Abstract
Factors that affect the absorption of cyclosporin A (CsA) were examined in gentamicin-induced acute renal failure (ARF) rats. In ARF rats, the area under the blood CsA concentration-time curve after oral administration was significantly decreased in comparison with that of control rats; 5.81 ± 0.55 vs 11.30 ± 1.59 mg h mL−1 (mean ± s.e.m.), respectively, and the relative bioavailabilities in ARF and control rats after oral administration were 15.2% and 43.4%, respectively. The flow rate of bile and the amount of bile acids in ARF rats were markedly decreased to about 61% of control, and 41% of control, respectively. The amount of CsA uptaken into the evened sac of jejunum, transferred to serosal side, and metabolized in tissues was significantly decreased in ARF rats without verapamil, while with 0.3 mM verapamil, the amount in ARF rats recovered to the levels of control rats. The absorption clearance of CsA in ARF rats was significantly decreased, however it was significantly improved by adding bile or bile acid. Adenosine triphosphate released from enterocytes in ARF rats was significantly decreased in the presence of 2.0 1−M CsA, 0.3 mM verapamil, or both, in comparison with control rats. From these findings, we concluded that a reduction of CsA bioavailability during ARF is caused by depression in bile excretion and renal function-dependent depression of uptake from intestinal tract via maybe P-gLycoprotein in enterocytes. They are main two factors that reduce the absorbed fraction of CsA in ARF rats.
INTRODUCTION
Cyclosporin A (CsA), a powerful immunosuppressant, has been shown to be effective in the prevention of rejection after solid organ transplantation and graft-versus-host disease (GVHD) after allogenic bone marrow transplantation Citation[[1]]. Management of therapy with CsA may be performed by measuring CsA blood concentration. However, several authors have reported a marked discrepancy between CsA measurements and the occurrence of adverse effects, rejection of solid organ,Citation[[2]] and risk of acute GVHD.Citation[3-4] The contradiction between blood levels and clinical events may be related to several disease states, which appear after solid organ transplantation or bone marrow transplantation.Citation[[5]] From the onset of CsA use in clinical practice, it has been believed that only renal function had no effects on the pharmacokinetics of CsA.Citation[[6]] However, several recent reports have suggested that renal function is closely related to the pharmacokinetics of CsA.Citation[7-8] In a series of our clinical reports, we found that renal dysfunction due to renal rejection, acute tubular necrosis, nephrotoxicity,Citation[[9]] and nephritic syndromeCitation[[10]] affected the pharmacokmetics of CsA. These types of renal dysfunction lead to a decrease in the area under the concentration versus time curve or through levels of CsACitation[[9]].
From the findings of a previous study using acute renal failure (ARF) rats,Citation[[11]] We confirmed that the relative bioavailability of CsA was 65% lower in ARF rats than in control rats. A reduction in absorbed fraction of CsA strongly contributed to the decrease in bioavailability after oral administration of CsA in ARF rats.Citation[[11]] We speculated that cholestatis, uremic toxin, intestinal edema because of ARF, and P-glycoprotein (P-gp) which is the MDR1 gene productCitation[[12]], might affect the intestinal absorption of CsA.
This study was conducted to characterize the factors that affect the absorbed fraction of CsA in ARF rats. We focused on the effects of ARF on function of bile excretion and expression of P-gp in the intestine.
MATERIALS AND METHODS
Chemicals
CsA was kindly donated by Sandoz (now Novartis Pharma). Cremophor EL for the preparation of cyclosporin injection, sodium carmellose for the preparation of oral solution, ursodeoxycholic acid, verapamil hydrochloride, and gentamicin sulfate were purchased from Nacalai Tesque. Other chemicals were of analytical grade and used without further purification. The assay of adenosine triphosphate (ATP) was performed by bioluminescence technique using an assay kit with luciferase. The assay kits used for bile acid, transaminase activity, creatinine, albumin, and blood urea nitrogen were purchased from commercially.
