Abstract
TGF-β1 has been demonstrated to be up-regulated in response to ischemic events both in animal models and in man. Demonstration of this up-regulation in the kidney following experimentally induced acute renal failure and in renal transplants complements similar findings in coronary and cerebral ischemia. Activation of TGF-β1 occurs as a direct consequence of hypoxia, angiotensin II signaling and loss of extra cellular matrix (ECM) integrity, all of which occur in renal ischemia-reperfusion injury. TGF-β1 thus up-regulates the synthesis of extracellular matrix components such as fibronectin and collagen IV providing a basis for the restoration of epithelial coverage in the regenerating tubule. TGF-β1 also regulates epithelial tubular cell proliferation and differentiation. This response is quickly closed down in response to recovery of the kidney. This review examines the evidence linking TGF-β1 activity to recovery from renal ischemia thereby constructing a hypothesis for the beneficial role of TGF-β1 in the post ischemic kidney.
INTRODUCTION
The mechanisms of recovery from acute renal failure and acute tubular necrosis caused by renal ischemia remains a relatively poorly understood topic. The diverse stimuli received by the kidney cells following ischemia require to be studied to unravel the important players in the recovery of nephron structure and function. Among them, the cytokine TGF-β1 may provide some answers in this respect given its role in extracellular matrix dynamics and its proven up-regulation in response to relevant stimuli such as Angiotensin II (AngII) and hypoxia. This review aims to build a picture of the basic mechanisms by which TGF-β1 acts as a finely controlled recovery signal in renal ischemia using evidence from both in vitro and in vivo work.
Acute renal failure (ARF) as a consequence of ischemia-reperfusion injury is caused by a number of circumstances, most notably renal artery stenosis, renal transplantation and major surgery, particularly that involving elderly patients.Citation[[1]], Citation[[2]]
The subcellular changes that contribute to changes in individual nephron and overall renal function following ARF have been well documented. However the mechanisms by which the kidney regenerates are much less coherently understood. The injury to the tubule caused by ischemia-reperfusion induced ARF is essentially biphasic. Firstly, there is an ischemic phase characterized by the lack of oxygen and nutrient availability, cellular ATP depletion and loss of mitochondrial viability. This then gives place to a number of deleterious processes in the reperfusion phase such as an increase oxygen free radicals production, and in cytosolic free calcium, high amplitude swelling of mitochondria, stripping of membrane phospholipids, loss of apical brush border structure in the proximal convoluted tubule, loss of electrolyte balance through deletion of basolateral Na+/K+ATPase activity and inflammatory reactions mediated by immune cell infiltration.Citation[[1]], Citation[[3]], Citation[[4]] The above outlined processes lead to an acute tubular necrosis, a process in which epithelial tubular cells of specific nephron segments death by both necrosis and apoptosis, resulting in sloughing of epithelia into the luminal space creating a physical obstruction in the tubule.Citation[[5]] Post-ischemic acute tubular necrosis is also accompanied by a marked decrease in renal blood flow and glomerular filtration rateCitation[[6]] in which the renal release of vasoactive substances such as angiotensin II, endothelin, platelet activating factor and adenosine play a major role.Citation[[7]], Citation[[8]], Citation[[9]], Citation[[10]]
Acute tubular necrosis, when reversible, is followed by recovery of renal function.Citation[[11]] With regard to the mechanisms involved in recovery from ARF, a new perspective has emerged regarding the relationship between the recovery of tubular integrity and the activity of several growth factors. Polypeptide growth factors are known to regulate a variety of growth and repair processes in kidney.Citation[[12]], Citation[[13]] Several laboratories have investigated a potential role for one or more of these agents in the regenerative process that occurs following ischemia by delineating the pattern of growth factor expression in post-ischemic injury. Among the best studied are epidermal growth factor (EGF),Citation[[14]], Citation[[15]] hepatocyte growth factor (HGF)Citation[[16]] and insulin-like growth factor-I (IGF-I).Citation[[17]] Taken together, these data suggest that EGF and HGF may mediate proliferative events that occur immediately after ischemic insult whereas IGF-I affects later processes involving tissue regeneration and/or differentiation of kidney epithelia. Recently interest has grown in the possible effects of some members of the TGF-β family of growth factors, upon the recovery from acute tubular necrosis.
The TGF-β family of growth factors is comprised of a large number of structurally related proteins, which are as a collective unit capable of regulating a wide range of cellular responses including proliferation, adhesion, differentiation, motility and cell death.Citation[[18]], Citation[[19]] Of specific interest in the recovery from ARF is the TGF-β1 isoform that has been proven to modulate extra cellular matrix (ECM) production both in vitroCitation[[20]], Citation[[21]], Citation[[22]] and in vivo.Citation[[23]], Citation[[24]], Citation[[25]] This review examines the evidence linking TGF-β1 activity to recovery from renal ischemia thereby constructing a hypothesis for the beneficial role of TGF-β1 in the post-ischemic kidney.
