, Granger D N. Regulation of E‐selectin expression in postischemic intestinal microvasculature. Am J Physiol Gastrointest Liver Physiol 2000; 278(6)G878–G885, [PUBMED], [INFOTRIEVE], [CSA]
- Sohn H Y, Krotz F, Zahler S, Gloe T, Keller M, Theisen K, Schiele T M, Klauss V, Pohl U. Crucial role of local peroxynitrite formation in neutrophil‐induced endothelial cell activation. Cardiovasc Res 2003; 57(3)804–815, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Cella G, Sbarai A, Mazzaro G, Vanzo B, Romano S, Hoppensteadt T, Fareed J. Plasma markers of endothelial dysfunction in chronic obstructive pulmonary disease. Clin Appl Thromb Hemost 2001; 7(3)205–208, [PUBMED], [INFOTRIEVE], [CSA]
- Janoff A, Carp H, Laurent P, Raju L. The role of oxidative processes in emphysema. Am Rev Respir Dis 1983; 127(2)S31–S38, [PUBMED], [INFOTRIEVE]
- Panda K, Chattopadhyay R, Chattopadhyay D, Chatterjee I B. Cigarette smoke‐induced protein oxidation and proteolysis is exclusively caused by its tar phase: prevention by vitamin C. Toxicol Lett 2001; 123(1)21–32, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Fischer B, Voynow J. Neutrophil elastase induces MUC5AC messenger RNA expression by an oxidant‐dependent mechanism. Chest 2000; 117(5 suppl 1)317S–320S, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Aoshiba K, Yasuda K, Yasui S, Tamaoki J, Nagai A. Serine proteases increase oxidative stress in lung cells. Am J Physiol Lung Cell Mol Physiol 2001; 281(3)L556–L564, [PUBMED], [INFOTRIEVE], [CSA]
- Wright D T, Fischer B M, Li C, Rochelle L G, Akley N J, Adler K B. Oxidant stress stimulates mucin secretion and PLC in airway epithelium via a nitric oxide‐dependent mechanism. Am J Physiol 1996; 271(5 Pt 1)L854–L861, [PUBMED], [INFOTRIEVE]
- Adler K B, Holden‐Stauffer W J, Repine J E. Oxygen metabolites stimulate release of high‐molecular‐weight glycoconjugates by cell and organ cultures of rodent respiratory epithelium via an arachidonic acid‐dependent mechanism. J Clin Invest 1990; 85(1)75–85, [PUBMED], [INFOTRIEVE], [CSA]
- Feldman C, Anderson R, Kanthakumar K, Vargas A, Cole P J, Wilson R. Oxidant‐mediated ciliary dysfunction in human respiratory epithelium. Free Radic Biol Med 1994; 17(1)1–10, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Decramer M, Dekhuijzen P N, Troosters T, van Herwaarden C, Rutten‐van Molken M, van Schayck C P, Olivieri D, Lankhorst I, Ardia A. The Bronchitis Randomized On NAC Cost‐Utility Study (BRONCUS): hypothesis and design. BRONCUS‐trial Committee. Eur Respir J 2001; 17(3)329–336, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Kasahara Y, Tuder R M, Cool C D, Lynch D A, Flores S C, Voelkel N F. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med 2001; 163(3 Pt 1)737–744, [PUBMED], [INFOTRIEVE], [CSA]
- Segura‐Valdez L, Pardo A, Gaxiola M, Uhal B D, Becerril C, Selman M. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest 2000; 117(3)684–694, [CROSSREF]
- Yokohori N, Aoshiba K, Nagai A. Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Chest 2004; 125(2)626–632, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Majo J, Ghezzo H, Cosio M G. Lymphocyte population and apoptosis in the lungs of smokers and their relation to emphysema. Eur Respir J 2001; 17(5)946–953, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Hodge S J, Hodge G L, Reynolds P N, Scicchitano R, Holmes M. Increased production of TGF‐beta and apoptosis of T lymphocytes isolated from peripheral blood in COPD. Am J Physiol Lung Cell Mol Physiol 2003; 285(2)L492–L499, [PUBMED], [INFOTRIEVE], [CSA]