Preparation of test solutions
The test solution of CsA for oral administration was prepared by suspending CsA (5 mg mL−1) in 0.3% sodium carmellose. The standard formulation for injection was prepared in a 7:13 mixture of absolute ethanol and Cremophor EL at a concentration of 50 mg mL−1 CsA. The test solutions for intravenous administration were prepared by diluting this standard formulation with 0.9% saline to a final concentration of 1.25 mg mL−1 CsA. The test solutions used in-vitro or in-situ studies were also prepared by diluting this test solution with Krebs-Henseleit buffer to a final concentration of 2.5 μg mL−1 CsA.
Animal Preparation and Induction of Acute Renal Failure by Gentamicin
Male Sprauge-Dawley (SD) rats, weighing 200–280 g, were purchased from Japan SLC. They were housed for at least 2 days in a clean room; general food and water were freely available. Acute renal failure (ARF) was induced by subcutaneous administration of 150 mg kg−1day−1 gentamicin in 0.9% saline for five consecutive days into the hind back without anesthesia. Control rats received injections of 0.9% saline into the hind back. Studies on rats with ARF or control were performed 72h after the last administration of gentamicin. On the day before the dosing experiments, rats were moved to the laboratory, restrained on an operating table under a slight diethyl ether anesthesia during the operation, and test solutions were administered. After the administration, rats were unrestrained.
Oral Administration
Rats fasted overnight (control and ARF, 200∼280 g) and received an oral dose of CsA (5 mg kg−1) given over a period of approximately 10 sec by use of an oral feeding tube. Blood samples were drawn without restriction from a neck vein at time 0 (before administration) and 1, 2, 4, 6, 8, 10, 12, and 24 h after administration.
Intravenous Administration
Rats fasted overnight (control and ARF, 200∼280 g) and received an intravenous dose of CsA (1.25 mg kg−1) given over a period of approximately 60 sec through the femoral vein. Blood samples were drawn without restriction from a neck vein at time 0 (before administration) and 0.25, 0.5, 1, 2, 4, 6, 9, and 12 h after administration.
Effect of ARF on Bile Secretion
To rats fasted overnight (control and ARF, 200∼280 g) under anesthesia by intraperitoneal injection of sodium pentobarbital (32 mg kg−1), a polyethylene cannula (i.d. 0.5 mm, o.d. 0.8 mm; Natsume, Tokyo) was surgically introduced into the bile tract to obtain bile samples at various times. After 30-min of lag time from the cannulation, bile samples were collected up to 7 hours. The flow rate of bile and the concentration of bile acid in collected sample were measured, and data were expressed as midpoint values of each interval.
Everted Sac Technique for CsA Uptake Study
The everted sac technique was performed as described Barr and Reigelman,Citation[[13]] with some modification. In brief, rats fasted overnight (200∼280g) were killed under heavy pentobarbital sodium anesthesia, and the jejunum (15-cm length from the Traiz ligament) were removed and everted with a stainless steel probe. Before the experiment, the everted sacs, reduced to about 10-cm length, were filled with 1.5 mL Krebs-Henselext buffer (pH 7.4) and treated with 95% O2 −5% CO2 gas. These everted sacs were treated with CsA (2.5 μg mL−1=2.0 μM) in the presence or absence of 0.3 mM verapamil, and incubated at 37°C for 30 min in 50-mL glass test-tubes containing 10 mL Krebs-Henseleit buffer (pH 6.0); 95% O2 −5% CO2 gas was supplied during the experiments. After incubation, the serosal and mucosal fluids were sampled, and intestinal tissue was homogenized with a 10-fold volume of the buffer solution.
Everted Sac Technique for ATP Measurements
With the same technique as described above, the everted sacs reduced to about 3-cm in length were filled with 0.5 mL Krebs-Henseleit buffer (pH 7.4) and treated with 95% O2 −5% CO2 gas. These shortened everted sacs were treated with CsA (2.5 μg mL−1=2.0 M) in the presence or absence of 0.3 mM verapamil, and incubated at 37°C for 30 min in 50-mL glass test-tubes containing 10 mL Krebs-Henseleit buffer (pH 6.0); 95% O2 −5% CO2 gas was supplied during the experiments. After incubation, the everted sacs were treated with 100 μL of 0.5% trichloroacetic acit for 1 min and ATP released from intestinal tissue to the outside buffer was measured.
