Abstract
Idiopathic nephrotic syndrome (NS) associated with focal segmental glomerulosclerosis (FSGS) and severe renal function impairment is usually refractory to the conventional treatment and progresses to end‐stage renal disease. Herein, we reported 10 patients with NS‐FSGS who had initially had CCr 34 ± 12 mL/min/1.73m2 (normal 120 mL/min/1.73m2), FE Mg 7.8 ± 2.6% (normal 2.2%), 24‐h urinary protein 3.1 g (normal < 200 mg) and been followed up for over 10 years. The initial intrarenal hemodynamic study revealed a marked elevation of efferent arteriolar resistance (RE 17289 ± 8636 dyne.s.cm− 5; normal 3000 dyne.s.cm− 5), intraglomerular hypertension (PG 57 ± 1 mm Hg; normal 52 mm Hg), hyperfiltration (FF 0.24; normal 0.2), marked reductions in GFR 35 ± 17 mL/min/1.73m2, renal plasma flow (RPF 159 ± 61 mL/min/1.73m2; normal 600 mL/min/1.73m2) and peritubular capillary flow (PTCF 123 ± 57 mL/min/1.73m2; normal 480 mL/min/1.73m2). Such a hemodynamic alteration indicated a hemodynamic maladjustment with a preferential constriction at RE. Treatment consists of multidrugs, namely angiotensin converting enzyme inhibitor, calcium channel blocker, antiplatelet and anticoagulant, with or without angiotensin II receptor antagonist. Following the treatment, correction of hemodynamic maladjustment has been achieved which is characterized by reductions in RE 6046 ± 2191 dyne.s.cm− 5, PG 52 ± mm Hg, FF 0.19 ± 0.1 and increments in RPF 341 ± 118 mL/min/1.73m2, PTCF 280 ± 106 mL/min/1.73m2 and GFR 64 ± 17 mL/min/1.73m2. Coinciding with hemodynamic improvement, there has been a steadily increased creatinine clearance and improvement in FE Mg 4.3 ± 2.6% and suppression of proteinuria 0.29 ± 0.4g/24 h after the period of follow‐up of greater than 10 years.
Introduction
Two crucial issues that need to be addressed in idiopathic nephrotic syndrome (NS) associated with focal segmental glomerulosclerosis (FSGS), that are relevant to the therapeutic unresponsiveness, are the delayed recognition of this entity and the unknown mechanism that induces nephronal damage, namely tubulointerstitial fibrosis.Citation[1], Citation[2], Citation[3], Citation[4] In respect to the former, a mild and early onset of NS‐FSGS can manifest clinically mimic minimal change disease (MCD) or mesangial proliferative nephrosis (MesP NS) and may be responsive to high‐dose prednisolone therapy. However, based upon the histopathologic picture, NS‐FSGS can be differentiated from the MCD or MesP‐NS by the presence of nephronal damage, namely tubulointerstitial fibrosis or glomerulosclerosis. In practice, to obtain the histopathologic information from the kidney biopsy is usually delayed and quite often pending on the therapeutic response to prednisolone or prednisolone plus immunosuppressant. In this regard, a simple, noninvasive and less time‐consuming test of tubular function by means of determining fractional excretion (FE) of magnesium (Mg) has been adopted to reflect the presence or absence of tubulointerstitial fibrosis, since there is a direct correlation between FE Mg and the magnitude of tubulointerstitial fibrosis.Citation[5] A normal value of FE Mg reflects an intact tubulointerstitial structure observed in MCD or MesP‐NS, and a high value of FE Mg reflects the presence of tubulointerstitial fibrosis and is in favor of nephrosis associated with tubulointerstitial disease such as NS‐FSGS. Such a diagnostic approach has greatly assisted us in early detection of NS‐FSGS and this practice has much privileged in early initiation of therapeutic prevention of renal disease progression in NS‐FSGS.