TGF-β1: SOME BASIC CONCEPTS
TGF-β1 is produced in most cells as a large latent complex in association with its N-terminal furan cleaved propeptide latency associated peptide (LAP).Citation[[26]] These latent complexes interact with an additional protein termed latent TGF-β binding protein (LTBP) which mediates the secretion and sequestration of the latent form through interactions with the ECM.Citation[[27]]
Thus TGF-β1 is maintained in the ECM in its inactive form awaiting activation in response to appropriate stimuli. This involves cleavage of the LAP pro-peptide either enzymatically or through conformational changes.Citation[[28]], Citation[[29]], Citation[[30]]
Underlining all mechanisms of activation is the observation that certain changes in the local ECM environment can lead to the increased release of active TGF-β1. This is potentially important in renal ischemic injury where necrotic epithelia slough into the tubule revealing a denuded basement membrane leading to degeneration of matrix integrity and increased bioavailability of active TGF-β1.
Once liberated, TGF-β1 binds to its cognate cell surface receptor complex leading to transmission of intracellular signals principally through serine/threonine kinase activation of a chain of proteins collectively known as smads.Citation[[19]] Signaling by these molecules terminates in the stimulation of promoter regions of AGAC nucleotide repeats termed smad binding elements (SBE's).Citation[[31]]
EXAMPLES OF TGF-β ACTIVITY IN ISCHEMIC EVENTS
Platelet mediated cardioprotection following ischemia-reperfusion injury has been attributed to the activity of TGF-β1.Citation[[32]] In this rat model a decrease in active TGF-β1 was noted following global ischemia. However, reperfusion of the heart with washed platelets or aggregated platelet supernatant attenuated myocardial dysfunction and preserved active TGF-β1 levels. Similar results were obtained when hearts were reperfused with recombinant TGF-β1, suggesting that the presence of active TGF-β1 permits the autoinduction and release of active TGF-β1 in the myocardium, which thus acts to preserve myocardial function. It has also been demonstrated that TGF-β1 preserves endothelial function after multiple brief coronary artery ischemia-reperfusion.Citation[[33]] Using isolated epicardial coronary artery rings obtained from dogs subjected to multiple ischemia-reperfusion events, it was noted that in ischemic animals the relaxation response to acetylcholine in pre-constricted rings was much poorer than in samples from control animals. Interestingly though in animals infused with TGF-β1 immediately prior to and following ischemic events the relaxation response was preserved. This demonstrates that TGF-β1 somehow protects endothelial activity in response to ischemia-reperfusion.
Increased TGF-β1 expression has also been shown to occur in brain tissue following ischemic stroke in humans.Citation[[34]] In this study brain samples from ten patients who had suffered ischemic stroke were examined and TGF-β1 expression localized to the border zone of the infarct. This coincides with the observed increase in microvasculature adjacent to the infarct site. TGF-β1 has also been reported to attenuate ischemia induced alterations in cerebrovascular responsiveness with results analogous to those mentioned above in coronary artery segments.Citation[[35]] In this case topical treatment with TGF-β1 (400 ng/mL) before and after cerebral ischemia in piglets significantly improved arteriolar dilatory responses with respect to that observed in ischemic controls.
In response to renal ischemia-reperfusion there have to date only been a limited number of publications examining the role of TGF-β1. In both a mouse model of acute tubular necrosisCitation[[36]] and the analogous events occurring in renal transplants in ratsCitation[[37]] an induction of TGF-β1 has been demonstrated concomitant with rises in various other cytokines and growth factors.
In regenerating rat tubules following renal ischemia levels of whole kidney TGF-β1 were elevated maximally at 24 h post ischemia when compared with sham operated control animals.Citation[[38]] Levels remained elevated throughout the following 14 days with a return to baseline levels by day 28.Citation[[38]] The expression was localized to regenerating cells of the outer medulla. A further study by the same groupCitation[[23]] attempted to examine the effect of this elevation in TGF-β1 with reference to its effect upon ECM related gene expression. Levels of plasminogen activator inhibitor 1 (PAI-1), tissue inhibitor of metalloproteinase 1 (TIMP-1), collagen IV and fibronectin were elevated in ischemic groups when compared with sham operated animals. Peak enhancement occurred between 12 h and 3 days post injury and remained elevated for up to 28 days. PAI-1 mRNA and protein were localized to the regenerating proximal tubule at 3 and 7 days post injury. TIMP-1 expression in contrast was concentrated in the distal portion of the nephron and collecting tubules. Localization of fibronectin was centred upon the regenerating basal lamina while collagen IV was detected in thickened tubulointerstitial. All of these changes were directly related to the activity of TGF-β1 as treatment of a group of animals with an anti-TGF-β1 antibody reversed these changes.
Thus in renal ischemia the current emphasis on TGF-β1 centres upon its role in provision of newly synthesized basal lamina. However this must be coupled with the fact that TGF-β1 can directly steer cells towards growth arrest. Taken together these observations place TGF-β1 in an important position as a “differentiation signal” in recovery of tubular structure and function.
HYPOXIA DIRECTLY ACTS AS A SIGNAL FOR TGF-B1 INDUCTION
Hypoxia is the causative agent in observed ATP depletion found in ischemia-reperfusion models of ARF. The withdrawal of oxygen thus represents in itself a crucial chemical signal that has begun to be investigated as a direct trigger for changes in gene expression.
