Abstract
Kidneys preserved for transplantation surgery sustain injuries caused by cold ischemia during storage. Additionally, kidneys harvested from non-heart-beating donors encounter the stress of warm ischemia. The aim of this study was to determine the specific cell types losing viability after warm and cold ischemia. In warm ischemia studies, the pedicles of left kidneys of Lewis rats were cross-clamped for up to 90 min. In cold ischemia studies, kidneys were flushed with cold University of Wisconsin solution and stored up to 48 h at 0–1°C. After warm or cold ischemia, kidneys were perfused via the renal arteries with Krebs-Henseleit bicarbonate (KHB) buffer at 37°C, followed by trypan blue to label the nuclei of nonviable cells. Warm ischemia for 90 min caused renal failure and led to injury of proximal tubular cells, e.g., loss of brush borders, cast formation and trypan blue labeling. Cold ischemia for 48 h also caused renal failure but, unlike warm ischemia, caused trypan blue labeling of glomerular podocytes and peritubular endothelial cells. In warm ischemia-induced injury, electron microscopy showed shedding of microvilli and marked swelling of proximal tubular cells, microvilli and mitochondria. In cold ischemia-induced injury, podocytes were blebbed and swollen, and their pedicels were detached from the basement membrane, but disruption in proximal tubules was milder. In conclusion, warm ischemia triggers injury primarily to proximal tubular cells, whereas cold ischemia damages glomerular podocytes and peritubular endothelial cells in addition to proximal tubules.
INTRODUCTION
Ischemia in the kidney leads to a series of complex cellular and molecular events that remain incompletely understood Citation[[1]], Citation[[2]], Citation[[3]]. Current methods to preserve kidneys for transplantation surgery usually entail periods of ex vivo cold ischemia. Prolonged cold ischemia can result in increased susceptibility to damage upon reperfusion and is strongly associated with delayed renal graft function and non-function Citation[[4]], Citation[[5]]. In clinical transplantation, kidneys from non-heart-beating donors are now being harvested because of the severe shortage of donor organs from heart-beating donors Citation[[6]], Citation[[7]]. For kidneys from non-heart-beating donors, a period of warm ischemia during cardiac arrest is inevitable prior to preservation by cold ischemic storage or hypothermic perfusion. In non-transplant settings, warm ischemia is well known to cause acute renal failure Citation[[8]], Citation[[9]].
Differences in cell killing within the kidney caused by warm and cold ischemia are not known. The goal of this study was to identify the specific cell types losing viability after warm ischemia and cold ischemic storage. It is shown that warm ischemia leads to loss of viability of proximal tubular cells, whereas cold ischemia triggers glomerular podocyte and peritubular vascular endothelial cell killing.
MATERIALS AND METHODS
Animals and Anesthesia
Adult male Lewis rats (220–250 g) were used and housed 4 to a cage. Each animal underwent anesthesia with an intraperitoneal injection of nembutal sodium solution (50 mg/kg, Abbott Laboratories, North Chicago, IL). All animals received humane care in compliance with guidelines approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill.
Induction of Warm Ischemia In Vivo
Animals were placed under a heat lamp to maintain body temperature during anesthesia. As described previously Citation[[10]], a midline laparotomy was performed, and the left kidney was dissected free from the surrounding tissue. A nontraumatic vascular clamp was placed across the renal pedicle to induce ischemia for 60 or 90 min with temporary closing of the abdomen. Five minutes before the end of the ischemic period, a contralateral nephrectomy was performed. Reperfusion of the left kidney was achieved by releasing the vascular clamp. The abdominal wall was closed with double-layer sutures, and the rats were allowed to recover. Rats that survived over two weeks were considered survivors of the study.
Cold Ischemia and Transplant Surgery
The surgical procedures of renal harvesting and grafting were described previously Citation[[11]]. Briefly, the left donor kidney with 1 cm of ureter was removed en bloc, including the renal artery with a 4 mm aortic segment and the renal vein with a patch of vena cava. The explant was flushed and preserved with University of Wisconsin (UW) solution (NPBI, Emmer-Compascuum, The Netherlands) at 0–1°C for 24 or 48 h. Both native kidneys of the recipient were removed, leaving the right ureter as long as possible. The donor kidney was then transplanted orthotopically to the abdominal aorta and inferior vena cava below the left renal artery by end-to-side anastomoses with 8–0 Ethilon monofilament nylon sutures. Ureter anastomosis was performed end-to-end close to the renal pelvis with 10–0 Ethilon monofilament sutures. The abdominal wall was closed, and the recipient rat was allowed to recover. Kidney recipients that survived over two weeks were considered survivors.