In respect to the latter issue concerning the pathogenetic mechanism of renal disease progression in NS‐FSGS, a hemodynamically mediated mechanism is likely to be relevant to it. A significant reduction in renal plasma flow and peritubular capillary flow, and elevated renal arteriolar resistances has been consistently documented in NS‐FSGS.Citation[6], Citation[7], Citation[8] Yenrudi and associatesCitation[9] have recently demonstrated that there is an inverse correlation between renal perfusion and a relative area of renal cortical interstitium in nephrosis. Similarly, Bohle and associatesCitation[10] also noted an inverse correlation between postglomerular capillary patency and the development of tubulointerstitial fibrosis in idiopathic nephrotic syndrome. Furthermore, an endothelial factor VIII defect in renal microcirculation also correlates with the severity of nephrosis.Citation[11] We have also observed that there has been a progressive reduction in renal perfusion as the disease severity progresses.Citation[12]
Such patients with advanced renal function impairment usually progress to end‐stage renal disease irrespective to the therapeutic regimen. Therefore, it is the purpose of this study to present a long‐term, successful response to the therapeutic strategy aiming to correct the hemodynamic alteration in 10 patients associated with NS‐FSGS and severe renal function impairment.
Materials and Methods
Ten patients associated with NS and FSGS who had initially presented with advanced renal function impairment and had a good compliance in follow‐up for 10 years or greater were included. All were subject to the following investigative procedures and treatment.
Renal Function Studies
Glomerular Function
Glomerular filtration rate was determined by measuring the 10‐hour endogenous creatinine clearance (CCr) or glomerular filtration rate (GFR) by the radioisotope technique using 99mTc‐labeled diethylene triamine pentaacetic acid (DTPA) and the value was converted to the body surface area of 1.73m2 by the method of calculation below:
Tubular Function
Indirect tubular transport was assessed by a 10‐hour urinary collection as previously described.Citation[13] Diuretic was not administered during or within 24 hours before the test. Briefly, after a regular supper, no additional food except drinking water, ad libitum was allowed. The patients were instructed to void at 7 PM, and then urine was collected from 7 PM to 5 AM. Clotted blood from venipuncture was drawn at the end of the test for the analysis of creatinine and magnesium levels. Urine samples were analyzed the same as blood samples by the Renal Metabolic Laboratory Unit. Analyses of 1) creatinine were determined by the method described by Faulkner and King and 2) magnesium was determined by Atomic Absorption Spectrophotometer (model 1100 G; Perkin Elmer, Norwalk, CT). A reflection of tubular transport was derived from the determination of FE Mg, which was calculated through the formula:The normal value of FE Mg is < 2.2 percent
Vascular Function (Intrarenal Hemodynamic Study)
Simultaneous assessments of effective renal plasma flow (RPF) using 131I‐labeled orthoiodohippuric acid (hippuran) and of glomerular filtration rate (GFR) using 99mTc‐labeled DTPA were determined.Citation[14] Intrarenal hemodynamics were calculated and based on modified Gomez’s equation. For calculation purpose, the effective filtration pressure across the glomerular capillaries (PF) is assumed to be 35 mmHg when the blood pressure is normal (BP 120/80 mmHg or less) and 40 mmHg when the blood pressure is high (BP > 120/80 mmHg).Citation[15] The hydrostatic pressure in Bowman’s space (Ht) is assumed to be 10 mmHg, the RPF and GFR are in mL/sec/1.73m2, the RBF is.
The peritubular capillary flow (PTCF) is derived from the substraction of GFR from RPF. From the above assumptions, the following equations are derived:
The intraglomerular hydrostatic pressure (PG)Where TP and FF are total plasma protein in gram and filtration fraction respectively.
