Abstract
Background. There is now good evidence to suggest that cytochrome P450 (CYP450) may act as an iron‐donating catalyst for the production of hydroxyl ion (OH·), which contributes to proximal tubular cell injury. However, it remains unclear which isoform of CYP450 is involved in this process. Cytochrome P4502E1 (CYP2E1) is a highly labile isoform which is not only involved in free radical generation, but has also been shown to be a source of iron in cisplatin‐induced renal injury. This study investigates the role of CYP2E1 in the proximal tubular cell injury induced by hydrogen peroxide (H2O2). Methods. Porcine proximal tubular cells (LLC‐PK1) were incubated with H2O2 (1 mM) for 4 h in the presence or absence of 0.1 mM of two CYP2E1 inhibitors; diallyl sulfide (DAS), or disulfiram (DSF), desferrioxamine (DFO) (0.1–0.4 mM), or catalase (CT) (78, 150, 300 U/mL). Cell death was determined by measuring LDH release. CYP2E1 activity was determined by p‐nitrophenol hydroxylation after 2 h incubation with H2O2. Results. Exposure of LLC‐PK1 to H2O2 significantly increased cell death. CT, DFO, DAS and DSF significantly reduced H2O2‐mediated cell death. Incubation with H2O2 increased CYP2E1 activation in time‐ and dose‐dependent manner, which was significantly reduced by CT, DFO, DAS and DSF. Conclusion. We propose that CYP2E1 activation occurs possibly due to OH· and contributes to H2O2‐mediated LLC‐PK1 cell necrosis by acting as a source of iron and perpetuating the generation of OH· via the Fenton reaction. Inhibition of CYP2E1 may be a novel approach for the prevention of tubular injury caused by oxidative stress.
Introduction
Reactive oxygen species [ROS] are important mediators of ischemic, toxic and immune‐based tissue injury.Citation[1], Citation[2], Citation[3], Citation[4] Acute tubular necrosis is a striking feature of ischemic acute renal failure (ARF) and it has been shown that the proximal tubular (PT) epithelial cells are the major sites of injury.Citation[5], Citation[6] ROS produce cellular injury via several mechanisms including peroxidation of membrane lipids, protein denaturation, and DNA damage.Citation[7] Moreover, inhibition of ROS generation can protect PT cells against oxidative stress by attenuation of the activation of DNA repair enzyme, poly (ADP‐ribose) polymerase.Citation[2], Citation[4]
The ability of oxidants to induce loss of cell viability in renal tubular cells has been studied extensively.Citation[5], Citation[8], Citation[9] Superoxide anion (O2· −) and hydrogen peroxide (H2O2) appear to be the primary species generated. These species generate more reactive species, including the highly reactive hydroxyl radical (OH·) via a reaction in which iron plays a catalytic role, commonly referred to as the “metal‐catalyzed Fenton reaction.”Citation[6] H2O2 appear to be the major reactive oxygen metabolite responsible for tissue injury, presumably because it is both stable and able to freely penetrate cellular targets and produce toxicity via the generation of other ROS.Citation[1], Citation[8] H2O2 has been implicated in the pathogenesis of acute renal injury in models of rhabdomyolysis, ischemia/reperfusion (I/R), and in toxic nephropathy in which the main site of injury is at the PT level.Citation[2], Citation[4], Citation[9]
It has been shown that iron chelates reduce cell injury in PT cells on exposure to H2O2.Citation[2], Citation[8] The source of the iron, which participates in H2O2‐mediated cellular injury, is not fully known. Several observations suggest that the source might be the extracellular heme protein such as hemoglobin and myoglobin, whereas others suggest that the iron storage protein ferritin and iron‐rich mitochondria are the principle sources of free iron.Citation[10], Citation[11], Citation[12] Several recent studies indicate that the iron rich enzyme, cytochrome P450 (CYP450) may also serve as a source of iron in I/R and H2O2‐induced PT cell injury.Citation[9], Citation[13] However, the exact isoform of CYP450 involved in these reactions is unknown. It was shown recently that the cytochrome P4502E1 (CYP2E1) isoform served as a significant source of catalytic iron in cisplatin‐induced nephrotoxicity.Citation[14]
The CYP2E1 isoenzyme is expressed mainly in liver and possibly in extrahepatic tissues such as kidney, lung, and lymphocytes.Citation[15] CYP2E1 is induced by various agents such as ethanol, acetone, isoniazid, pyridine, and isopropanol. It also can be induced by conditions such as starvation and diabetes mellitus.Citation[15] It is involved in the metabolism of low‐molecular weight compounds such as ethanol, vinyl chloride, and carbon tetrachloride.Citation[16], Citation[17], Citation[18] It has been shown previously that CYP2E1 induction is involved in hepatotoxicity subsequent to ROS generation.Citation[19], Citation[20], Citation[21] However, its role in oxidant induced PT injury has not been previously shown. Therefore, this study was undertaken to evaluate the role of this CYP450 isoform in oxidant induced PT cell injury and the possibility of attenuating the cell injury by inhibiting this enzyme. We chose a simple previously employed model that utilizes H2O2 as the agent of injury and PT epithelial (LLC‐PK1) cells as the target.