Ex Vivo Kidney Perfusion and Determination of Cell Killing by Trypan Blue Uptake
After warm and cold ischemia, some kidneys were perfused via the renal artery with Krebs-Henseleit-bicarbonate (KHB) buffer, pH 7.4, saturated with 95% O2 and 5% CO2 at 37°C. The initial flow rate was 1 ml/min/g, which was gradually increased to 4–6 ml/min/g over 5 min. During the next 5 min, trypan blue (500 µM) was added to label the nuclei of nonviable cells Citation[[12]], Citation[[13]], Citation[[14]]. Kidneys were then fixed by infusion of 2% paraformaldehyde and 2% glutaraldehyde in KHB buffer, processed by dehydration and then embedded in paraffin. Sections were cut at 5 µm and stained with hematoxylin and eosin (H&E) and with eosin alone. Nonviable cells were counted from blue-stained nuclei in eosin-stained sections in glomeruli with visible vascular poles and were expressed as the percentage of the total number of nuclei counted in H&E-stained sections. Nonviable cells were also counted in 5 cortical fields selected at random for each specimen.
Immunohistochemical Staining
Paraffin sections (5-µm) were stained immunohistochemically using primary antibodies against ezrin (Berkeley Antibody Company, Richmond, CA) to identify glomerular podocytes Citation[[15]] and antibodies to Factor VIII and desmin (Dako Corporation, Carpinteria, CA) to detect vascular endothelial cells and glomerular mesangial cells, respectively Citation[[16]].
Measurement of Urinary Albumin
Urine samples were collected one day after transplantation and stored at −80°C. Albumin was determined using a Sigma Diagnostic Kit (631-2-100P, Sigma Diagnostic Inc., St. Louis, MO) according to the manufacturer's instructions.
Electron Microscopy
Kidneys were fixed by infusion of 2% paraformaldehyde and 2% glutaraldehyde in KHB buffer. For scanning electron microscopy (SEM), fixed kidneys were cut into 2 × 6 × 8 mm3, placed in 1% of osmium tetroxide in 0.1 M sodium cacodylate buffer (pH 7.3) for 1 h at room temperature, and then washed in distilled water. Subsequently, the tissue was dehydrated in ethanol and dried at the critical point in CO2. The dried tissue was cut, mounted on an aluminum stub and coated with Au–Pd for viewing in a JEOL 820 scanning electron microscope (JEOL Ltd., Tokyo, Japan). For transmission electron microscopy (TEM), pieces of 1-mm diced kidney tissues were washed in 0.2 sodium cacodylate buffer (pH 7.3) for two days and post-fixed in 1% OsO4 at 4°C overnight. The samples were then washed in distilled water and dehydrated in ethanol. After Epon plastic embedding, 1-µm sections were cut and stained with toluidine blue. Selected samples containing glomeruli were thin sectioned and examined with a Tecnai 12 transmission electron microscope.
Statistics
Results were expressed as means ± the standard error of mean (SEM) with values for n of 6–10 per group. Statistical significance of differences between means was determined using a paired t-test, analysis of variance, or Fisher's Exact test, as appropriate. A p value of less than 0.05 was considered significant.
RESULTS
Renal Function and Survival After Warm and Cold Ischemia
In preliminary experiments, the times of warm and cold ischemia that led to renal failure were determined. Seven of 8 rats survived 14 days after 60 min of warm ischemia, whereas 2 of 8 survived after 90 min of warm ischemia (p<0.05). Thus, 90 min of warm ischemia caused renal failure in 75% of rats. After 24 h of cold storage in UW solution followed by transplantation, 6 of 8 rats survived. After 48 h of cold ischemic storage and transplantation, no rat survived longer than one week after surgery. Thus, 48 h of cold storage in the UW solution caused lethal damage to rat renal grafts.
We also evaluated creatinine clearance after periods of warm and cold ischemia that most rats survived. In rats subjected to 60 min of warm ischemia, creatinine clearance was 1.5 ± 0.2 ml/min/kg one day later, which was 27% of preischemic values (5.6 ± 0.3 ml/min/kg, p<0.05). In rats receiving kidney grafts after 24 h of cold storage, creatinine clearance at day 1 postoperatively was 1.3 ± 0.2 ml/min/kg, which was only 23% of the normal value (p<0.05).