Efferent arteriolar resistance (RE)
Mode of Therapy
All 10 patients were treated with multidrug regimen consisting of vasodilators, namely angiotensin‐converting enzyme inhibitor (enalapril 10–40 mg/day), calcium channel blocker (isradipine 10–20 mg/day), dipyridamole 50–100 mg/day plus baby aspirin gr.1/ day, and with or without AII receptor antagonist (Losartan 50–100 mg/day). In addition, vitamin C 1000–3000 mg/day and Vitamin E (400–800 IU) were supplemented. Prednisolone 1–2 mg/kg/day was given to all of them.
Statistical Analysis
Values in text are expressed as mean + SEM. The difference between pre‐and posttreatment values was performed by the Student’s paired t‐test. The difference was statistically significant when the p value was less than 0.05.
Result
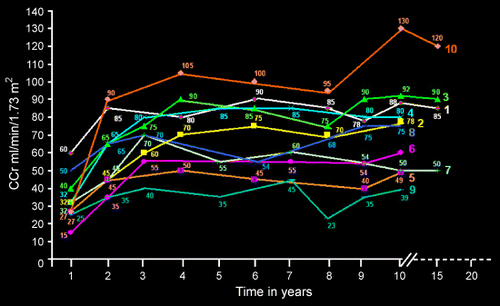
The initial results of renal function studies were as follows: the mean CCr was 34 ± 12 mL/min/1.73m2, FE Mg was 7.8 ± 2.6 percent, total urinary protein was 3.1 ± 1.4 g/24 h. The initial intrarenal hemodynamic study revealed GFR 35 ± 17 mL/min/1.73m2 (normal 120 mL/min/1.73m2), RPF 159 ± 61 mL/min/1.73m2 (normal 600 mL/min/1.73m2), PTCF 123 ± 57 mL/min/1.73m2 (normal 480 mL/min/1.73m2), PG 57 ± 1 mm Hg (normal < 52 mm Hg), RE 17289 ± 8636 dyne.s.cm− 5 (normal 3000 dyne.s.cm− 5), and FF 0.24 ± 0.1 (normal < 0.2). Following the treatment, a serial determination of CCr demonstrated a progressive increment as depicted in . The follow‐up renal function studies revealed a mean CCr of 70 ± 20 mL/min/1.73m2, p < .001; a mean value of FE Mg of 4.3 ± 2.6 per cent, p < .001; and a mean value of urinary protein of 0.2 ± 0.4 g/24 h, p < .001. The intrarenal hemodynamics revealed GFR 64 ± 17 mL/min/1.73m2, p < .001; RPF 341 ± 118 mL/min/1.73m2, p < .001; PTCF 280 ± 106 mL/min/1.73m2, p < .001; PG 52 ± 1 mm Hg; p < .001, RE 6046 ± 2191 dyne.s.cm− 5; p < .001, and FF 0.19 ± 0.1, p NS.
Figure 1. Serial determination of posttreatment CCr in NS‐FSGS. (View this art in color atwww.dekker.com.)
Discussion
The recognition of nephrosis associated with FSGS in these 10 patients is somewhat delayed when the renal function on admission, has already shown a rather severe impairment which is reflected by a low CCr and a high value of FE Mg. This specific category of patients with NS‐FSGS is usually refractory to the conventional therapy (prednisolone and immunosuppressant). Such a patient has toxic factor(s) in the serum, which induces high endothelial cell cytotoxicity.Citation[16] The presence of oxidative stressCitation[13] and enhanced proinflammatory cytokine (increased TNFα) with defective anti‐inflammatory cytokine (interleukin‐10)Citation[17] may be responsible for such enhanced endothelial cell cytotoxicity. The preceding observation correlates with the clinically progressive reduction in renal plasma flow and peritubular capillary flow as the disease severity progresses. The intrarenal hemodynamic alteration documented in these patients revealed a marked reduction in RPF and PTCF, which reflects the severe degree of glomerular and postglomerular endothelial dysfunction and thus supports the in‐vitro endothelial cytotoxicity test. This intrarenal hemodynamic finding would explain the therapeutic failure under conventional therapy in which the abnormal intrarenal hemodynamics and renal perfusion deficit has been left unattended.