Single-pass Perfusion Technique
To rats fasted overnight (control and ARF, 200–280 g) under anesthesia by intraperitoneal injection of sodium pentobarbital (32 mg kg−1), the length of 15-cm from the proximal end of jejunum was surgically looped for the absorption experiment. After the contents in jejunal lumen were washed out 3 times with saline solution, the saline solution left in the lumen was expelled with air at 1mL min−1. Then the perfusion of drug solutions (2.5 μg mL CsA with or without rat bile or bile acid) in Krebus-Henseleit buffer (pH 7.4−9.O) was performed by single-pass infusion from the proximal to distal end at a rate of 1.1 mL min−1 using an infusion pump (Ismatic, Switzerland). After 10-min of time lag, the perfused solution was collected every 20 min for 60 min. To correct the volume change due to the absorption of moisture in the perfusates, inulin (0.32 mg mL−1) was added to all perfusates, and inulin concentration was measured by colorimetric determination. Then absorption clearance was given as a mean of 3 deter minations per rat.
Pharmacokinetic Analysis
Pharmacokinetic parameters for dosing study were obtained by moment analysis described by Yamaoka et al. using a computer program written in a BASIC language.Citation[[14]] The elimination rate constants (β, h−1) at terminal phase after oral and intravenous administration were determined by linear regression of at least three data points from the terminal portion of plots of blood concentration against time. Oral clearance (CL/F) and total body clearance (CLt) were determined by dividing the oral dose by AUCpo and the intravenous dose by AUCiv, where AUCpo and AUCiv are the areas under the plots of whole-blood concentration against time after oral and intravenous administration, respectively; they were calculated using the linear trapezoidal rule up to the last measured blood concentration, and extrapolated to infinity by β and the last value of concentration. The volume of distribution at steady-state (Vdss) and the apparent volume of distribution at steady-state (Vdss/F) for CsA were obtained from the equations:where MRTivand MRTpo are, respectively, the mean residence times after intra venous and oral administration.
The absorption clearance (CLa) of CsA from intestinal lumen was calculated using the following equations:where Qin and Qout represent perfusion rates in inlet or outlet, respectively, and Cin and Cout represent the concentrations of CsA in the perfusates in inlet or outlet, respectively.
CSA Assay
CsA concentrations in whole blood, serosal fluid, mucosal fluid, incubation fluid, and tissue homogenates, and gentamicin concentration in plasma were measured by fluorescence polarization immunoassay (FPIA) with a monoclonal antibody using the abbot TDx assay system.Citation[[15]] The calibration curves for respective samples were prepared by fitting the measuring standard samples with several known amounts of CsA. The coefficients of variation for repeated analysis of a known concentration (0.3 μg mL−1) in samples were below 5%.
Statistics
Results were expressed as mean ± standard error of the mean (s.e.m.). Test for differences of the mean values between two groups and among more than three groups were performed by the unpaired t-test and one-way ANOVA, respectively, and differences were considered significant at below 0.05.
RESULTS
The concentrations of plasma gentamicin in rats with gentamicin-induced ARF at 72 h after the last dosing of gentamicin exhibited detection limits in plasma by FPIA (n = 6). The mean values of creatinine and blood urea nitrogen in ARF rats were, respectively, 2.2-fold and 1.7-fold higher than in control rats (n = 5, P < 0.001). There were no significant differences in transaminase activity or albumin between control and ARF rats (n = 5).
The mean whole-blood concentration-time profiles obtained in this study after oral and intravenous administration of CsA to control and ARF rats are shown in . The whole-blood concentrations of CsA in ARF rats after intravenous administration were significantly higher than those in control rats (b), although oral administration to ARF rats resulted in whole-blood concentrations of CsA that were significantly lower than in control rats (a).
Figure 1. Whole blood concentrations of CsA after (a) intravenous administration of 1.25 mg kg−1 and (b) oral administraion of 5 mgkg−1:• control rats; ▴ ARF rats. Each symbol with bar represents the mean ± s.e.,. of 5 to 6 experiments. *p < 0.005. **p < 0.01 compared with control by unpaired t-test.