Cell Killing After Lethal Warm and Cold Ischemia
The renal cortex of normal untreated kidneys showed characteristic glomerular and tubular architecture by light microscopy (A) with virtually no trypan blue labeling (B). After 90 min of warm ischemia, proximal tubular injury was prominent, characterized by swelling of tubular epithelial cells, bleb formation, loss of brush borders, and accumulation of debris in lumens (C). The inner surface of tubules was rough, and red blood cells congested the peritubular vessels. Glomerular structure appeared to be normal (C). Trypan blue labeling revealed that some proximal tubular cells had become nonviable (D). No trypan blue-labeled cells were observed in glomerular and peritubular areas.
Figure 1. Light micrographs of renal cortex. Kidneys were perfused with KHB buffer plus trypan blue and prepared for light microscopy, as described in MATERIALS AND METHODS. A and B are kidneys before ischemia. C and D are kidneys after 90 min of warm ischemia. E and F are kidneys after 48 h of cold storage in UW solution. A, C and E were stained with H&E. B, D and F were stained with eosin only to visualize trypan blue-stained nuclei as indicators of loss of cell viability. Blue arrows identify proximal tubular wall thinning with loss of brush borders. White arrows identify luminal debris or blebs. Black arrows identify trypan blue-labeled nonviable cells. Scale bar is 50 µm. (See color insert at end of issue.)
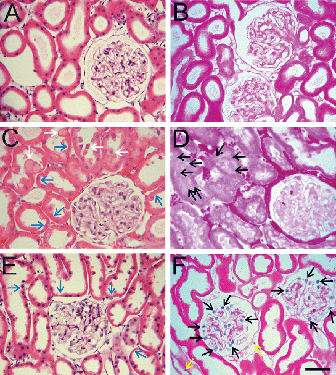
After 48 h of cold storage in UW solution, the walls of most proximal tubules were thinner than normal, and some tubules had lost brush borders (E). The glomeruli appeared normal. However, trypan blue labeling revealed loss of viability of many cells in the glomeruli (F). Quantitation of the trypan blue-labeled nuclei showed that 22 ± 3% of glomerular cells were nonviable. In addition, some trypan blue labeling in peritubular areas was observed (9 ± 3/high power field). Tubular epithelial cells did not label with trypan blue.
After acute warm ischemia, injury to the inner cortex-outer medullary zone is expected to occur Citation[[17]], Citation[[18]]. Light microscopic sections of normal, non-ischemic kidneys revealed characteristic proximal and distal tubular structure (A) with no trypan blue labeling (B). After 90 min of warm ischemia, casts formed in many distal tubules and erythrocyte congestion in peritubular areas was prominent (C). Some trypan blue-labeled tubular cells were observed, but peritubular trypan blue labeling was absent (D).
Figure 2. Light micrographs of inner cortex-outer medulla. Kidneys were perfused with trypan blue, as described in . A and B are kidneys before ischemia. C and D are kidneys after 90 min of warm ischemia. E and F are kidneys after 48 h of cold storage in UW solution. A, C and E were stained with H&E. B, D and F were stained with eosin only to visualize trypan blue-stained nuclei as indicators of loss of cell viability. Blue arrows identify peritubular erythrocyte trapping. White arrows identify luminal debris or casts. Black arrows identify trypan blue-labeled nonviable cells. Scale bar is 50 µm. (See color insert at end of issue.)
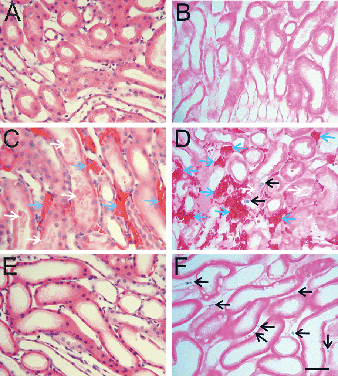
After 48 h of cold storage, the most prominent change in H&E sections of outer cortex and medulla was thinning of tubular walls and dilation of tubular lumens (E). Tubular casts and erythrocyte congestion were absent. Trypan blue labeling was observed in peritubular spaces but not in proximal and distal tubules (F).