Materials and Methods
Chemicals
The cell culture solutions and media, catalase, disulfiram (DSF), diallyl sulfide (DAS), dimethyl sulfoxide (DMSO), 4‐nitrocatechol 10 M HCl, potassium phosphate, potassium chloride, Dulbecco's modified Eagle's medium (DMEM), and H2O2 (30.7%) were purchased from Sigma Chemical Co. (Poole, Dorset, UK). Sterile culture flasks and plates (6 and 96 well) were obtained from Corning Incorporated (New York, USA). Ethyl alcohol, Methyl alcohol, Isopropyl alcohol, and agarose gel were obtained from BDH (Poole, Dorset, UK).
Cell Culture
LLC‐PK1, which are immortalized renal PT epithelial cells derived from pig, were obtained from European Collection of Cell Culture, (Salisbury, Wiltshire, UK). Cells were routinely grown in 75 cm2 flasks in DMEM in a humidified atmosphere of 95% air and 5% CO2 and sub‐cultured at 3–4 days intervals.Citation[22] DMEM was supplemented with 10% fetal calf serum (FCS) and 1% [v/v] antibiotics (penicillin and streptomycin). Cells were used for experiments within eight passages to ensure cell line stability. For LDH assay, cells were cultured in 6‐well plated at a density of 4 × 104 cells/well in a final volume of 2 ml. For CYP2E1 activity, LLC‐PK1 cells were plated in 75 cm2 flasks at a density of 4 × 106 cells/flask in volumes of 12 mL of the medium. In all experiments, cells were studied as a 70–80% confluent monolayer, and incubated in a humidified atmosphere at 37°C and 5% CO2.
Cell Treatment
A stock of 100 mM H2O2 was prepared freshly in DMEM medium. In all experiments, cells were treated with a final concentration of 1 mM H2O2. For the free radical scavenger experiments, 78, 150, and 300 U/mL of CT, was added along with H2O2 (1 mM) to assess protective effects.Citation[23] The CYP2E1 inhibitors, DAS and DSF were dissolved in DMSO (99.9%) and in two separate experiments, cells were pre‐treated with 0.1 mM of the inhibitors for 2 h followed by 1 mM of H2O2.Citation[13] Cells were pre‐treated with DFO (0.1–0.4 mM) dissolved in DMEM medium for 30 min prior to H2O2 treatment.Citation[19]
Measurement of Lactate Dehydrogenase (LDH) Release
Cell death was assessed by measuring the index of LDH release in the supernatant leakage from LLC‐PK1 cells. LDH release was measured using the cytotoxicity kit obtained from Roche (Lewes, Sussex, UK). Briefly, 100 µL of media was mixed with the reagent and incubated at room temperature for 5–10 min. The absorbance was measured at 490 nm using a microplate reader (MRX II Dynex technologies, Billingshurst, Sussex, UK. The results are expressed as a percentage of the positive control, which was cell incubated with 2% [v/v] of Triton X100 for 10 min.