There have been a number of reports relating in vitro induction of TGF-β1 directly with hypoxia. One study in synovial fibroblastCitation[[39]] has revealed hypoxia appears to directly up-regulate expression of TGF-β1 and concomitantly that of the TGF-β1 target gene vascular endothelial derived growth factor. Studies in dermal fibroblastsCitation[[40]] and the HepG2 cell lineCitation[[41]] have also revealed a marked increase in TGF-β1 mRNA when cells are maintained in hypoxic culture conditions. Such data suggest that hypoxia induced increases in TGF-β1 responses are likely to be biologically significant in response to ischemic events.
The activity of inducible nitric oxide synthase (iNOS) a gene thought to play a cell-damaging role in the post-ischemic kidneyCitation[[42]] has been described more fully with reference to the mechanisms of gene activation post-ischemia. Expression of iNOS is up-regulated following renal ischemia.Citation[[43]] Its activation in IFN-gamma stimulated macrophages has been demonstrated to possess the functional requirement of a hypoxia response element in its promoter to facilitate induction by the iron chelator Desferrioxamine.Citation[[44]] This draws a mechanistic link between the hypoxia-induced changes in gene expression observed in mammalian cells and in the yeast Saccharomyces cerevisiae where the iron sensitive induction of gene expression has been well-delineated.Citation[[45]] This mechanism is reliant on the cells capacity to sense hypoxia through an inability to maintain heme biosynthesis. The resultant drop in heme levels restricts the activity of the protein ROX-1 a trans-repressor of genes associated with the hypoxic state, thus leading to hypoxia response genes becoming de-repressed, allowing the cell to mount an active response to the drop in oxygen supply. The iron sensitive mechanism of iNOS regulation thus demonstrates a well-conserved mechanism, which may well, be operative in the oxygen-regulated activity of TGF-β1.
TGF-β1 AUTOINDUCTION
The capacity of active TGF-β1 to propagate induction from its own locus demonstrates the great potential for activation possessed by this system. The AP-1 site has been demonstrated as the region of the TGF-β1 promoter stimulated in autoinductionCitation[[46]] presumably by a signaling mechanism distinct from those mediated by smads or through an intracellular synapse between the pathways.
INDUCTION OF TGF-β1 IN ARF: ROLE OF ANGIOTENSIN II
Loss of activity of the Na+/K+ATPase on the basolateral side of the proximal tubular epithelia results in urinary sodium wasting and a decrease in extracellular volume. The decrease in renal perfusion that accompanies acute tubular necrosis results in activation of the Renin–Angiotensin system in an attempt to restore extracellular volume sodium balance and renal perfusion.Citation[[47]] This “axis” operates as a closed, self-regulating system that rapidly shuts itself down in response to an improvement in blood pressure, renal perfusion and sodium balance.
Another action of angiotensin II however is to induce expression of growth factors important in recovery from ischemic injury. While on the one hand its induction by platelet derived growth factor (PDGF) induces a proliferative response,Citation[[48]] it also induces TGF-β1 expression which is an anti-proliferative pro-differentiation signal,Citation[[49]] thus providing a dual signal for both repopulation and functional differentiation of the tubule. The link between angiotensin II and TGF-β1 induction has been proven in vitro in rat mesangial cells to rely upon the activation of protein kinase C.Citation[[50]] This induction occurs in parallel with rises in the expression of extracellular matrix components such as collagen IV, the primary collagen of the basal lamina and a gene known to be directly up-regulated by TGF-β1 signaling.Citation[[51]] Inhibition of proliferation and induction of hypertrophy by the AngII/ TGF-β1 axis has also been demonstrated in vitro in experiments where cells maintained in serum-free media became hypertrophic through a TGF-β1 mediated mechanism.Citation[[49]]
The majority of work to date however linking AngII and TGF-β1 has centered upon the potentially damaging role of TGF-β1 in ECM accumulation in models of interstitial fibrosis. In these studies the administration of angiotensin converting enzyme (ACE) inhibitorsCitation[[52]], Citation[[53]] has been employed in an attempt to dampen the expression of TGF-β1 and thus prevent excessive ECM accumulation, i.e. block the fibrotic process.
However it has been reported that inhibition of ACE activity can in itself lead to ARF in patients without renal artery stenosis.Citation[[54]], Citation[[55]] A further report has shown inhibition of ACE activity in cases of multiple myeloma related ARF can precipitate the level of injury,Citation[[56]] thus the positive role of AngII in ischemic pathologies cannot be ignored.
TGF-β1-INDUCED UP-REGULATION OF ECM-RELATED GENES IS CRUCIAL IN STRUCTURAL/FUNCTIONAL RECOVERY OF THE TUBULE
Reproliferating epithelia in the post-ischemic tubule require an intact basal lamina in order to achieve a number of functions. Firstly, as a prerequisite for cell spreading and repopulation of the tubule and secondly, in order to adopt correct functional polarity and thus differentiate into epithelia characterized by bi-directional transport capacity and a defined brush border.
Thus regeneration of the basal lamina requires a rebalance in the expression of genes involved in maintenance of the equilibrium of matrix turnover. To this effect one of the clearest actions of TGF-β1 centres upon the up-regulation of genes which code for both components of the ECM and equally for proteinase inhibitory proteins.