Identification of Nonviable Cell Types
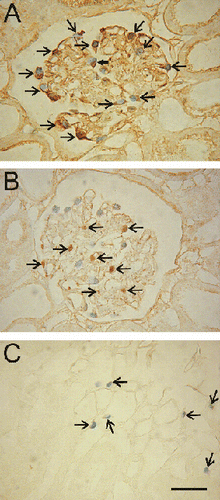
Immunohistochemistry was used to identify the types of cells losing viability after cold ischemia. After 48 h of cold ischemia, trypan blue-labeled glomerular cells were positive to ezrin, a marker of podocytes (A), but were negative for desmin, a mesangial cell marker (B). Antibodies against Factor VIII were used to identify vascular endothelial cells. Factor VIII staining revealed that trypan blue-labeled peritubular cells were endothelial cells (C). However, trypan blue-labeled glomerular cells were negative for Factor VIII (data not shown).
Figure 3. Identification of trypan blue-labeled podocytes and peritubular endothelial cells. Kidneys were stored for 48 h in UW solution, reperfused with KHB buffer containing trypan blue, and prepared for immunocytochemistry, as described in MATERIALS AND METHODS. A: A section was stained with primary antibody against ezrin, a podocyte marker. Trypan blue-labeled nuclei in the glomerulus displayed brown staining for ezrin (arrows). B: The section was stained with primary antibody against desmin, a glomerular mesangial cell marker. Desmin-positive cells (arrows) did not label with trypan blue. C: The section was stained with primary antibody against Factor VIII, a vascular endothelial cell marker. Brown staining for Factor VIII (arrows) coincided with trypan blue labeling of medullary peritubular cells. Scale bar is 50 µm. (See color insert at end of issue.)
Urinary Concentration of Albumin
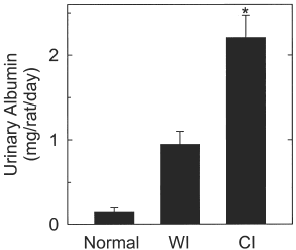
Urinary albumin from normal, untreated rats was minimal (). After 90 min of warm ischemia, albumin in the first 24 h urine increased over 6-fold. After 48 h of cold ischemia, urinary albumin increased more than 14-fold, which was more than twice as great as after 90 min of warm ischemia (, p<0.05). The findings show that lethal cold ischemia caused more albumin excretion than lethal warm ischemic injury, possibly due to podocyte damage.
Figure 4. Urinary excretion of albumin after warm and cold ischemia to rat kidneys. Urine was collected for albumin measurements for 24 h before ischemia, during the first 24 h after 90 min of warm ischemia and during the first 24 h following 48 h of cold ischemic storage and renal transplantation, as described in MATERIALS AND METHODS. In the cold ischemia (CI) study, kidneys were transplanted after being stored for 48 h in cold UW solution, urine samples were collected during the first 24 h after transplantation. *, p<0.05 compared to warm ischemia.
Ultrastructural Analysis of Podocytes and Proximal Tubules
SEM of normal kidneys showed the characteristic multiple interdigitating foot processes (pedicels) of normal podocytes (A). After 90 h of warm ischemia, the cell bodies of podocytes appeared slightly swollen, and their pedicels were not disrupted (C). After 48 h of cold ischemia, however, podocytes were markedly swollen and distorted with large protruding blebs. Many pedicels had detached or disappeared, and those that remained were shortened and thickened (E).
Figure 5. Scanning electron micrographs of podocytes and cortical tubules. Kidneys were prepared for SEM, as described in MATERIALS AND METHODS. A and B are normal kidneys. C and D are kidneys after 90 min of warm ischemia. E and F are kidneys after 48 h of cold storage in UW solution. A, C and E show glomerular podocytes. B, D and F show proximal tubules. In C, note swelling of podocytes, as compared to normal kidney (A). In E, podocyte alterations after cold ischemia were more marked than after cold ischemia (C). Note large blebs protruding from podocyte cell bodies and the swelling and disorganization of foot processes (pedicels). In D, note swollen tubular walls and debris accumulation in tubular lumens, as compared to normal kidney (B). After 48 h of cold ischemia, tubular walls became thinner with blebbing and partial loss of brush borders on the apical surface (F). Bars are 5 µm for A, C and E and 10 µm for B, D and E.