Preparation of Microsomes from LLC‐PK1 Cells
Preparation of microsomes was conducted as described previously. Briefly, 3–4 × 106 cells were lysed in ice buffer A [0.25 M potassium phosphate, 0.15 M potassium chloride, 0.25 M sucrose, 1 mM EDTA, pH 7.5]. The lysate was sonicated for 5 min, using a Jencons‐plus sonicator (Leighton Buzzard, Bedfordshire, UK) and centrifuged at 12,000 rpm for 15 min at 4°C. The supernatant was spun a further 60 min at 38,000 rpm. The resulting microsomal pellet was re‐suspended in 100 µL of buffer B [0.1 M sodium phosphate, 2 mM MgCl2, pH 7.4].Citation[24] The protein concentration determined was by the Bradford method.Citation[26]
Measurement of Cytochrome P4502E1 Activity
Cytochrome P4502E1 activity was assessed by using the substrate p‐nitrophenol as described by Watt et al.Citation[27] Briefly, the LLC‐PK1 cell monolayer was detached using cell scraper, and re‐suspended in 1 mM of buffer A [0.25 M potassium phosphate, 0.15 M potassium chloride, 0.25 M sucrose, 1 mM EDTA, pH 7.5]. The sonicate was centrifuged as 18,000 r.p.m at 4°C, and re‐suspended in 5 mL of buffer A, which was further centrifuged for 60 min at 38,000 r.p.m at the same temperature. The microsomes were suspended in 100 µL of buffer B [0.1 M sodium phosphate, 2 mM MgCl2, pH 7.4]. The reaction was initiated with 10 µL of 10mM NADPH and incubated at 37°C for 60 min. The reaction was stopped with 30 µL of 20% [w/v] trichloroacetic acid and centrifuged for 5 min. 10 µL of 10 N NaOH was added to the supernatant and the wavelength was recorded at 450 nm against the standard 160—2.5, p‐nitrocatechol and expressed in mM/mg/min.
Statistical Analysis
Results are presented as mean ± SD. Means were obtained from four experiments performed in duplicates. Paired t tests were used for statistical analysis differences. Statistical significance was assured when P < 0.05.
Results
Attenuation of H2O2‐Mediated LLC‐PK1 Cell Injury by CYP2E1 Inhibitors, Catalase and Desferrioxamine
Incubation of LLC‐PK1 for 4 h with 1 mM of H2O2 caused significant LDH release compared to controls (3.5 ± 5.1% vs. 62.2 ± 19.2% P < 0.001). Compared to LLC‐PK1 treated with H2O2, pre‐treatment with CYP2E1 inhibitors, DAS or DSF significantly reduced LDH release to 19.2 ± 4.4% and 21.7 ± 2.1% respectively (P < 0.005 vs. H2O2) In addition, catalase (78,150, 300 U/mL) and DFO (0.1, 0.2, 0.3 and 0.4 mM) reduced the LDH release in dose dependent manner with maximal effect seen at 300 U/mL (13.6 ± 12) and 0.4 mM (15.9 ± 3.7) for catalase and DFO respectively ().
Figure 1. (A) Cytotoxicity of H2O2 on LLC‐PK1 cells; Cells were treated with 1 mM of H2O2 with and without CT (300 U/mL), CYP2E1 inhibitors, DAS (0.1 mM), DSF (0.1 mM) and DFO (0.4 mM) and incubated for 4 h at 37°C. Cell death was assessed by measurement of LDH release and expressed as a percentage of the positive control (LLC‐PK1 cells incubated with 2% [v/v] Triton X100). Data shown are mean value ± SD of duplicate from four experiments. Significance wasevaluated with student t‐test. *P < 0.001 versus control, +P < 0.005 vs. H2O2, **P < 0.001 vs. H2O2. (B) Effect of CYP2E1 inhibitors, catalase and desferrioxamine on increased CYP2E1 activity caused by H2O2; LLC‐PK1 cells were incubated with H2O2 (1 mM) for 2 h with and without CYP2E1 inhibitors, DAS and DSF (0.1 mM), catalase (300 U/mL) and DFO (0.4 mM). CYP2E1 activity was assessed from the isolated microsomes and expressed as mM/mg/min. Data shown are mean value ± SD of four experiments. *P < 0.01 vs. control, +P < 0.05 vs. H2O2.