Collagen IV represents the principal building block of the basal lamina and a number of reports have shown that TGF-β1 induces up-regulation of collagen IV production. Parallel studies in the NIH-3T3 cell line, (a fibroblast like cell line from murine embryo) and in cultures of NRK-49F normal rat kidney cells have shown that in response to maximal doses of 5 and 10 ng/ml TGF-β1 respectively, the maximal induction of mRNA for collagen IV occurred between 12 and 18 h post-treatment.Citation[[20]] Activation of collagen IV in mesangial cells in response to TGF-β1 has also been shown to reach a maximal level within the first 24 h post-treatment.Citation[[21]]
While acting to up-regulate ECM component synthesis, TGF-β1 has also been shown to up-regulate the expression of factors such as PAI-1 and TIMP-1Citation[[23]] which operate to inhibit ECM degradation.
Thus, in the examples above listed, it can be seen that TGF-β1 induced by various stimuli, acts to up-regulate the expression of genes implicated in the net gain of ECM, a key factor in both the spreading and differentiation of tubular epithelia and subsequent restoration of tubular structure and function.
NEGATIVE FEEDBACK CONTROL OF TGF-β1 SIGNALING: THE DIFFERENCE BETWEEN RECOVERY AND FIBROSIS
The activity of TGF-β1 and its primary effect upon the expression of ECM related genes requires to be adequately controlled in order to prevent progression from ischemia-reperfusion injury to fibrosis. In renal ischemia we can point to a number of factors which operate to ensure that such a progression does not occur. Firstly, consider the differences in the role of TGF-β1 in renal ischemia and renal fibrosis. In the first case evidence points to a beneficial role, while in the second emphasis is placed on the crucial and direct role of TGF-β1 in the chronic progression of the fibrotic process. In animal models of both pathologies, the starting point is a direct stress or injury to the kidney. However, at this point the pathologies diverge as in the case of ischemia-reperfusion injury the kidney is allowed to recuperate while in fibrosis the damage increases with time, a result of the chronic nature of the injury inflicted. Both regimes induce TGF-β1 expression but in the case of ischemia-reperfusion injury this induction is transient and responsive to improvements in kidney structure and function whereas in fibrosis TGF-β1 is believed to become the perpetuating factor in the tissue damage. Thus, in ischemia-reperfusion there must exist specific mechanisms that down-regulate TGF-β1 signaling.
TGF-β1 AUTOINIHIBITION WORKS AT BOTH THE INTRACELLULAR AND EXTRACELLULAR LEVEL
As discussed previously the TGF-β1 signal is primarily transmitted through smad activation and consequent up-regulation of target genes through smad complex interactions with specific promoter sites termed SBE's. Interestingly, one such gene codes for the inhibitory smad7. Transfection of this gene to mammalian cells blocks TGF-β1 responsiveness.Citation[[57]] The mechanism by which this occurs centres upon the competition of smad7 with smad2/3 for binding to the activated TGF-β1 receptor complex.
The activity of membrane bound, TGF-β1 receptor complex associated endoglin, may represent a mechanism by which TGF-β1 signaling can be blocked or modulated at the point of binding to receptors at the cell surface. Endoglin is the candidate gene for the vascular disease hereditary hemorrhagic telangiectasiaCitation[[58]] and its expression has been demonstrated in endothelial cellsCitation[[59]] macrophages,Citation[[60]] erythroid precursors,Citation[[61]] syncytiotrophoblastsCitation[[62]] mesangial cellsCitation[[63]] and vascular smooth muscle cells.Citation[[64]] It has been shown to form heteromeric complexes with the TGF-β1 receptor complex by co-immunoprecipitation assay.Citation[[65]]
This interaction acts to at least modulate the response to TGF-β1 as witnessed in the observation that TGF-β1 induction of fibronectin and PECAM-1 expression are diminished in the U-937 monocyte cell line transfected with endoglin.Citation[[66]] In the same report overexpression of endoglin also inhibited TGF-β1 induced growth arrest and inhibition of c-myc expression. Moreover in non-transfected cells of this lineage, treatment with TGF-β1 induced up-regulated mRNA expression of endoglin proving it to be a TGF-β1 responsive gene possibly involved in negative feedback mechanisms. Endoglin also regulates TGF-β1-induced mouse fibroblast migration and adhesionCitation[[67]] and ECM synthesis (Rodriguez-Barbero and López-Novoa, Unpublished results).
Given that endoglin harbors an extracellular RGD motif, an essential functional feature of some extracellular matrix proteins such as fibronectin, the hypothetical role of endoglin in post-ischemic TGF-β1 signal modulation can be further guessed at. The possibility that endoglin plays an important role in cell–cell contact and that these associations may alter the signaling capacity of adjacent TGF-β1 receptor complexes remains an intriguing possibility. The acidic phosphoprotein osteopontin is another RGD bearing non matrix component which has been shown to be up-regulated in the proximal and distal tubule segments of the post-ischemic nephron.Citation[[68]] We have recently shown that endoglin is up-regulated in kidneys with chronic renal failure,Citation[[69]] unilateral ureteral ligationCitation[[70]] and renal ischemia (Docherty, Eleno, and López-Novoa, unpublished data). Thus, there appears to be sufficient reason in investigating the role that endoglin may play in the modulation of TGF-β1 signaling in the recovering kidney.