- Wagner P D. Vascular endothelial growth factor and the pathogenesis of emphysema. Am J Med 2003; 114(5)413–414, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Kanazawa H, Asai K, Hirata K, Yoshikawa J. Possible effects of vascular endothelial growth factor in the pathogenesis of chronic obstructive pulmonary disease. Am J Med 2003; 114(5)354–358, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Kasahara Y, Tuder R M, Taraseviciene‐Stewart L, Le Cras T D, Abman S, Hirth P K, Waltenberger J, Voelkel N F. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest 2000; 106(11)1311–1319, [PUBMED], [INFOTRIEVE], [CSA]
- Tuder R M, Zhen L, Cho C Y, Taraseviciene‐Stewart L, Kasahara Y, Salvemini D, Voelkel N F, Flores S C. Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockade. Am J Respir Cell Mol Biol 2003; 29(1)88–97, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Silkoff P E, Martin D, Pak J, Westcott J Y, Martin R J. Exhaled nitric oxide correlated with induced sputum findings in COPD. Chest 2001; 119(4)1049–1055, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Montuschi P, Kharitonov S A, Barnes P J. Exhaled carbon monoxide and nitric oxide in COPD. Chest 2001; 120(2)496–501, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Agusti A G, Villaverde J M, Togores B, Bosch M. Serial measurements of exhaled nitric oxide during exacerbations of chronic obstructive pulmonary disease. Eur Respir J 1999; 14(3)523–528, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Rutgers S R, van der Mark T W, Coers W, Moshage H, Timens W, Kauffman H F, Koeter G H, Postma D S. Markers of nitric oxide metabolism in sputum and exhaled air are not increased in chronic obstructive pulmonary disease. Thorax 1999; 54(7)576–580, [PUBMED], [INFOTRIEVE]
- Yoshida M, Taguchi O, Gabazza E C, Kobayashi T, Yamakami T, Kobayashi H, Maruyama K, Shima T. Combined inhalation of nitric oxide and oxygen in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1997; 155(2)526–529, [PUBMED], [INFOTRIEVE], [CSA]
- Roger N, Barbera J A, Farre R, Cobos A, Roca J, Rodriguez‐Roisin R. Effect of nitric oxide inhalation on respiratory system resistance in chronic obstructive pulmonary disease. Eur Respir J 1996; 9(2)190–195, [PUBMED], [INFOTRIEVE]
- Baigorri F, Joseph D, Artigas A, Blanch L. Inhaled nitric oxide does not improve cardiac or pulmonary function in patients with an exacerbation of chronic obstructive pulmonary disease. Crit Care Med 1999; 27(10)2153–2158, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF], [CSA]
- Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, Kirk M, Freeman B A. Nitric oxide regulation of superoxide and peroxynitrite‐dependent lipid peroxidation. Formation of novel nitrogen‐containing oxidized lipid derivatives. J Biol Chem 1994; 269(42)26066–26075, [PUBMED], [INFOTRIEVE]
- Sadeghi‐Hashjin G, Folkerts G, Henricks P A, Verheyen A K, van der Linde H J, van Ark I, Coene A, Nijkamp F P. Peroxynitrite induces airway hyperresponsiveness in guinea pigs in vitro and in vivo. Am J Respir Crit Care Med 1996; 153(5)1697–1701, [CSA]
- Mador M J, Bozkanat E. Skeletal muscle dysfunction in chronic obstructive pulmonary disease. Respir Res 2001; 2(4)216–224, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF], [CSA]
- Polkey M I, Kyroussis D, Hamnegard C H, Mills G H, Green M, Moxham J. Diaphragm strength in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1996; 154(5)1310–1317, [PUBMED], [INFOTRIEVE], [CSA]
- Couillard A, Maltais F, Saey D, Debigare R, Michaud A, Koechlin C, LeBlanc P, Prefaut C. Exercise‐induced quadriceps oxidative stress and peripheral muscle dysfunction in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2003; 167(12)1664–1669, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF], [CSA]