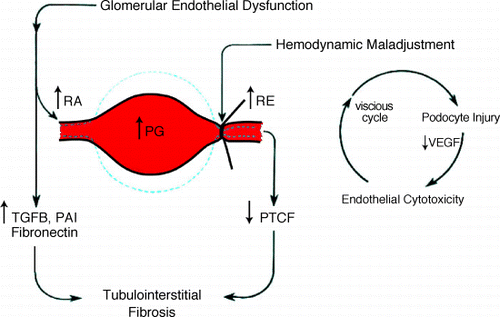
The intrarenal hemodynamic alteration is typically a pattern of hemodynamic maladjustment commonly encountered in other forms of chronic renal diseaseCitation[18], Citation[19] and is characterized by a preferential constriction at the efferent arteriole (). Such a constriction exerts 3 significant hemodynamic impacts. Proximal to the efferent arteriolar constriction, 1) it exaggeratedly increases the value of CCr or GFR and often misleads the actual glomerular function, which may be abnormally low. 2) it elevates the intraglomerular hydrostatic pressure and induces overdistension of the glomerular capillary loop by which it causes detachment of the podocytes from the basement membrane due to its nonproliferative and nondistensible characteristics.Citation[20], Citation[21], Citation[22] Such injury to the podocytes would decrease the production of vascular endothelial growth factor which is essential to the proliferation and regeneration of the glomerular and postglomerular endothelial cells.Citation[23] Further injury to the glomerular endothelial cell would aggravate in a vicious cycle manner, the greater degree of hemodynamic maladjustment, the further injury to the podocytes and eventually injury to the endothelial cell. Podocytes injury and podocytopenia has been well documented in NS‐FSGS and IgA nephropathy.Citation[22], Citation[23], Citation[24] Dital to the efferent arteriolar constriction, 3) it exaggeratedly reduces the PTCF, which mainly supplies the tubulointerstitial structure. Therefore, a sustained reduction in PTCF if allowed uncorrected, would not only induce ischemic injury but also activate the profibrogenic pathway and eventually culminate in the development of tubulointerstitial fibrosis.Citation[25], Citation[26], Citation[27]
Figure 2. Hemodynamic maladjustment in NS‐FSGS. (View this art in color atwww.dekker.com.)
The preceding information of hemodynamic maladjustment in relevant to the pathogenetic mechanism of renal disease progression is supported by the therapeutic intervention with multidrugs regimens aiming to relax the arteriolar constriction. The result of the therapeutic response in our patients associated with NS‐FSGS and advanced renal impairment has proven that this is to be the case. A correction of hemodynamic maladjustment results in a relaxation of the arteriolar constriction, which is concomitantly observed with the increments in RPF, PTCF, GFR and with the decrement in PG and FF. Coinciding with the hemodynamic improvement, there has been a steady increase in CCr and a progressive decline in FE Mg. The progressive improvement in CCr reflects a restoration of renal function, which is normally not seen in the patients under conventional therapy. The decline in FE Mg is likely to reflect a suppression of inflammatory and profibrogenic process in the tubulointerstitial compartment, which concurs with the beneficial effect of vasodilators on the regression of kidney disease in experimental animal models.Citation[28], Citation[29]
From the practical point of view, we have observed that the dose of vasodilator to correct the hemodynamic maladjustment required is much higher than needed for maximal blood pressure control. It also requires a longer duration of treatment with such vasodilators. The titration of the doses of vasodilators is mainly dependent upon the values of CCr, FE Mg and total urinary protein. FE Mg appears to be a very sensitive and practicable marker for clinical severityCitation[5] as well as for the hemodynamic status,Citation[30] since it correlates directly with the magnitude of tubulointerstitial fibrosis and inversely with the PTCF.