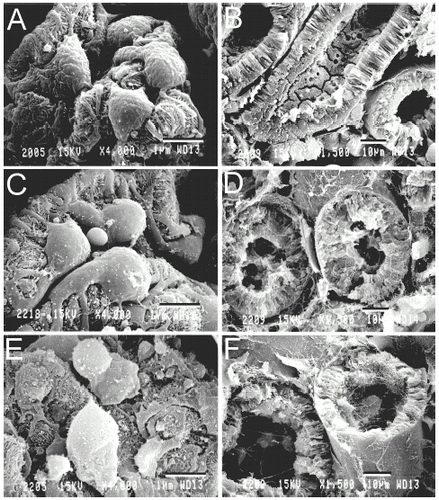
Cortical proximal tubules were also examined by SEM. In comparison to normal kidneys (B), 90 min of warm ischemia caused proximal tubular swelling. Additionally, brush borders were missing, and debris was present in tubular lumens (D). In kidneys after 48 h of cold ischemia, SEM showed less marked disruption of brush borders with enlargement of tubular lumens and thinning of tubular walls (F). In addition, debris was also observed in tubular lumens (F).
TEM of normal kidneys revealed the characteristic filtration apparatus of the glomerulus, including fenestrated endothelium, basement membrane and podocytes (A). After 90 min of warm ischemia, podocytes displayed slight swelling, but their cytoplasmic density remained unchanged, and their mitochondria appeared normal (C). By contrast, podocytes of kidneys after 90 min of cold ischemia were severely disrupted (E). Individual podocytes were markedly swollen. Cytoplasmic density was greatly decreased, mitochondria were swollen and rounded, and the nuclear chromatin was aggregated peripherally (E). These changes indicated loss of cell viability. Additionally, the pedicels were swollen and often detached from the basement membrane.
Figure 6. Transmission electron micrographs of glomeruli and proximal tubules. Kidneys were prepared for TEM, as described in MATERIALS AND METHODS. A and B are normal kidneys. C and D are kidneys after 90 min of warm ischemia. E and F are kidneys after 48 h of cold storage in UW solution. A, C and E show the glomerular filtration apparatus. B, D and F show proximal tubules. In C, note mild swelling of podocytes after warm ischemia compared to normal kidney (A). Podocyte changes were more marked after cold ischemia and included swelling of cells, pedicels and mitochondria, decreased cytoplasmic density, peripheral chromatin aggregation, and detachment of foot process from the basement membrane (E). In D, note marked swelling of microvilli and mitochondria, decreased cytoplasmic density, and partial loss of brush borders in proximal tubules after warm ischemia compared to normal kidney (B). Cold ischemia caused milder disruption of brush borders and thinning of tubular walls (F). Bars are 2 µm.
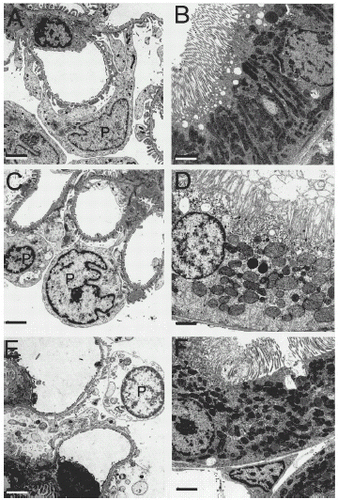
Proximal tubules were also examined by TEM. Proximal tubules of normal kidneys displayed intact brush borders and normal cytoplasm (B). After 90 min of warm ischemia, proximal tubular brush borders were distorted and, in some instances, absent (D). Mitochondria became rounded and swollen, and cytoplasmic density was decreased (D). By contrast, after 48 h of cold ischemia, mitochondrial swelling and brush border alterations were relatively mild (F). Tubular walls became thinner than normal, but cytoplasmic density remained normal. Peritubular endothelial cells frequently appeared nonviable, as demonstrated by aggregated nuclear chromatin, cytoplasmic rarefaction and large amplitude mitochondrial swelling (data not shown).
DISCUSSION
Differences in Renal Injury After Warm and Cold Ischemia
In the present study, warm and cold ischemia caused different patterns of kidney injury. Warm ischemic injury leading to kidney failure was associated with profound tubular damage. Proximal tubular cells were swollen and many lost viability. Brush borders were disrupted or entirely absent, which may have contributed to tubular cast formation. Glomerular injury, however, was not a prominent feature after warm ischemia. In contrast to warm ischemia, cold ischemia caused prominent glomerular and peritubular endothelial damage. As documented by trypan blue labeling, numerous podocytes lost cell viability. Associated with podocyte damage was increased urinary excretion of albumin, which was more than twice as great after cold ischemia than warm ischemia. Peritubular endothelial cell killing also was prominent after cold ischemia. Additionally, proximal tubular alterations occurred after cold ischemia, which included thinning of tubular walls and dilation of tubular lumens. However, these changes to tubular cells were less marked after cold ischemia than warm ischemia, and killing of tubular cells did not occur.