- Couillard A, Koechlin C, Cristol J P, Varray A, Prefaut C. Evidence of local exercise‐induced systemic oxidative stress in chronic obstructive pulmonary disease patients. Eur Respir J 2002; 20(5)1123–1129, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF]
- Agusti A G, Sauleda J, Miralles C, Gomez C, Togores B, Sala E, Batle S, Busquets X. Skeletal muscle apoptosis and weight loss in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002; 166(4)485–489, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF], [CSA]
- Franco A A, Odom R S, Rando T A. Regulation of antioxidant enzyme gene expression in response to oxidative stress and during differentiation of mouse skeletal muscle. Free Radic Biol Med 1999; 27(9–10)1122–1132, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF], [CSA]
- Rabinovich R A, Ardite E, Troosters T, Carbo N, Alonso J, Gonzalez de Suso J M, Vilaro J, Barbera J A, Polo M F, Argiles J M, Fernandez‐Checa J C, Roca J. Reduced muscle redox capacity after endurance training in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 164(7)1114–1118, [PUBMED], [INFOTRIEVE], [CSA]
- Culpitt S V, Rogers D F, Shah P, De Matos C, Russell R E, Donnelly L E, Barnes P J. Impaired inhibition by dexamethasone of cytokine release by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2003; 167(1)24–31, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF], [CSA]
- Barnes P J, Adcock I M. How do corticosteroids work in asthma?. Ann Intern Med 2003; 139(5 Pt 1)359–370, [PUBMED], [INFOTRIEVE]
- Ito K, Barnes P J, Adcock I M. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin‐1beta‐induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol 2000; 20(18)6891–6903, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF]
- Romieu I, Trenga C. Diet and obstructive lung diseases. Epidemiol Rev 2001; 23(2)268–287, [PUBMED], [INFOTRIEVE], [CSA]
- Watson L, Margetts B, Howarth P, Dorward M, Thompson R, Little P. The association between diet and chronic obstructive pulmonary disease in subjects selected from general practice. Eur Respir J 2002; 20(2)313–318, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF]
- Denny S I, Thompson R L, Margetts B M. Dietary factors in the pathogenesis of asthma and chronic obstructive pulmonary disease. Curr Allergy Asthma Rep 2003; 3(2)130–136, [PUBMED], [INFOTRIEVE], [CSA]
- Tabak C, Feskens E J, Heederik D, Kromhout D, Menotti A, Blackburn H W. Fruit and fish consumption: a possible explanation for population differences in COPD mortality (The Seven Countries Study). Eur J Clin Nutr 1998; 52(11)819–825, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF], [CSA]
- Walda I C, Tabak C, Smit H A, Rasanen L, Fidanza F, Menotti A, Nissinen A, Feskens E J, Kromhout D. Diet and 20‐year chronic obstructive pulmonary disease mortality in middle‐aged men from three European countries. Eur J Clin Nutr 2002; 56(7)638–643, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF], [CSA]
- Forli L, Pedersen J I, Bjortuft O, Blomhoff R, Kofstad J, Boe J. Vitamins A and E in serum in relation to weight and lung function in patients with advanced pulmonary disease. Int J Vitam Nutr Res 2002; 72(6)360–368, [PUBMED], [INFOTRIEVE]
- Sargeant L A, Jaeckel A, Wareham N J. Interaction of vitamin C with the relation between smoking and obstructive airways disease in EPIC Norfolk. European Prospective Investigation into Cancer and Nutrition. Eur Respir J 2000; 16(3)397–403, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF]