![Figure 1. (A) Cytotoxicity of H2O2 on LLC‐PK1 cells; Cells were treated with 1 mM of H2O2 with and without CT (300 U/mL), CYP2E1 inhibitors, DAS (0.1 mM), DSF (0.1 mM) and DFO (0.4 mM) and incubated for 4 h at 37°C. Cell death was assessed by measurement of LDH release and expressed as a percentage of the positive control (LLC‐PK1 cells incubated with 2% [v/v] Triton X100). Data shown are mean value ± SD of duplicate from four experiments. Significance wasevaluated with student t‐test. *P < 0.001 versus control, +P < 0.005 vs. H2O2, **P < 0.001 vs. H2O2. (B) Effect of CYP2E1 inhibitors, catalase and desferrioxamine on increased CYP2E1 activity caused by H2O2; LLC‐PK1 cells were incubated with H2O2 (1 mM) for 2 h with and without CYP2E1 inhibitors, DAS and DSF (0.1 mM), catalase (300 U/mL) and DFO (0.4 mM). CYP2E1 activity was assessed from the isolated microsomes and expressed as mM/mg/min. Data shown are mean value ± SD of four experiments. *P < 0.01 vs. control, +P < 0.05 vs. H2O2.](/cms/asset/42f73fd3-3ca5-4620-b0ee-515b5403487f/irnf_a_11379237_uf0001_b.gif)
CYP2E1 Activity in LLC‐PK1 Cells Treated with H2O2
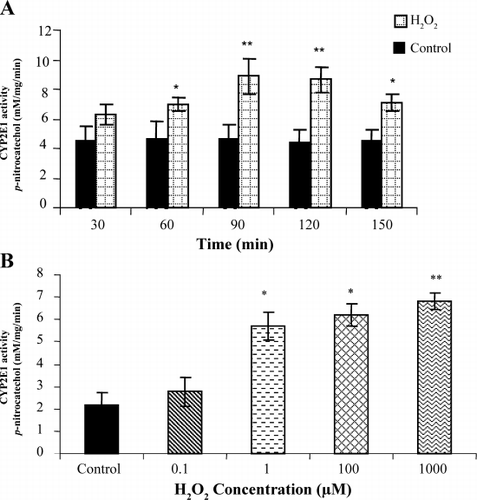
Incubation of LLC‐PK1 cells with H2O2 produced a significant increase in CYP2E1 activity. The activity was found to be time dependent after exposure of cells to 1 mM of H2O2. After 60 min of incubation with H2O2, the activity of CYP2E1 increased to 6.9 ± 1.1 (P < 0.05 vs. control), whereas after 90 min and 120 min, the activity increased further to 8.9 ± 0.8 and 8.7 ± 0.7 respectively (P < 0.01 vs. control). This increase in activity preceded LDH release (data not shown). Prolonged incubation (150 min) was associated with decline in CYP2E1 activity compared to 120 min, due to ongoing cell death (). Additionally, CYP2E1 activity was found to be dose‐dependent after exposing cells to increasing concentration of H2O2 ().
Figure 2. (A) Effect of H2O2 on CYP2E1 activity in LLC‐PK1 cells (increasing time); LLC‐PK1 cells were incubated with H2O2 for increasing time periods. At the end of incubation time, microsomes were isolated as described in Materials and Methods section and used for CYP2E1 activity assessment, which was assessed by measuring the hydroxylation of CYP2E1 substrate, p‐nitrophenol and expressed as mM/mg/min. Data shown are mean value ± SD of four experiments. Significance was evaluated with student t‐test. *P < 0.05 vs. control, **P < 0.01 vs. control. (B) Effect of H2O2 in on CYP2E1 activity in LLC‐PK1 cells (increasing concentration); LLC‐PK1 cells were incubated with H2O2 in dose dependent manner for 2 h. At the end of incubation time, CYP2E1 activity was assessed from the isolated microsomes and expressed as mM/mg/min. Data shown are mean value ± SD of four experiments. Significance was evaluated with student t‐test. *P < 0.05 vs. control, **P < 0.01 vs. control.
Inhibition of H2O2‐Mediated CYP2E1 Activation by CYP2E1 Inhibitors, Catalase and Desferrioxamine
The increase in CYP2E1 activity caused by incubation of LLC‐PK1 cells with 1 mM of H2O2 for 2 hrs was significantly reduced by pre‐incubating cells with 0.1 mM of DAS and DSF. Incubation of cells with 1 mM of H2O2 for 2 h activated CYP2E1 by 6.7 ± 1.5 mM/mg/min (P < 0.01 vs. control). This elevation in CYP2E1 activity was reduced by DAS and DSF to 2.6 ± 0.5, and 2.5 ± 0.6 mM/mg/min respectively (P < 0.05 vs. H2O2) (). This increase in CYP2E1 activity was also significantly reduced to 2.7 ± 1.2 mM/mg/min by DFO (P < 0.01 vs. H2O2). In addition, H2O2‐induced increase CP2E1 activity was inhibited in a dose‐dependent manner by catalase at 78, 150, 300 U/mL to 5.8 ± 0.7, 4.2 ± 0.5, and 2.5 ± 0.2 mM/mg/min respectively. However; the significant reduction in activity was caused by the doses of 150 (P < 0.05 vs. H2O2), and 300 U/mL of CT (P < 0.01 vs. H2O2).