We compared warm and cold ischemia under conditions where both stresses led to lethal kidney failure. To duplicate the clinical transplant situation, a model of warm ischemia to one kidney with a contralateral nephrectomy was chosen. In this model, 90 min of warm ischemia caused lethal injury to 75% of rats, whereas 60 min of warm ischemia was lethal only 12% of the time, consistent with previous reports Citation[[10]], Citation[[19]]. To assess injury after cold storage, we stored kidneys in UW solution at 0–1°C and then transplanted them into recipient rats whose native kidneys were removed. After 24 h of cold ischemia, 75% of recipient rats survived, but no renal grafts survived after 48 h of cold storage. Our findings are consistent with earlier studies showing that rat kidneys are more sensitive to cold ischemic injury than human and canine kidneys Citation[[20]]. Although UW solution preserves canine kidneys successfully for 72 h Citation[[21]], successful preservation of rat kidneys for 48 h by simple cold storage in UW solution alone has not been possible.
Tubular Injury After Ischemia
Previous studies on renal ischemia have focused on renal tubular cells Citation[[22]]. Tubular epithelial cells are characterized by high rates of ATP-dependent solute transport supported by mitochondrial oxidative phosphorylation. Thus, tubular cells are sensitive to decreased oxygen availability, especially under normothermic conditions when metabolism is not suppressed. In the present study, marked tubular injury occurred after prolonged warm ischemia, including cellular swelling, loss of brush borders, mitochondrial swelling and loss of cell viability. Thus, tubular damage represented the main feature of warm ischemia-induced renal injury.
In kidneys subjected to prolonged cold ischemia, loss of viability of tubular cells was virtually absent. Additionally, structural alterations to tubular epithelial cells were less marked than after prolonged warm ischemia. Inhibition of tubular metabolism by hypothermia may contribute to the relative protection during cold storage. Nonetheless, we observed brush border abnormalities, wall thinning and luminal debris after cold ischemia. Thus, tubular changes are a component of cold ischemia-induced injury.
Podocyte Injury in Renal Ischemia–Reperfusion
Unlike proximal tubular cells, little attention has been paid to podocytes in postischemic renal injury. Podocytes are highly differentiated and specialized cells that form an integral component of the glomerular filtration apparatus and regulate the glomerular filtration rate Citation[[23]]. Podocytes do not replicate postnatally. When podocytes are lost for any reason, new cells cannot replace them Citation[[24]]. Therefore, podocyte damage likely leads to sustained impairment of renal function Citation[[25]], Citation[[26]]. Acutely, podocyte injury increases glomerular permeability to protein, which may contribute to formation of obstructive tubular casts and increased urinary excretion of protein. Swelling of podocyte foot processes after cold ischemia may also produce a mechanical barrier causing a decreased glomerular filtration rate. Our observation of podocyte, killing after prolonged cold ischemia suggests that protection of podocytes should be a therapeutic goal in kidney preservation for transplantation. Podocyte killing did not occur after prolonged warm ischemia, which was the most prominent difference between warm and cold ischemia.
Endothelial Killing in Peritubular Areas
Vascular endothelial cell injury and up-regulation of adhesion molecule expression are implicated in ischemia/reperfusion injury to kidneys Citation[[27]], Citation[[28]], Citation[[29]]. The present study showed selective peritubular endothelial cell killing in both the cortex and the medulla after prolonged cold ischemia. Endothelial cell injury likely produces vasoconstriction and increased leukocyte infiltration, leading to chronic ischemia in the reperfused tissue Citation[[30]]. Protection of endothelial cells is beneficial to postischemic renal function after cold ischemia Citation[[31]]. Therefore, protection against endothelial cell damage should remain an important goal in hypothermic kidney preservation.
Although endothelial cell killing was not observed in lethal warm ischemia-induced renal injury, marked congestion of red blood cells in the areas of inner cortex and outer medulla was observed. This congestion, also called “erythrocyte trapping”, is strongly associated with postischemic renal damage and acute renal failure Citation[[32]], Citation[[33]]. The mechanisms responsible for the development of this phenomenon are still unclear. However, warm ischemia-induced red blood trapping was not related to endothelial killing, because unlike what occurred after cold ischemia, endothelial cells did not label with trypan blue after warm ischemia.