- Grievink L, Smit H A, Ocke M C, van 't Veer P, Kromhout D. Dietary intake of antioxidant (pro)‐vitamins, respiratory symptoms and pulmonary function: the MORGEN study. Thorax 1998; 53(3)166–171, [PUBMED], [INFOTRIEVE]
- Tager M, Piecyk A, Kohnlein T, Thiel U, Ansorge S, Welte T. Evidence of a defective thiol status of alveolar macrophages from COPD patients and smokers. Chronic obstructive pulmonary disease. Free Radic Biol Med 2000; 29(11)1160–1165, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF], [CSA]
- Bridgeman M M, Marsden M, Selby C, Morrison D, MacNee W. Effect of N‐acetyl cysteine on the concentrations of thiols in plasma, bronchoalveolar lavage fluid, and lung tissue. Thorax 1994; 49(7)670–675, [PUBMED], [INFOTRIEVE]
- Linden M, Rasmussen J B, Piitulainen E, Tunek A, Larson M, Tegner H, Venge P, Laitinen L A, Brattsand R. Airway inflammation in smokers with nonobstructive and obstructive chronic bronchitis. Am Rev Respir Dis 1993; 148(5)1226–1232, [PUBMED], [INFOTRIEVE], [CSA]
- Rahman I, Morrison D, Donaldson K, MacNee W. Systemic oxidative stress in asthma, COPD, and smokers. Am J Respir Crit Care Med 1996; 154(4 Pt 1)1055–1060, [PUBMED], [INFOTRIEVE], [CSA]
- Rahman I, Swarska E, Henry M, Stolk J, MacNee W. Is there any relationship between plasma antioxidant capacity and lung function in smokers and in patients with chronic obstructive pulmonary disease?. Thorax 2000; 55(3)189–193, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF]
- Smith C A, Harrison D J. Association between polymorphism in gene for microsomal epoxide hydrolase and susceptibility to emphysema. Lancet 1997; 350(9078)630–633, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF]
- Harrison D J, Cantlay A M, Rae F, Lamb D, Smith C A. Frequency of glutathione S‐transferase M1 deletion in smokers with emphysema and lung cancer. Hum Exp Toxicol 1997; 16(7)356–360, [PUBMED], [INFOTRIEVE], [CSA]
- Yamada N, Yamaya M, Okinaga S, Nakayama K, Sekizawa K, Shibahara S, Sasaki H. Microsatellite polymorphism in the heme oxygenase‐1 gene promoter is associated with susceptibility to emphysema. Am J Hum Genet 2000; 66(1)187–195, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF], [CSA]
- Kurys E, Kurys P, Kuzniar A, Kieszko R. Analysis of antioxidant enzyme activity and magnesium level in chronic obstructive pulmonary disease (COPD). Ann Univ Mariae Curie Sklodowska [Med] 2001; 56: 261–266, [CSA]
- Blackwell T S, Blackwell T R, Holden E P, Christman B W, Christman J W. In vivo antioxidant treatment suppresses nuclear factor‐kappa B activation and neutrophilic lung inflammation. J Immunol 1996; 157(4)1630–1637, [PUBMED], [INFOTRIEVE]
- Davreux C J, Soric I, Nathens A B, Watson R W, McGilvray I D, Suntres Z E, Shek P N, Rotstein O D. N‐acetyl cysteine attenuates acute lung injury in the rat. Shock 1997; 8(6)432–438, [PUBMED], [INFOTRIEVE]
- Muller B, Kleschyov A L, Malblanc S, Stoclet J C. Nitric oxide‐related cyclic GMP‐independent relaxing effect of N‐acetylcysteine in lipopolysaccharide‐treated rat aorta. Br J Pharmacol 1998; 123(6)1221–1229, [PUBMED], [INFOTRIEVE], [CSA]
- Muller T, Gebel S. The cellular stress response induced by aqueous extracts of cigarette smoke is critically dependent on the intracellular glutathione concentration. Carcinogenesis 1998; 19(5)797–801, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF]
- Ortolani O, Conti A, De Gaudio A R, Masoni M, Novelli G. Protective effects of N‐acetylcysteine and rutin on the lipid peroxidation of the lung epithelium during the adult respiratory distress syndrome. Shock 2000; 13(1)14–18, [PUBMED], [INFOTRIEVE]