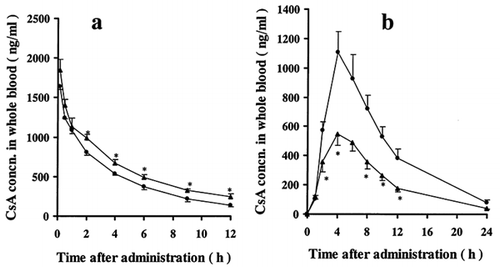
lists the pharmacokinetic parameters calculated by moment analysis from the data shown in . The mean AUCpo in control and ARF rats were 11.30 ± 1.59 mghL−1and 5.81 ± 0.55 mghL−1, respectively. The mean CL/F in control and ARF rats were 0.48 ± 0.07 Lh−1kg−1 and 0.90 ± 0.07 Lh−1kg−1, respectively, and the mean Vss/F were 4.5 ± 0.49 Lkg−1 and 8.4 ± 0.68 Lkg−1, respectively. There were significant increases in these pharmacokinetic parameters in ARF rats after oral administration. There was, however, no difference in the mean elimination rate constant after oral administration, βpo, between control and ARF rats. On the other hand, the mean AUCiv in control and ARF rats were 6.50 ± 0.51 mghL−1and 9.59 ± 0.76 mghL−1, respectively, and the mean CLt in control and ARF rats were 0.20 ± 0.01 L h−1kg−1 and 0.13 ± 0.01 L h−1kg−1, respectively. In addition, the mean elimination rate constants after intravenous administration, βiv, in control and ARF were 0.194 ± 0.013 h−1and 0.114 ± 0.01 h−1, respectively. The AUCiv was significantly increased, while the CLt and βiv were significantly decreased in ARF rats after intravenous administration. The relative bioavailabilities of CsA in control and ARF rats were 43.4% and 15.2% respectively.
Table 1. Pharmacokinetic parameters of CsA after intravenous and oral administration to control and ARF rats
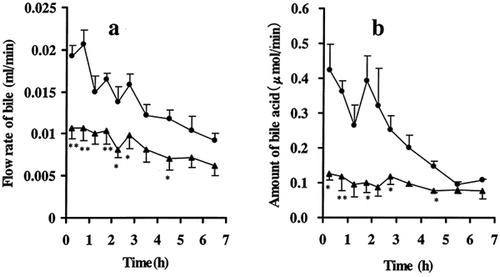
shows the effects of ARF on bile flow and amount of bile acid. Over the indicated study period, marked decreases in the bile flow and amount of bile acid were found. The areas under the curve for the bile flow (a) and the amount of bile acid (b) in ARF rats were reduced to about 61% and 41% of in control rats, respectively.
Figure 2. Effects of ARF on (a) the flow rate of bile and (b) the amount of bile acid • control rats; ▴ ARF rats. Each symbol with bar represents the mean ± s.e.m. of 5 to 6 experiments.*p < 0.05, **p < 0.01 compared with control by unpaired t-test.
The effects of ARF on intestinal uptake of CsA with and without verapamil, an inhibitor for P-glycoprotein, using everted sac from jejunum, are listed in . In the absence of verapamil, the remaining amount of CsA in mucosal side in ARF rats was about 1.2-fold higher than that in control rats (P < 0.01), while there was no difference in the presence of 0.3 mM verapamil (16.23 ± 0.13 μg vs 16.82 ± 0.33 μg). In ARF rats with verapamil, however, the remaining amount of CsA in mucosal side was significantly decreased, from 21.77 ± 0.13 μg to 16.82 ± 0.33 μg (P < 0.05). In the absence of verapamil, the amounts of CsA transferred to serosal sides, uptaken into intestinal tissue, and the remaining amount in the intestinal tissue, were significantly decreased in ARF rats (P < 0.01). In the presence of verapamil, however, there were no differences between control and ARF rats. In addition, the amount of metabolized CsA in the intestinal tissue in ARF rats was also significantly decreased (P < 0.05) in the absence of verapamil, from 2.40 ± 0.35 μg to 0.83 ± 0.20 μg. The addition of verapamil improved the intestinal uptake and tissue metabolism of CsA in ARF rats.