OTHER MECHANISMS WHICH MAY ACT TO LIMIT TGF-β1 ACTIVITY POST-ISCHEMIA
Improvements in renal blood flow and nephron function could effectively reduce TGF-β1 signaling. Shutdown of the renin–angiotensin signaling system in response to improved electrolyte control would give rise to a reduction in the production of AngII. This in theory would operate to lower the rate of TGF-β1 induction providing a physiological sensor linking TGF-β1 activity to functional competence of the kidney.
An intact basal lamina would lead to increased ECM sequestration of TGF-β1. In this instance restoration of tubular structure and the renewed integrity of the basal lamina would ensure the increased sequestration of nascent TGF-β1 in its latent form anchored in the ECM.
If TGF-β1 like the iNOS gene harbors regulatory regions influenced by hypoxia then in a situation analogous to that seen in the yeast Saccharomyces Cerevisiae these responses should again become repressed in the oxygenated environment.
CONCLUSION
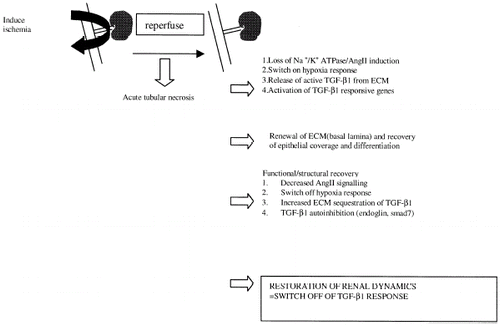
In this hypothesis and review paper we have attempted to illustrate the potential importance of TGF-β1 signaling in the recovery from acute renal failure. Evidence linking its expression in the post-ischemic state to its potential functional significance has been presented. The suggestion that TGF-β1 signaling is sensitive to oxygen deprivation, loss of matrix integrity and organ dysfunction place it in a potentially important position in the regenerative capacity of the post-ischemic kidney. Moreover, the fine control of its activity by a number of parameters fundamental in the recuperation of normal kidney function suggest that in the case of renal ischemic injury we ought to consider TGF-β1 activity in a positive light. presents a hypothetical mode of action for TGF-β1 in recovery from acute renal failure resultant from ischemia. This area of study may prove important in the understanding of growth factor activity in the regenerating kidney, ultimately leading to the design of new strategies to improve post-ischemic renal function.
Figure 1. Hypothetical model of TGF-β1 activity in recovery from renal ischemia.
ACKNOWLEDGMENT
N.G.D. is supported by a foreign study award from the John Mather Trust (Glasgow, Scotland) and by the Instituto Reina Sofia de Investigación nefrológica. Studies from our own laboratory have been supported by a grant from CICYT (SAF 0095/98).
REFERENCES
- Stein J.H., Lifschitz M.D., Barnes L.D. Current Concepts on the Pathophysiology of Acute Renal Failure. Am. J. Physiol. 1978; 234: F171–F181
- Macías-Núñez J.F., López-Novoa J.M., Martínez-Maldonado M. Acute Renal Failure in the Aged. Sem. Nephrol. 1996; 16: 330–338
- Hostetter T.H., Brenner B.M. Renal Circulatory and Nephron Function in Experimental ARF. Acute Renal Failure, B.M. Brenner, J.M. Lazarus. Churchill, New York 1988; 67–89
- García-Criado F.J., Eleno N., Valdunciel J.J., Santos F., Reverte M., Lozano F.S., Ludeña M.D., Gómez-Alonso A., López-Novoa J.M. Protective Effect of Exogenous Nitric Oxide on the Renal Function and Inflammatory Response in a Model of Ischemia-Reperfusion. Transplantation 1998; 66: 82–990
- Venkatachalam M.A., Bernard B.D., Donohoe J.F., Levinsky N.G. Ischemic Damage and Repair in the Rat Proximal Tubule: Differences Among the S1, S2 and S3 Segments. Kidney Int. 1978; 14: 31
- Daugharty T.M., Brenner B.M. Reversible Hemodynamic Defect in Glomerular Filtration Rate After Ischemic Injury. Am. J. Physiol. 1975; 228: 1436
- Mason J., Kain H., Shiigai T., Welsch J. The Early Phase of Experimental Acute Renal Failure. V. The Influence of Suppressing the Renin–Angiotensin System. Pflügers Arch. 1979; 380: 233
- López-Novoa J.M. Potential Role of Platelet Activating Factor in Acute Renal Failure. Kidney Int. 1999; 55: 1672–1682
- López-Farré A., Gomez-Garre D.N., Bernabeu F., López-Novoa J.M. A Role for Endothelin in the Maintenance of Postischemic Acute Renal Failure. J. Physiol. 1991; 444: 513–522
- Gould J., Morton M.J., Sivaprasadarao A., Bowmer C.J., Yates M.S. Renal Adenosine A1 Receptor Binding Characteristics and mRNA Levels During the Development of Acute Renal Failure in the Rat. Br. J. Pharmacol. 1997; 120: 947–953