Discussion
The role of CYP2E1 isoform in H2O2‐mediated proximal tubular cytotoxicity has not been previously examined. Our results indicate that prior treatment of the cells with CYP2E1 specific inhibitors result in a marked attenuation of H2O2‐induced cell injury in parallel with significant inhibition of H2O2‐stimulated CYP2E1 activity. Furthermore, catalase, which converts H2O2 to H2O and O2, afforded cytoprotection in parallel with prevention of CYP2E1 activation. Moreover, iron chelation with DFO prevented both CYP2E1 activation and cell injury. Interestingly, H2O2‐stimulated CYP2E1 was both dose and time dependent and preceded cell injury by 2 hours. It was also seen at a sub‐lethal H2O2 concentration suggestive of cause and effect relationship. In addition, CYP2E1 activation seen within two hours of exposure is indicative of stimulation of pre‐formed enzyme rather than de‐novo synthesis.
The mechanism by which CYP450 mediates H2O2 cytotoxicity in LLC‐PK1 cells is not clear. However, it has been shown before that CYP450 acts as a source of iron in the kidney tissue,Citation[28] and in reperfusion of injury in rabbit lung.Citation[29] Iron is known to catalyze the Fenton reaction whereby H2O2 is converted to OH·, which is regarded a potent mediator of I/R injury.Citation[28] In this regard, prevention of iron release by iron chelate is protective against cell injury caused by H2O2.Citation[2], Citation[30], Citation[31], Citation[32] Moreover, infusion of iron chelate DFO during the postischemic reperfusion phase of renal I/R significantly improves glomerular filtration rate and reduces histological signs of renal injury.Citation[2], Citation[33] Our data extends these observations by identifying the isoform of P450 involved in H2O2‐mediated injury of renal PT cells.
The role of other isoforms of CYP450 was not investigated in the current study. Our results however are suggestive of CYP2E1 being an important isoform involved in the oxidant injury. Selective inhibition of CYP2E1 by two chemically dissimilar and cell permeable inhibitors (DAS and DSF) markedly attenuated H2O2‐mediated cell necrosis. Chelation of H2O2 by catalase prevented not only cell injury but also CYP2E1 activation. This finding implicates activation of CYP2E1 as a pivotal reaction in H2O2‐induced cell death. Moreover, DFO an iron chelate, which inhibits the catalytic Fenton reaction and OH· generation, protected against necrosis and prevented CYP2E1 activation suggestive of a role for OH· in the activation of CYP2E1.
Interestingly, the CYP2E1 enzyme has also been shown to act as an oxidant, which on activation, can result in ROS generation and perpetuation of oxidant injury. It is a loosely coupled enzyme, which produces ROS such as O2· − and H2O2 in high amounts relative to other CYP450 isoforms,Citation[34], Citation[35] because CYP2E1 is rapidly reduced even in the absence of substrate.Citation[36] Previous studies have demonstrated the contribution of CYP2E1 induction in the generation of ROS and the initiation of liver diseaseCitation[37] or cytotoxicity in Hep G2 cells.Citation[38] CYP2E1 was found to display high NADPH oxidase activity with production of O2· − and H2O2 during NADPH oxidation.Citation[34], Citation[35] It has been shown that CYP2E1 induction is involved in hepatotoxicity by ROS generation.Citation[19], Citation[20], Citation[21] Inhibition of CYP2E1 by DAS completely blocked lipid peroxidation and prevented liver pathology in rats due to ethanol.Citation[39] Furthermore, in rats, DAS ameliorated ethanol‐induced changes in hepatic fatty acid composition, which is CYP2E1‐dependent lipid peroxidation process.Citation[40] Our data support these findings in LLC‐PK1 cell death caused by H2O2.