Clinical Implications
When kidneys are harvested from non-heart-beating cadaver donors, varying periods of warm ischemia occur prior to cold ischemia Citation[[6]]. The present study shows that warm ischemia causes a different pattern of renal damage than cold ischemia. These different types of injury may be additive when kidneys are exposed to both warm and cold ischemia. Indeed, a previous study showed that 20 min of warm ischemia combined with 24 h of cold ischemia, but not warm or cold ischemia alone, caused graft failure after kidney transplantation in rats Citation[[11]]. Thus, targeting of the different types of warm and cold ischemic injury will likely be required to achieve optimal preservation of kidneys from non-heart-beating cadaver donors.
ACKNOWLEDGMENT
This work was supported, in part, by the Collaborative Funding Assistance Program of the North Carolina Biotechnology Center and NIH Grant DK37034. Imaging facilities were supported, in part, by NIH Center Grants 5-P30-DK34987 and 1-P50-AA11605.
REFERENCES
- Bonventre J.V., Kelly K.J. Adhesion Molecules and Acute Renal Failure. Adv. Nephrol. Necker. Hosp. 1996; 25: 159–176
- Weight S.C., Bell P.R., Nicholson M.L. Renal Ischaemia–Reperfusion Injury. Br. J. Surg. 1996; 83: 62–170
- Rabb H., O’Meara Y.M., Maderna P., Coleman P., Brady H.R. Leukocytes, Cell Adhesion Molecules and Ischemic Acute Renal Failure. Kidney Int. 1997; 51: 1463–1468
- Lee C.M., Carter J.T., Randall H.B., Hiose R., Stock P.G., Melzer J.S., Dafoe D.C., Freise C.E., Alfrey E.J. The Effect of Age and Prolonged Cold Ischemia Times on the National Allocation of Cadaveric Renal Allografts. J. Surg. Res. 2000; 91: 83–88
- Peters T.G., Shaver T.R., Ames J.E., Santiago-Delpin E.A., Jones K.W., Blanton J.W. Cold Ischemia and Outcome in 17 937 Cadaveric Kidney Transplants. Transplantation 1995; 59: 191–196
- Kootstra G. The Asystolic, or Non-Heart Beating, Donor. Transplantation 1997; 63: 917–921
- Pacholczyk M.J., Lagiewska B., Szostek M., Chmura A., Morzycka-Michalik M., Rowinska-Stryjecka D., Walaszewski J., Rowinski W. Transplantation of Kidneys Harvested from Non-Heart-Beating Donors: Early and Long-Term Results. Transpl. Int. 1996; 9(Suppl. 1)S81–S83
- Burke T.J., Burnier M., Langberg H., Shanley P., Schrier R.W. Renal Response to Shock. Ann. Emerg. Med. 1986; 15: 1397–1400
- Weisberg L.S., Allgren R.L., Genter F.C., Kurnik B.R. Cause of Acute Tubular Necrosis affects Its Prognosis. The Auriculin Anaritide Acute Renal Failure Study Group. Arch. Intern. Med. 1997; 157: 1833–1838
- Yin M., Kurvers H.A., Tangelder G.J., Booster M.H., Buurman W.A., Kootstra G. Ischemia-Reperfusion Injury of Rat Kidney Relates More to Tubular Than to Microcirculatory Disturbances. Ren. Fail. 1996; 18: 211–223
- Yin M., Buurman W.A., Daemen J.W., Kootstra G. The PAF Antagonist TCV-309 Reduces Graft PMN Infiltration and Enhances Early Function of 24-Hour-Preserved Rat Kidneys with Long Warm Ischemia. Transplantation 1996; 61: 1443–1446
- Caldwell-Kenkel J.C., Thurman R.G., Lemasters J.J. Selective Loss of Nonparenchymal Cell Viability After Cold Ischemic Storage of Rat Livers. Transplantation 1988; 45: 834–837
- Currin R.T., Toole J.G., Thurman R.G., Lemasters J.J. Evidence That Carolina Rinse Solution Protects Sinusoidal Endothelial Cells Against Reperfusion Injury After Cold Ischemic Storage of Rat Livers. Transplantation 1990; 50: 1076–1078
- Currin R.T., Caldwell-Kenkel J.C., Lichtman S.N., Bachmann S., Takei Y., Kawano S., Thurman R.G., Lemasters J.J. Protection by Carolina Rinse Solution, Acidotic pH, and Glycine Against Lethal Reperfusion Injury to Sinusoidal Endothelial Cells of Rat Livers Stored for Transplantation. Transplantation 1996; 62: 1549–1558