- Finn W.F. Enhanced Recovery from Postischemic Acute Renal Failure. Micropuncture studies in the rat. Circ. Res. 1980; 46: 440–448
- Schena F.P. Role of Growth Factors in Acute Renal Failure. Kidney Int. Suppl. 1998; 66 S11–S15
- Hammerman M.R. Potential Role of Growth Factors in the Prophylaxis and Treatment of Acute Renal Failure. Kidney Int. 1998; 64: S19–S22
- Nouwen E.J., Verstrepen W.A., De Broe M.E. Epidermal Growth Factor in Acute Renal Failure. Ren. Fail. 1994; 16: 49–60
- Schaudies R.P., Nonclercq D., Nelson L., Toubeau G., Zanen J., Heuson-Stiennon J.A., Laurent G. Endogenous EGF as a Potential Renotrophic Factor in Ischemia-Induced Acute Renal Failure. Am. J. Physiol. 1993; 265: F425–F434
- Sugimura K., Goto T., Tsuchida K., Takemoto Y., Kim T., Kishimoto T. Production and Activation of Hepatocyte Growth Factor in Acute Renal Failure. Ren. Fail. 2001; 23: 597–603
- Tsao T., Wang J., Fervenza F.C., Vu T.H., Jin I.H., Hoffman A.R., Rabkin R. Renal Growth Hormone–Insulin-Like Growth Factor-I System in Acute Renal Failure. Kidney Int. 1995; 47: 1658–1668
- Attisano L., Wrana J.L., Lopez-Casillas F., Massague J. TGF-Beta Receptors and Actions. Biochim. Biophys. Acta. 1994; 26: 71–80
- Heldin C.H., Miyazono P., Dijke P. TGF-β Signaling from Cell Membrane to Nucleus Through Smad Proteins. Nature 1997; 390: 465–471
- Mielder D., Grande J., Killen P. Transforming Growth Factor-Beta 1 Induces Collagen IV Gene Expression in NIH-3T3 Cells. Lab. Invest. 1993; 69: 387–395
- Suzuki S., Ebihara I., Tomino Y., Kode H. Transcriptional Activation of Matrix Genes by TGF-β1 in mesangial cells. Exp. Nephrol. 1993; 1: 229–237
- Kuncio G.S., Alvarez S., Li P., Killen P. Transforming Growth Factor β modulation of the alpha1 (IV) collagen gene in murine PT cells. Am. J. Physiol. 1996; 271: F120–F125
- Basile D.P., Daniel R.M., Hammerman M.R. Extracellular Matrix Related Genes in Kidney After Ischemic Injury: Potential Role for TGF-β in repair. Am. J. Physiol. 1998; 275: F894–F903
- Sun Y., Zhang J.Q., Zhong J., Ramires F.J. Angiotensin II, Transforming Growth Factor β and Repair in the Infarcted Heart. J. Mol. Cell. Cardiol. 1998; 30: 559–569
- Mao J., Jui H., Zhao S., Junard A., Scammel-La Fleur T., Dixon I.M. Elevation of Expression of Smads 2, 3 and 4, Decorin and TGF-B in the Chronic Phase of Myocardial Infarct Scar Healing. J. Mol. Cell. Cardiol. 1999; 31: 667–678
- Miyazono K., Heldin C.H. Latent High Molecular Weight Complex of TGF-β1. J.Biol.Chem. 989; 263: 6407–6415
- Saharinen J., Taipale J., Keski-Oja B. Association of the Small Latent Transforming Growth Factor β with an Eight Cysteine Repeat of its Binding Protein LTBP-1. EMBO. J. 1996; 15: 245–253
- Shultz-Cherry S., Chen H., Mosher D.F., Misenheimer T.M., Krutzch H.C., Roberts D.D., Murphy Ullrich J.E. Regulation of Transforming Growth Factor Beta Activation Through Discrete Sequences of Thrombospondin. J. Biol. Chem. 1995; 270: 7304–7310
- Rowley D.A., Becken E.T., Stach R.M. Autoantibodies Produced by Young Ipr Mice Carry Transforming Growth Factor Beta and Suppress Cytotoxic Lymphocyte Responses. J. Exp. Med. 1995; 181: 1875–1880
- Roberts A.B. Molecular and Cell Biology of TGF-β. Miner Electrolyte Metab 1998; 24: 111–119
- Raftery L.A., Sutherland D.J. TGF-Beta Family Signal Transduction in Drosophila Development: from Mad to Smads. Dev. Biol. 1999; 210: 251–268
- Mehta J.L., Yang B.C., Strates B.S., Mehta P. Role of TGF-β1 in Platelet-Mediated Cardioprotection During Ischemia-Reperfusion in Isolated Rat Hearts. Growth Factors 1999; 16: 179–190
- Kenny D., Coughlan M.G., Pagel P.S., Kampine J.P., Warltier D.C. Transforming Growth Factor Beta 1 Preserves Endothelial Function After Multiple Brief Coronary Artery Occlusions and Reperfusion. Am. Heart. J. 1994; 127: 1456–1461
- Krupinski J., Kumar P., Kumar S., Kaluza J. Increased Expression of TGF-β1 in Brain Tissue After Ischemic Stroke in Humans. Stroke 1996; 27: 852–857