Taken as whole, these data are consistent with the suggestion that an increase in CYP2E1 activity plays a role in oxidant‐induced PT injury. Thus, inhibitors of CYP2E1 activity may be useful in conditions associated with oxidative stress of the kidney. This study should stimulate further investigations to elucidate the effects of potent safe inhibitors of CYP2E1 in renal disorders associated with oxidative stress. Certainly, one of the inhibitors used in our study (DSF) is commonly used to prevent the carbon tetrachloride‐mediated hepatotoxicity and help reduce alcohol consumption via the inhibition of CYP2E1.Citation[41], Citation[42]
In conclusion, this study demonstrates that the cellular death caused by H2O2 in LLC‐PK1 cells is partially mediated and perpetuated by hydroxyl ion‐mediated activation of CYP2E1. Our data is also consistent with activated CYP2E1 being a source of iron in oxidant renal cell injury. We therefore propose that inhibitors of CYP2E1 provide a novel approach for the therapy of I/R injury and other renal conditions associated with oxidative stress.
Acknowledgments
This work was sponsored by Department of Pharmacology and Toxicology, Faculty of Medicine and Medical Sciences, Umm Al‐Qura University, Makkah, Kingdom of Saudi Arabia. PKC is supported by the National Kidney Research Fund (Project Grant R41/2/2000).
| Abbreviations | ||
| CYP2E1: | = | cytochrome P4502E1 |
| CT: | = | catalase |
| DSF: | = | disulfiram |
| DMEM: | = | Dulbecco's modified Eagle's medium |
| DMSO: | = | dimethylsulfoxide |
| LDH: | = | lactate dehydrogenase |
| mg: | = | milligram |
| min: | = | minute |
| mM: | = | millimolar |
| rpm: | = | round per minute |
| U/mL: | = | unit per milliliter |
References
- Nagy I. Z. On the true role of oxygen free radicals in the living state, aging, and degenerative disorders. Ann. N. Y. Acad. Sci. 2001; 928: 187–199, [PUBMED], [INFOTRIEVE], [CSA]
- Chatterjee P. K., Cuzzocrea S., Brown P. A., Zacharowski K., Stewart K. N., Mota‐Filipe H., Thiemermann C. Tempol, a membrane‐permeable radical scavenger, reduces oxidant stress‐mediated renal dysfunction and injury in the rat. Kidney Int. 2000; 58(2)658–673, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Bauer V., Bauer F. Reactive oxygen species as mediators of tissue protection and injury. Gen. Physiol. Biophys. 1999; 18: 7–14, [PUBMED], [INFOTRIEVE]
- Chatterjee P. K., Cuzzocrea S., Thiemermann C. Inhibitors of poly (ADP‐ribose) synthetase protect rat proximal tubular cells against oxidant stress. Kidney Int. 1999; 56(3)973–984, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Humes H. D. Role of calcium in pathogenesis of acute renal failure. Am. J. Physiol. 1986; 250: F579–F589, [PUBMED], [INFOTRIEVE]
- Baliga R., Ueda N., Walker P. D., Shah S. V. Oxidant mechanisms in toxic acute renal failure. Drug Metab. Rev. 1999; 31(4)971–997, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Oldham K. M., Bowen P. E. Oxidative stress in critical care: is antioxidant supplementation beneficial?. J. Am. Diet. Assoc. 1998; 98(9)1001–1008, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Andreoli S. P., McAteer J. A. Reactive oxygen molecule‐mediated injury in endothelial and renal tubular epithelial cells in vitro. Kidney Int. 1990; 38: 785–794, [PUBMED], [INFOTRIEVE]
- Paller M. S, Jacob H. S. Cytochrome P‐450 mediates tissue‐damaging hydroxyl radical formation during reoxygenation of the kidney. Proc. Natl. Acad. Sci. U.S.A. 1994; 91: 7002–7006, [PUBMED], [INFOTRIEVE], [CSA]
- Ueda N., Guidet B., Shah S. V. Gentamicin‐induced mobilization of iron from renal cortical mitochondria. Am. J. Physiol. 1993; 265: F435–F439, [PUBMED], [INFOTRIEVE]