- Weinbroum A A, Kluger Y, Ben Abraham R, Shapira I, Karchevski E, Rudick V. Lung preconditioning with N‐acetyl‐L‐cysteine prevents reperfusion injury after liver no flow‐reflow: a dose‐response study. Transplantation 2001; 71(2)300–306, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF], [CSA]
- Blesa S, Cortijo J, Martinez‐Losa M, Mata M, Seda E, Santangelo F, Morcillo E J. Effectiveness of oral N‐acetylcysteine in a rat experimental model of asthma. Pharmacol Res 2002; 45(2)135–140, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF]
- Victor V M, De la Fuente M. N‐acetylcysteine improves in vitro the function of macrophages from mice with endotoxin‐induced oxidative stress. Free Radic Res 2002; 36(1)33–45, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF], [CSA]
- Victor V M, Rocha M, De la Fuente M. Regulation of macrophage function by the antioxidant N‐acetylcysteine in mouse‐oxidative stress by endotoxin. Int Immunopharmacol 2003; 3(1)97–106, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF]
- Floyd R A, Hensley K, Forster M J, Kelleher‐Anderson J A, Wood P L. Nitrones as neuroprotectants and antiaging drugs. Ann N Y Acad Sci 2002; 959: 321–329, [PUBMED], [INFOTRIEVE], [CSA]
- Symonds G, Renzetti A.D., Jr., Mitchell M M. The diffusing capacity in pulmonary emphysema. Am Rev Respir Dis 1974; 109(3)391–394, [PUBMED], [INFOTRIEVE]
- Paredi P, Kharitonov S A, Leak D, Shah P L, Cramer D, Hodson M E, Barnes P J. Exhaled ethane is elevated in cystic fibrosis and correlates with carbon monoxide levels and airway obstruction. Am J Respir Crit Care Med 2000; 161(4 Pt 1)1247–1251, [PUBMED], [INFOTRIEVE], [CSA]
- Boman G, Backer U, Larsson S, Melander B, Wahlander L. Oral acetylcysteine reduces exacerbation rate in chronic bronchitis: report of a trial organized by the Swedish Society for Pulmonary Diseases. Eur J Respir Dis 1983; 64(6)405–415, [PUBMED], [INFOTRIEVE]
- Oral N‐acetylcysteine and exacerbation rates in patients with chronic bronchitis and severe airways obstruction. British Thoracic Society Research Committee. Thorax 1985; 40(11)832–835
- Grassi C, Morandini G C. A controlled trial of intermittent oral acetylcysteine in the long‐term treatment of chronic bronchitis. Eur J Clin Pharmacol 1976; 09(5–6)393–396, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF]
- Hansen N C, Skriver A, Brorsen‐Riis L, Balslov S, Evald T, Maltbaek N, Gunnersen G, Garsdal P, Sander P, Pedersen J Z, Ibsen T B, Rasmussen F V. Orally administered N‐acetylcysteine may improve general well‐being in patients with mild chronic bronchitis. Respir Med 1994; 88(7)531–535, [PUBMED], [INFOTRIEVE], [CSA]
- Long‐term oral acetylcysteine in chronic bronchitis. A double‐blind controlled study. Eur J Respir Dis Suppl 1980; 111: 93–108
- Pela R, Calcagni A M, Subiaco S, Isidori P, Tubaldi A, Sanguinetti C M. N‐acetylcysteine reduces the exacerbation rate in patients with moderate to severe COPD. Respiration 1999; 66(6)495–500, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF], [CSA]
- Rasmussen J B, Glennow C. Reduction in days of illness after long‐term treatment with N‐acetylcysteine controlled‐release tablets in patients with chronic bronchitis. Eur Respir J 1988; 1(4)351–355, [PUBMED], [INFOTRIEVE]
- Crapo J D, Day B J. Modulation of nitric oxide responses in asthma by extracellular antioxidants. J Allergy Clin Immunol 1999; 104(4 Pt 1)743–746, [PUBMED], [INFOTRIEVE], [CSA]
- Bowler R P. Regulation of Extracellular Superoxide Dismutase, in Department of Cellular and Structural Biology. University of Colorado Health Sciences Center, Denver 2001; 111