References
- Glassock R. J. Therapy of idiopathic nephrotic syndrome in adults. Am. J. Nephrol. 1993; 13: 422–428, [PUBMED], [INFOTRIEVE], [CSA]
- Cameron J. S., Turner D. R., Ogg C. S., Chantler C., Williams D. G. The long‐term prognosis of patients with focal segmental glomerulosclerosis. Clin. Nephrol. 1978; 10: 213–218, [PUBMED], [INFOTRIEVE]
- Wehrmann M., Bohle A., Held H., Schumm G., Kendziorra H., Pressler H. Long‐term prognosis of focal sclerosing glomerulonephritis: an analysis of 250 cases with particular regard to tubulointerstitial changes. Clin. Nephrol. 1990; 33: 115–122, [PUBMED], [INFOTRIEVE]
- Baldwin D. S. Chronic glomerulonephritis: non immunologic mechanisms of progressive glomerular damage. Kidney Int. 1982; 21: 109–120, [PUBMED], [INFOTRIEVE]
- Futrakul P., Yenrudi S., Futrakul N., Sensirivatana R., Kingwatanakul P., Jungthirapanich J., Cherdkiadtikul T., Laohapaibul A., Watana D., Singkhwa V., Futrakul N., Pongsin P. Tubular function and tubulointerstitial disease. Am. J. Kidney Dis. 1999; 33: 886–891, [PUBMED], [INFOTRIEVE]
- Futrakul P., Poshyachinda M., Mitrakul C. Focal sclerosing glomerulonephritis: a kinetic evaluation of hemostasis and the effect of anticoagulant therapy: a controlled study. Clin. Nephrol. 1978; 10: 180–186, [PUBMED], [INFOTRIEVE]
- Scandling J. S., Black V. M., Deen W. M., Myers B. D. Glomerular perm selectivity in healthy and nephrotic humans. Adv. Nephrol. 1992; 21: 159–176
- Futrakul P., Futrakul N., Sitprija V. Enhanced renal perfusion improves function in severe nephrosis with focal segmental glomerulosclerosis. Nephrology 1995; 1: 51–57
- Yenrudi S., Laohapaibul A., Kittidiwit W., Suteparuk S., Futrakul N. A correlation between renal morphology and renal circulation in pediatric nephrotic syndrome. Ren. Fail. 2001; 23: 85–90, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Bohle A., Mackensen‐Haen S., Wehrmann M. Significance of postglomerular capillaries in the pathogenesis of chronic renal failure. Kidney Blood Press. Res. 1996; 19: 191–195, [PUBMED], [INFOTRIEVE], [CSA]
- Futrakul N., Kittidiwit W., Yenrudi S. Reduced endothelial factor VIII staining in renal microcirculation correlates with hemodynamic alteration in nephrosis. Ren Fail. 2003; 25: 759–764, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Futrakul P., Sitprija V., Yenrudi S., Poshyachinda M., Sensirivatana R., Watana D., Singklwa V., Jungthirapanich J., Futrakul N. Glomerular endothelial dysfunction determines disease progression: a hypothesis. Am. J. Nephrol. 1997; 17: 533–540, [PUBMED], [INFOTRIEVE], [CSA]
- Futrakul N., Tosukhowong P., Valyapongpichit Y., Tipprukmas N., Futrakul P., Patumraj S. Oxidative stress and hemodynamic maladjustment in chronic renal disease: a therapeutic implication. Ren. Fail. 2002; 24: 433–445, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Futrakul P., Poshyachinda M., Futrakul N., Chaiwatanarat T., Sensirivatana R., Thamaree S., Watana D., Kingwatanakul P. Intrarenal hemodynamic alterations and tubular functions in nephrotic syndrome associated with focal segmental glomerulosclerosis (FSGS): a pathogenetic and therapeutic implication. Current Therapy in Nephrology, V. E. Andreucci, A. Dal Canton. Wichtig, Milan 1993; 107–114