- Agrawal R., Sharma P. K., Rao G. S. Release of iron from ferritin by metabolites of benzene and superoxide radical generating agents. Toxicology 2001; 168(3)223–230, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Trenam C. W., Blake D. R., Morris C. J. Skin inflammation: reactive oxygen species and the role of iron. J. Invest. Dermatol. 1992; 99(6)675–862, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Baliga R., Zgang Z., Shah S. Role of cytochrome P‐450 in hydrogen peroxide‐induced cytotoxicity to LLC‐PK1 cells. Kidney Int. 1996; 50: 1118–1124, [PUBMED], [INFOTRIEVE]
- Liu H., Baliga M., Baliga R. Effect of cytochrome P450 2E1 inhibitors on cisplatin‐induced cytotoxicity to renal proximal tubular epithelial cells. Anticancer Res. 2002; 22(2A)863–868, [PUBMED], [INFOTRIEVE], [CSA]
- Klaassen C. D. Casarett and Doull's toxicology. The Basic Science of Poisons5th Ed. McGraw‐Hill, London 1995; 737–740
- Diaz‐Gomez M. I., Castro G. D., Delgado D. E., Layno A. M.A., Costantini M. H., Castro J. A. Cytochrome P450 reductase‐mediated anaerobic biotransformation of ethanol to 1‐hydroxyethyl‐free radicals and acetaldehyde. Toxicology 2000; 154: 113–122, [CSA], [CROSSREF]
- Holt S., Roy G., Mitra S., Upton P. B., Bogdanffy M. S., Swenberg J. A. Deficiency of N‐methylpurine‐DNA‐glycosylate expression in nonparenchymal cells, the target cell for vinyl chloride and vinyl fluoride. Mutat. Res. 2000; 460: 105–115, [PUBMED], [INFOTRIEVE]
- Zangar R. C., Benson J. M., Burnett V. L., Springer D. L. Cytochrome P450 2E1 is the primary enzyme responsible for low‐dose carbon tetrachloride metabolism in human liver microsomes. Chem. Biol. Interact. 2000; 125: 233–243, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Sakurai K., Cederbaum A. I. Oxidative stress and cytotoxicity induced by ferric‐nitrilotriacetate in HepG2 cells that express cytochrome P450 2E1. Mol. Pharmacol. 1998; 54: 1024–1035, [PUBMED], [INFOTRIEVE], [CSA]
- Castillo T., Koop D. R., Kamimura S., Triadafilopoulos G., Tsukamoto H. Role of cytochrome P‐450 2E1 in ethanol‐, carbon tetrachloride‐ and iron‐dependent microsomal lipid peroxidation. Hepatology 1992; 16: 992–996, [PUBMED], [INFOTRIEVE]
- Dai Y., Rashba‐Step J., Cederbaum A. I. Stable expression of human cytochrome P4502E1 in HepG2 cells: characterization of catalytic activities and production of reactive oxygen intermediates. Biochemistry 1993; 32: 6928–6937, [PUBMED], [INFOTRIEVE]
- Agarwal A. J., Balla G., Croatt A. J., Vercellotti G. M., Nath K. A. Renal tubular epithelial cells mimic endothelial cells upon exposure to oxidized LDL. Am. J. Physiol. 1996; 271: F814–F823, [PUBMED], [INFOTRIEVE]
- Yusof M., Yildiz D., Ercal N. n‐acetyl‐l‐cysteine protects against delta‐aminolevulinic acid‐induced 8‐hydroxydeoxyguanosine formation. Toxicol. Lett. 1999; 106: 41–47, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Saito H., Ohi H., Sugata E., Murayama N., Fujita Y., Higuchi. Purification and characterization of a cytochrome P450 from liver microsomes of Xenopus laevis. Arch. Biochem. Biophys. 1997; 345(1)56–64, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Mari M., Cederbaum A. I. Induction of catalase, alpha, and microsomal glutathione S‐transferase in CYP2E1 overexpressing HepG2 cells and protection against short‐term oxidative stress. Hepatology 2001; 33(3)652–661, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal. Biochem. 1976; 72: 248–254, [PUBMED], [INFOTRIEVE]
- Watt K. C., Plopper C. G., Buckpitt A. R. Measurement of cytochrome P450 2E1 activity in rat tracheobronchial airways using high‐performance liquid chromatography with electrochemical detection. Anal. Biochem. 1997; 248: 26–30, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Liu H., Shah S. V., Baliga R. Cytochrome P‐450 as a source of catalytic iron in minimal change nephrotic syndrome in rats. Am. J. Physiol., Renal. Physiol. 2001; 280(1)F88–F94