- Armstead W.M., Mirro R., Zuckerman S.L., Shibata M., Leffler C.W. Transforming Growth Factor Beta Attenuates Ischemia Induced Alterations in Cerebrovascular Reponses. Am. J. Physiol. 1993; 264: H381–H385
- Goes N., Urmson J., Ramassar V., Halloran P.F. Ischemic Acute Tubular Necrosis Induces an Extensive Local Cytokine Response. Evidence for Induction of Interferon Gamma, Transforming Growth Factor Beta-1, Granulocyte-Macrophage Colony Stimulating Factor, Interleukin-2 and Interleukin-10. Transplantation 1995; 59: 565–572
- Pual L.C., Saito K., Davidoff A., Benediktsson H. Growth Factor Transcripts in Rat Renal Transplants. Am. J. Kidney Dis. 1996; 28: 44–50
- Basile D.P., Rovak J.M., Martin D.R., Hammerman M.R. Increased Transforming Growth Factor-beta 1 Expression in Regenerating Rat Renal Tubules Following Ischemic Injury. Am. J. Physiol. 1996; 270: F500–F509
- Berse B., Hunt J.A., Diegel R.J., Morganelli P., Yeo K., Brown F., Fava R.A. Hypoxia Augments Cytokine (Transforming Growth Factor-Beta (TGF-β) and IL-1) Induced Vascular Endothelial Growth Factor Secretion by Human Synovial Fibroblasts. Clin. Exp. Immunol. 1999; 115: 176–182
- Patel B., Khaliq A., Jarvis-Evans J., McLeod D., Mackness M., Boulton M. Oxygen Regulation of TGF-β1 mRNA in Human Hepatoma (HepG2) Cells. Biochem. Mol. Biol. Int. 1994; 34: 639–644
- Falanga V., Qian S.W., Daniepour D., Katz M.H., Roberts A.B., Sporn M.B. Hypoxia Up-Regulates the Synthesis of TGF-β1 by Human Dermal Fibroblasts. J. Invest. Dermatol. 1991; 97: 634–637
- Ling H., Edelstein C., Gengaro P., Meng X., Lucia S., Knotek M., Wansiripaisan A., Shi Y., Schrier R. Attenuation of Renal Ischemia-Reperfusion Injury in Inducible Nitric Oxide Synthase Knockout Mice. Am. J. Physiol. 1999; 277: F383–F390
- Valdivielso J.M., Crespo C., Alonso J.R., Martínez-Salgado C., Eleno N., Arévalo M., Pérez-Barriocanal F., López-Novoa J.M. Renal Ischemia in the Rat Stimulates Glomerular Nitric Oxide Synthesis. Am. J. Physiol. 2001; 280: R771–R779
- Melillo G., Taylor L.S., Brooks A., Musso T., Cox G.W., Varesio L. Functional Requirement of the Hypoxia Response Element in the Activation of the Inducible Nitric Oxide Synthase Promoter by the Iron Chelator Desferroxamine. J. Biol. Chem. 1997; 18: 12236–12243
- Zitomer R.S., Carrico P., Deckert J. Regulation of Hypoxic Gene Expression in Yeast. Kidney Int. 1997; 51: 507–513
- Hahn A.W.A., Resink T.J., Bernhardt J., Ferracin F., Buhler F.R. Stimulation of Autocrine PDGF AA-Homodimer and Transforming growth Factor Beta-1 in Vascular Smooth Muscle Cells. Biochem. Biophy. Res. Commun. 1991; 178: 1451–1458
- Laragh J.H. The Renin System in Hypertension: A Research Journey. Angiotensin Converting Enzyme Inhibitors: Mechanism of Action and Clinical Applications, Z.P Horovitz. Urdan & Shwarzenberg, Baltimore-Munich 1981; 403–436
- Nafrilan A.J., Pratt R.E., Dzau V.J. Induction of PDGF A-Chain and c-myc gene Expressions by Angiotensin II in Cultured Rat Vascular Smooth Muscle Cells. J. Clin. Invest. 1991; 83: 1419–1424
- Gibbons G.H., Pratt R.E., Dzau V.J. Vascular Smooth Muscle Cell Hypertrophy Versus Hyperplasia: Autocrine Transforming Growth Factor Beta-1 Determines Growth Responses to Angiotensin II. J. Clin. Invest. 1992; 90: 456–461
- Weiss R.H., Ramirez A. TGF-Beta and Angiotensin II Induced Mesangial Matrix Protein Secretion is Mediated by Protein Kinase C. Nephrol. Dial. Transplant. 1998; 13: 2804–2813
- Motojima M., Kakuchi J., Yoshioka T. Association of TGF-Beta Signaling in Angiotensin II induced PAI-1 mRNA Up-Regulation in Mesangial Cells:Role of PKC. Bichim. Biophys. Acta. 1999; 449: 217–226
- Anderson S., Rennke H.G., Brenner B.M. Therapeutic Advantage of Converting Enzyme Inhibitors in Arresting Progressive Renal Disease Associated with Systemic Hypertension in the Rat. J. Clin. Invest. 1986; 77: 1993–2000
- Zoja C., Abbate M., Corna D., Cattanio M., Donadelli R., Bruzzi I., Oldroyd S., Benigni A., Remuzzi G. Pharmacologic Control of Angiotensin II Ameliorates Renal Disease while Reducing Renal TGF-β in Experimental Mesangioproliferative Glomerulonephritis. Am. J. Kidney. Dis. 1998; 31: 453–463