Table 2. Effects of ARF on intestinal uptake CsA with and without verapamil using everted sac from jejunum
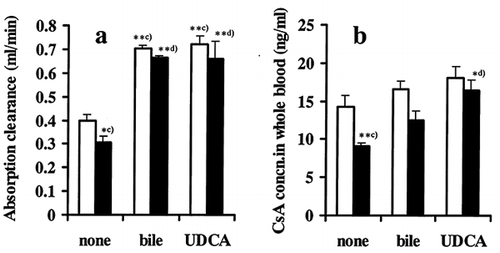
shows the effect of bile and ursodeoxycholic acid (UDCA) as a bile acid on absorption clearance and whole blood levels of CSA at steady-state using the single-pass perfusion method. Absorption clearance was significantly decreased in ARF rats in the absence of bile or bile acid, and it was significantly improved by adding bile or bile acid in the perfusate. There were no differences in the absorption clearance between control and ARF in the presence of bile or bile acid (Figure Citation[[3]]a). Simultaneously, the whole-blood levels of CsA at steady state were significantly decreased in ARF rats in the absence of bile or bile acid, and they were increased by adding bile or bile acid (Figure Citation[[3]]b) in both control and ARF.
Figure 3. Effect of bile or bile acid on (a) absorption clearance of CsA and (b) blood concentration of CsA at steady state with and without ARF in in-situ single-pass perfusion method: LI, control rats; E, ARF rats. Bile from normal rat was diluted with perfusate to a final concentration of 0.013 mL min−1, and UDCA represents ursodeoxycholic acid (3.7 mg mL−1) as a bile acid. Each column with bar represents the mean ± s.e.m. of 3 to 5 experiments. Significant difference: c), against control without bile and UDCA; d), against ARF without bile and UDCA. *p < 0.05, **p < 0.01 by one-way ANOVA.
To further investigate the effects of ARF on intestinal absorption, the amount of ATP released from intestinal tissues prepared as everted sac was measured because ATP is the source for all sorts of carrier-mediated ATPase including P-gp in the intestinal tissue. As listed in , the amount of ATP released from the everted sac to the mucosal fluid (10 mL buffer) in ARF rats was the same as that of control rats. In the presence of CsA (2.0 μM) or verapamil (0.3 mM), the amounts of ATP released from everted sacs were significantly decreased in both control and ARF. In addition, combined presence of CsA (2.0 μM) and verapamil (0.3mM) provided the most significant decreases in the amount of ATP. Moreover, the amount of ATP released from the everted sac in ARF rats was relatively retarded in comparison with that of control rats.
Table 3. Effects of ARF on ATP released from everted sac of small intestine
DISCUSSION
In our previous study, we concluded that a reduction in the fraction of CsA absorbed (one of availabilities) in the intestinal tract strongly contributed to a decrease in AUC after oral administration in glycerol-induced ARF rats.Citation[[11]] Hence, this previous study also allowed us to begin defining the factors affecting the oral bioavailability of CsA. So in this study, to further elucidate the factors affecting the fraction of CsA absorbed in ARF rats, we focused on the roles of bile secretion and P-gp (MDR-1 gene product) located in the enterocytes.Citation[[12]]
From the findings of a single dose study, the relative bioavailabilities of CsA in control and ARF rats induced by gentamicin were 43.4% and 15.2%, respectively. About a 65% decrease in the relative CsA bioavailability in ARF rats was found in comparison with control rats (). The pharma cokinetic parameters, CL/F and Vdss/F in ARF rats, were significantly increased, whereas CLt and Vdss were significantly reduced. These observations suggest that the reduced bioavailability (F) strongly contribute to the pharmacokinetics of CsA after oral administration in the presence of ARF. In addition, the findings of intravenous study showing a significant increase in MRT and significant decrease in CLt orβiv in ARF rats suggest that ARF prolongs the residence of CsA in blood circulation. All the pharmacokinetic parameters obtained in gentamicin-induced ARF rats are in the same range as those obtained in glycerol-induced ARF rats in our previous studies.Citation[[11]]