- Guasch A., Sibley R. K., Huie P., Myers B. D. Extent and course of glomerular injury in human membranous glomerulopathy. Am. J. Physiol. 1992; 263: F1034–F1043, [PUBMED], [INFOTRIEVE]
- Futrakul N., Panichakul T., Chaisuriya P., Sirisinha S., Patumraj S., Futrakul P. Endothelial cell cytotoxicity and renal hypoperfusion in idiopathic nephrotic syndrome. Nephron 2000; 86: 241–242, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Futrakul N., Butthep P., Patumraj S., Tipprukmas N., Futrakul P. Enhanced tumor necrosis factor in the serum and renal hypoperfusion in nephrosis associated with focal segmental glomerulosclerosis. Ren. Fail. 2000; 22: 213–217, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Futrakul P., Poshyachinda M., Yenrudi S., Seleekul P., Sensirivatana R., Futrakul N. Intrarenal hemodynamic abnormality in severe form of glomerulonephritis: therapeutic benefit with vasodilators. J. Med. Assoc. Thai. 1992; 75: 375–385, [PUBMED], [INFOTRIEVE], [CSA]
- Futrakul N., Laohapaibul A., Futrakul P. Glomerular endothelial dysfunction and hemodynamic maladjustment in vesicoureteric reflex. Reflex .Ren. Fail. 2003; 25: 479–483, in press[CSA], [CROSSREF]
- Kriz W., Elger M., Nagata M., Kretzlar M., Uiker S., Koeppen‐Hagemann I., Tenschert S., Lemley K. V. The role of podocytes in the development of glomerulosclerosis. Kidney Int. 1994; 45(suppl 45)64–72
- Rennke H. G. How does glomerular epithelial injury contribute to progressive glomerular damage?. Kidney Int. 1994; 45(suppl 45)58–63
- Fogo A. Nephrotic syndrome: molecular and genetic basis. Nephron 2000; 85: 8–13, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Fan L., Wakayama T., Yokoyama S., Amano O., Iseki S. Down‐regulation of vascular endothelial growth factor and its receptors in the kidney in rats with puromycin aminonucleoside nephrosis. Nephron 2002; 90: 95–102, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Lemley K. V., Lafayetti R. A., Safai M., Derby G., Blouch K., Squarer A., Myers B. D. Podocytopenia and disease severity in IgA nephropathy. Kidney Int. 2002; 61: 1475–1485, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Fogo A. B. The role of angiotensin II and plasminogen activator inhibitor‐1 in progressive glomerulosclerosis. Am. J. Kidney Dis. 2000; 35: 179–188, [PUBMED], [INFOTRIEVE]
- Bottinger E. P., Bitzer M. TGF‐B signaling in renal disease. J. Am. Soc. Nephrol. 2002; 13: 260–269, [CSA], [CROSSREF]
- Ruiz‐Ortega M., Egido J. Angiotensin II modulates cell growth‐related events and synthesis of matrix proteins in renal interstitial fibroblasts. Kidney Int. 1997; 52: 1497–1510
- Komine N., Khang S., Wead L. M., Blantz R. C., Gubbar F. B. Effect of combining an ACE inhibitor and angiotensin II receptor blocker on plasma and kidney tissue angiotensin II levels. Am. J. Kidney Dis. 2002; 39: 159–164, [PUBMED], [INFOTRIEVE]
- Remuzzi A., Gagliardini E., Donadino C., Fassi A., Sanzalli F., Lepre M. S., Remuzzi G., Benigni A. Effect of angiotensin II antagonism on the regression of kidney disease in the rat. Kidney Int. 2002; 62: 885–894, [PUBMED], [INFOTRIEVE], [CROSSREF]
- Futrakul N., Yenrudi S., Futrakul P., Cherdkiadtikul T., Laohapaibul A., Futrakul S., Sensirivatna R. Peritubular capillary flow and tubular function in idiopathic nephrotic syndrome. Nephron 2000; 85: 181–182, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]