- Bridoux F., Hazzan M., Pallot J.L., Fleury D., Lemaitre V., Kleinknecht D., Vanhille P. Acute Renal Failure After the Use of Angiotensin-Converting Enzyme Inhibitors in Patients Without Renal Artery Stenosis. Nephrol. Dial. Transplant. 1992; 7: 100–104
- Bitar R., Flores O., Reverte M., López-Novoa J.M., Macias J.F. Beneficial Effect of Verapamil Added to Chronic Ace Inhibitor Treatment on Renal Function in Hypertensive Elderly Patients. Int Urol. Nephrol. 2000; 32: 165–169
- Rabb H., Gunasekaron H., Gunasekaron S., Saba S.R. Acute Renal Failure from Multiple Myeloma Precipitated by ACE Inhibitors. Am. J. Kid. Dis. 1999; 33: E5
- Alsuhito N., Mozhgan A., Moren A., Nakayama T., Chritian J., Rainer H., SusumuI K., Masahiro K., Nils E.H., Heldin C.L., ten Dijke P. Identification of Smad7 a TGF-β Inducible Antagonist of TGF-β signaling. Nature 1997; 389: 631–635
- McAllister K.A., Grogg K.M., Johnson D.W., Gallione C.J., Baldwin M.A., Jackson C.E., Helmbold E.A., Markel D.S., McKinnon W.C., Murrell J. Endoglin, a TGF-Beta Binding Protein of Endothelial Cells, is the Gene for Hereditary Hemorrhagic Telangiectasia Type 1. Nat. Genet. 1994; 8: 345–351
- Chiefet S., Bellon T., Cales C., Vera S., Bernabeu C., Massague J., Letarte M. Endoglin is a Component of the Transforming Growth Factor Beta Receptor System in Human Endothelial Cells. J. Biol. Chem. 1992; 267: 19027–19030
- Lastres P., Bellon T., Cabanas C., Sanchez-Madrid F., Acevedo A., Gougos A., Letarte M., Bernabeu C. Regulated Expression on Human Macrophages of Endoglin an Arg-Gly-Asp-Containing Surface Antigen. Eur. J. Immunol. 1992; 22: 393–397
- Pierelli L., Scambia G., Bonanno G., Rutella S., Puggioni P., Battaglia A., Mozzetti S., Marone M., Menichella G., Rumi C., Mancuso S., Leone G. CD34+/CD105+ Cells are Enriched in Primitive Circulating Progenitors Residing in the G0 Phase of the Cell Cycle and Contain all Bone Marrow and Cord Blood CD34+/CD38low/-precursors. Br. J. Hematol. 2000; 108: 610–620
- Gougos A., St Jaques S., Greaves A., O'Connell P.J., d'Apice A.J., Buhring H.J., Bernabeu C., van Mourik J.A., Letarte M. Identification of Distinct Epitopes of Endoglin, an RGD-Containing Glycoprotein of Endothelial Cells, Leukemic Cells and Syncytiotrophoblasts. Int. Immun. 1992; 4: 83–92
- Rodríguez-Barbero A., Obreo J., Eleno N., Rodríguez-Peña A., Düwel A., Jerkic M., Bernabéu C., López-Novoa J.M. Expression of Endoglin mRNA and Protein in Human and Rat Mesangial Cells and its Up-Regulation by TGF-β1. Biochem. Biophys. Res. Commun. 2001; 282: 142–147
- Adam P.J., Clesham G.J., Weissberg P.L. Expression of Endoglin mRNA and Protein in Human Vascular Smooth Muscle Cells. Biochem. Biophys. Res. Commun. 1998; 247: 33–37
- Hidetoshi Y., Hidenori I., Grimsby S., Moren A., ten Dijke P., Miyazono K. Endoglin forms a Heteromeric Complex with the Signaling Receptors for Transforming Growth Factor β. J. Biol. Chem. 1994; 269: 1995–2001
- Lastres P., Letamendia A., Hongwei Z., Rius C., Almendro N., Raab U., López L., Langa C., Fabra A., Letarte M., Bernabeu C. Endoglin Modulates Cellular Responses to TGF-β1. J. Cell. Biol. 1996; 3: 1109–1118
- Guerrero-Esteo M., Lastres P., Letamendía A., Pérez-Alvarez M.J., Langa C., López L.A., Fabra A., García-Pardo A., Vera S., Letarte M., Bernabeú C. Endoglin Over-Expression Modulates Cellular Morphology, Migration, and Adhesion of Mouse Fibroblasts. Eur. J. Cell. Biol. 1999; 78: 614–623
- Persy V.P., Verstrepen W.A., Ysebaert D.K., De Greef K.F., De Broe M.E. Differences in Osteopontin Up-Regulation Between Proximal and Distal Tubules after Renal Ischemia/Reperfusion. Kidney Int. 1999; 56: 601–611
- Rodriguez-Peña A., Prieto M., Duwel A., Rivas J.V., Eleno N., Perez-Barriocanal F., Arevalo M., Smith J.D., Vary C.P., Bernabeu C., Lopez-Novoa J.M. Up-Regulation of Endoglin, a TGF-Beta-Binding Protein, in Rats with Experimental Renal Fibrosis Induced by Renal Mass Reduction. Nephrol. Dial. Transplant. 2001; 16: 34–39
- Arevalo M., Perez-Barriocanal F., Eleno N., Rodriguez-Peña A., Lopez-Novoa J.M. Up-Regulation of Endoglin Induced by Unilateral Ureteral Ligation in Rats. J. Am. Soc. Nephrol. 2000; 11: A3231