- Hugo C., Nangaku M., Shankland S.J., Pichler R., Gordon K., Amieva M.R., Couser W.G., Furthmayr H., Johnson R.J. The Plasma Membrane–Actin Linking Protein, Ezrin, Is a Glomerular Epithelial Cell Marker in Glomerulogenesis, in the Adult Kidney and in Glomerular Injury. Kidney Int. 1998; 54: 1934–1944
- Hegele R.G., Behar M., Katz A., Silverman M. Immunocytochemical Characterization of Cells in Rat Glomerular Culture. Clin. Invest. Med. 1989; 12: 181–186
- Beck F.X., Ohno A., Dorge A., Thurau K. Ischemia—Induced Changes in Cell Element Composition and Osmolyte Contents of Outer Medulla. Kidney Int. 1995; 48: 449–457
- Heyman S.N., Brezis M., Epstein F.H., Spokes K., Rosen S. Effect of Glycine and Hypertrophy on Renal Outer Medullary Hypoxic Injury in Ischemia Reflow and Contrast Nephropathy. Am. J. Kidney Dis. 1992; 19: 578–586
- Jablonski P., Howden B.O., Rae D.A., Birrell C.S., Marshall V.C., Tange J. An Experimental Model for Assessment of Renal Recovery from Warm Ischemia. Transplantation 1983; 35: 198–204
- Southard J.H., Marsh D.C., McAnulty J.F., Belzer F.O. The Importance of O2-Derived Free Radical Injury to Organ Preservation and Transplantation. Transplant. Proc. 1987; 19: 1380–1381
- Ploeg R.J., Goossens D., Mcanulty J.F., Southard J.H., Belzer F.O. Successful 72-Hour Cold Storage of Dog Kidneys with UW Solution. Transplantation 1988; 46: 191–196
- Solez K. The Morphology of Acute Renal Failure. Acute Renal Failure, 3rd Ed., J.M. Lazarus, B.M. Brenner. Churchill Livingstone, New York 1993
- Kriz W., Elger M., Nagata M., Kretzler M., Uiker S., Koeppen-Hageman I., Tenschert S., Lemley K.V. The Role of Podocytes in the Development of Glomerular Sclerosis. Kidney Int. 1994; 45(Suppl.)S64–S72
- Kriz W., Gretz N., Lemley K.V. Progression of Glomerular Diseases: Is the Podocyte the Culprit?. Kidney Int. 1998; 54: 687–697
- Schwartz M.M. The Role of Podocyte Injury in the Pathogenesis of Focal Segmental Glomerulosclerosis. Ren. Fail. 2000; 22: 663–684
- Kriz W., Elger M., Hosser H., Hahnel B., Provoost A., Kranzlin B., Gretz N. How Does Podocyte Damage Result in Tubular Damage?. Kidney Blood Press. Res. 1999; 22: 26–36
- Zizzi H.C., Zibari G.B., Granger D.N., Singh I., Cruz L.D., Abreo F., McDonald J.C., Brown M.F. Quantification of P-Selectin Expression After Renal Ischemia and Reperfusion. J. Pediatr. Surg. 1997; 32: 1010–1013
- Sheridan A.M., Bonventre J.V. Cell Biology and Molecular Mechanisms of Injury in Ischemic Acute Renal Failure. Curr. Opin. Nephrol. Hypertens. 2000; 9: 427–434
- Satoh S., Stowe N.T., Inman S.R., Sankari B.R., Magnusson M.O., Novick A.C. Renal Vascular Response to Vasodilators Following Warm Ischemia and Cold Storage Preservation in Dog Kidneys. J. Urol. 1993; 149: 186–189
- Wilhelm S.M., Simonson M.S., Robinson A.V., Stowe N.T., Schulak J.A. Cold Ischemia Induces Endothelin Gene Upregulation in the Preserved Kidney. J. Surg. Res. 1999; 85: 101–108
- Kuzu M.A., Koksoy C., Alacayir I., Yazar O., Kuterdem E. Thromboxane Synthase Inhibitor, UK 38485, Prevents Renal Injury in the Rabbit Isolated Perfused Kidney Exposed to Cold Ischemia. Transplantation 1995; 59: 1096–1099
- Nygren A., Hellberg O., Hansell P. Red-Cell Trapping in the Rat Renal Microcirculation Induced By Low Osmolar Contrast Media and Mannitol. Invest. Radiol. 1993; 28: 1033–1038
- Jacobsson J., Odlind B., Tufveson G., Wahlberg J. Effects of Cold Ischemia and Reperfusion on Trapping of Erythrocytes in the Rat Kidney. Transpl. Int. 1988; 1: 75–79