Usually, it is well known that bile is an important factor affecting the absorption of CsA from the intestinal lumen to blood circulation. The influence of bile on cyclosporin absorption was shown in a study using a whole blood HPLC assay in an orthotopic liver transplant patient regulated by a T-tube. The bioavailability after T-tube clamping showed a considerable increase in AUC in comparison with an open T-tube.Citation[[16]] Lindholm et al. showed in healthy volunteers a slight increase (1.25-fold) in CsA absorption, when 500 mg of bile acids and a light breakfast were given.Citation[[17]] Bile, therefore, seems to serve as an emulsifier, allowing CsA to be transferred from the intestinal lumen to blood circulation. Biochemically, bile acid monosulphate can be synthesized by the isolated perfused rat kidney and evidence for renal synthesis of bile acid sulphates was found in patients with cholestatis.Citation[[19]] In addition, bile acid sulphuryltransferases have been found in kidney tissues of rats and in human liver and small intestine,Citation[[20]] and hydroxylation of steroids are known to occur in the kidney.Citation[[21]] Therefore, renal dysfunction seems to depress metabolic transformation of bile in its metabolic pathway. These may also indicate that there is kidney-liver or kidney-small intestine connection in bile metabolism under renal dysfunction. However, the functions of bile secretion from the bile tract into intestine under renal dysfunction have not been clarified. From the data shown in , it was concluded that the absolute quantity of bile or bile acid in ARF rats was insufficient to serve as an emulsifier for CsA. The detailed mechanisms of retarded flow rate of bile or bile acid are unknown, however, we speculate the inhibition of bile acid synthesis from cholesterol, or failure in systemic circulation occurring from systemic edema due to renal dysfunction.
The MDR1 gene product, P-gp, presents in the enterocyte brush border can limit the bioavailability of CsA. P-gp in the enterocytes acts to prolong the duration of absorption of CsA, and it is functionally linked with enterocyte CYP3A4.Citation[[12]] Lown et al. reported that the enterocyte concentrations of P-gp vary up to 10-fold among renal transplants, and patients with higher enterocyte concentrations of P-gp tend to have higher oral clearance.Citation[[22]] In addition, enterocytes in renal transplant patients have about 10-fold higher expression of P-gp than in normal subjects.Citation[[22]] These observations suggest a possibility that the expression of P-gp concentration in enterocytes may be enhanced by renal dysfunction. Our results in the uptake study using everted sac method as listed in clearly show that the uptake of CsA into enterocytes is retarded in the presence of ARF. Moreover, our results in clearly exhibit that the existence of P-gp-mediated transport amplified by ARF contributes to the absorption of CsA from the intestinal tract, because P-gp-mediated transport requires Alp. Our results strongly support the observations described by Lown et al.Citation[[22]]
In in-situ single-pass perfusion, we examined the effects of bile and a bile acid, UDCA on the absorption clearance. The amount of rat bile and UDCA added to the perfusate were calculated from the mean flow rate of bile and the concentration of bile acids obtained in control rats in . Significant decreases in the absorption clearance and CsA blood levels without additives in ARF rats clearly show that ARF is closely related to the absorption of CsA from the intestinal tract. Marked improvement in the absorption clearance and CsA blood levels by adding bile or UDCA in ARF rats indicates that the absorption of CsA from intestinal tract is dependent on an emulsifier. The effects of bile or UDCA on the absorption clearance of CsA in ARF rats may be independent of the effect of expression of P-gp in ARF rats.
In summary, our data show that a decrease of approximately 65% in the relative bioavailability of CsA after oral administration to ARF rats in comparison with control rats can be attributed to at least two factors, renal dysfunction-dependent depression of the bile flow rate and bile acid; and renal dysfunction-dependent uptake which is due tomaybe expression of P-gp in enterocytes. Moreover, our findings that verapamil or UDCA improve the uptake of CsA into enterocytes or its absorption clearance are applicable to the oral pharmacokinetics of CsA, and management of immunosuppressive therapy in organ transplantation.