- Bysani G. K., Kennedy T. P., KY N., Rao N. V., Blaze C. A., Hoidal J. R. Role of cytochrome P‐450 in reperfusion injury of the rabbit lung. J. Clin. Invest. 1990; 86: 1434–1441, [PUBMED], [INFOTRIEVE]
- Hsieh H. J., Cheng C. C., Wu S. T., Chiu J. J., Wung B. S., Wang D. L. Increase of reactive oxygen species (ROS) in endothelial cells by shear flow and involvement of ROS in shear‐induced c‐fos expression. J. Cell. Physiol. 1998; 175(2)156–162, [PUBMED], [INFOTRIEVE]
- Barbouti A., Doulias P. T., Zhu B. Z., Frei B., Galaris D. Intracellular iron, but not copper, plays a critical role in hydrogen peroxide‐induced DNA damage. Free Radic. Biol. Med. 2001; 31(4)490–498, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Wang Y., Luo Z. The effects of deferoxamine on bovine pulmonary endothelial cell injury induced by hydrogen peroxide. Hunan Yike Daxue Xuebao 1997; 22(3)203–205, [PUBMED], [INFOTRIEVE], [CSA]
- Libra S., Pagano D., Curella G., Litrico V., Cancelliere M., Audibert D., LaTerra S., Gruttadauria S., Pappalardo A., D'Alessandro M. Experimental research on the use of deferoxamine in the prevention of renal damage from acute ischemia. Ann. Ital. Chir. 1999; 70(4)569–573, [PUBMED], [INFOTRIEVE], [CSA]
- Liu H., Baliga M., Baliga R. Effect of cytochrome P450 2E1 inhibitors on cisplatin‐induced cytotoxicity to renal proximal tubular epithelial cells. Anticancer Res. 2002; 22(2A)863–868, [PUBMED], [INFOTRIEVE], [CSA]
- Cederbaum A. I., Wu D., Mari M., Bai J. CYP2E1‐dependent toxicity and oxidative stress in HepG2 cells. Free Radic. Biol. Med. 2001; 31(12)1539–1543, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Guengerich F. P., Johnson W. W. Kinetics of ferric cytochrome P450 reduction by NADPH‐cytochrome P450 reductase: rapid reduction in the absence of substrate and variations among cytochrome P450 systems. Biochemistry 1997; 36(48)14741–14750, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Morimoto M., Hagbjork A. L., Nanji A. A., Ingelman‐Sundberg M., Lindros K. O., Fu P. C., Albano E., French S. W. Role of cytochrome P4502E1 in alcoholic liver disease pathogenesis. Alcohol 1993; 10(6)459–464, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Chen Q., Cederbaum A. I. Cytotoxicity and apoptosis produced by cytochrome P450 2E1 in Hep G2 cells. Mol. Pharmacol. 1998; 53(4)638–648, [PUBMED], [INFOTRIEVE], [CSA]
- French M., S. W., Reitz R. C., Koop D., Klopfenstein B., Estes K., Clot P., Ingelman‐Sundberg M., Albano E. Lipid peroxidation, CYP2E1 and arachidonic acid metabolism in alcoholic liver disease in rats. J. Nutr. 1997; 127(Suppl. 5)907S–911S, [PUBMED], [INFOTRIEVE]
- Morimoto M., Reitz R. C., Morin R. J., Nguyen K., Ingelman‐Sundberg M., French S. W. CYP‐2E1 inhibitors partially ameliorate the changes in hepatic fatty acid composition induced in rats by chronic administration of ethanol and a high fat diet. J. Nutr. 1995; 125(12)2953–2964, [PUBMED], [INFOTRIEVE]
- Emery M. G., Jubert C., Thummel K. E., Kharasch E. D. Duration of cytochrome P‐450 2E1 (CYP2E1) inhibition and estimation of functional CYP2E1 enzyme half‐life after single‐dose disulfiram administration in humans. J. Pharmacol. Exp. Ther. 1999; 291(1)213–219, [PUBMED], [INFOTRIEVE], [CSA]
- Lauriault V. V., Khan S., O'Brien P. J. Hepatocyte cytotoxicity induced by various hepatotoxins mediated by cytochrome P‐450IIE1: protection with diethyldithiocarbamate administration. Chem. Biol. Interact. 1992; 81: 271–289, [PUBMED], [INFOTRIEVE], [CROSSREF]