REFERENCES
- Ptachcinski R J, Venkataramanan R, Burckart G J. Clinical pharmacokinetics of cyclosporin. Clin Pharmacokinet 1986; 11: 107
- Fahr A. Cyclosporin clinical pharmacokinetics. Clin Pharmacokinet 1993; 24: 472
- Gratwohl A, Speck B, Wenk M. Cyclosporine in human bone marrow transplantation: serum concentration, graft-versus-host disease and nephrotoxicity. Transplantation 1983; 36: 40
- Lindholm A, Ringden O, Lonnqvist B. The role of cyclosporine dosage and plasma levels in efficacy and toxicity in bone marrow transplant recipients. Transplantation 1987; 43: 680
- Shibata N, Yamaji A, Park K, Tomoyoshi T, Sako H, Abe H, Kodama M, Nakane Y, Hodohara K, Hosoda S. A simple method for predicting the cyclosporin A erythrocyte-to-plasma distribution ratio in blood and its clinical assessment. Biol Pharm Bull 1994; 17: 709
- Shaw L M, Bowers L B, Demers L, Freeman D, Moyer T, Sanghvi A, Seltman H, Venkataramanan R. Critical issue in cyclosporine monitoring: report of task force on cyclosporine monitoring. Clin Chem 1987; 33: 1269
- Shibata N, Hoshino N, Minouchi T, Yamaji A, Park K, Tomoyoshi T, Abe H, Kodama M. Relationship between area under the concentration versus time curve of cyclosporin A, creatinine clearance, hematocrit value, and other clinical factors in Japanese renal transplant patients. Int J Clin Pharmacol Ther 1998; 36: 202
- Schroeder T J, Sridhar N, Pesce A J, Alexander J W, First M R. Clinical correlations of cyclosporine-specific and -nonspecific assays in stable renal transplants, acute rejection, and cyclosporine nephrotoxicity. Ther Drug Monit 1993; 15: 190
- Shibata N, Shimakawa H, Yamaji A. Retrospective pharmacokinetic data analysis of cyclosporin A using routine monitoring data in renal transplant patients: consideration on its pharmacokinetic profiles and physiological factors during renal dysfunction. Jpn J Hosp Pharm 1993; 19: 270
- Morimoto J, Shibata N, Hoshino N, Miniuchi T, Yamaji A, Kim C J, Park K I, Kiso Y. Clinical utility of converting cyclosporine into tacrolimus in a living-related renal transplant patient with secondary nephrotic syndrome. Jpn J Ther Drug Monit 1999; 16: 135
- Shibata N, Ohmae T, Hoshino N, Minouchi T, Yamaji A. Influence of glycerol-induced acute renal failure on the pharmacokinetics of cyclosporin in Rats. J Pharm Pharmacol 1999; 51: 397
- Watkins P B. The barrier function of CYP3A4 and p-glycoprotein in the small bowel. Advanced Drug Delivery Reviews 1997; 29: 161
- Barr W H, Reigelman S. Intestinal drug absorption and metabolism: comparison of methods and models to study physiological factors of in-vitro and in-vivo intestinal absorption. J Pharm Sci 1970; 59: 154
- Yamaoka K, Nakagawa T. Uno T: Moment analysis for disposition kinetics of several cephalosporin antibiotics in rats. J Pharm Pharmacol 1983; 35: 19
- Abbott Laboratories Diagnostic Division. TDx System Operation Manual. Abbott Laboratories, North Chicago 1984
- Mehta M U, Venkataramanan R, Burckart G J, Ptachcinski R J, Delamos B. Effect of bile on cyclosporin absorption in liver transplant patients. Br J Clin Pharmacol 1988; 25: 579
- Lindholm A, Henricsson S, Dahlqvist R. The effect of food and bile acid administration on the relative bioavailability of cyclosporin. Br J Clin Pharmacol 1990; 29: 541
- Summerfield J A, Gollan J L, Billing B H. Synthesis of bile acid monosulfates by isolated perfused rat kidney. Biochem J 1976; 156: 339
- Thomassen P A. Urinary bile acids in late pregnancy and in recurrent cholestasis of pregnancy. Eur J Clin Invest 1979; 9: 425
- Bremmelgaard A. Bile acids in plasma of patients without kidney. Scand J Clin Lav Invest 1983; 43: 603
- Cowen A E, Korman M G, Hofmann A F, Turcotte J, Carter J A. Radioimmunoassay of sulfated lithocholates. J Lipid Res 1977; 18: 698
- Lown K S, Mayo R R, Leichtman A B, Hsiao H L, Turgeon D K, Ren P S, Brown M B, Guo W, Rossi S J, Benet L Z, Watkins P B. Role of intestnal P-glycoprotein (MDR1) in inter patient variation in the oral bioavailability of cyclosporine. Clin Pharmacol Ther 1997; 62: 248