Abstract
The synthesis, structure and properties of tetrathiapentalene-based (TTP) organic conductors are reviewed. Among various TTP-type donors, bis-fused tetrathiafulvalene, 2,5-bis(1,3-dithiol-2-ylidene)-1,3,4,6-tetrathiapentalene (BDT-TTP) and its derivatives afford many metallic radical cation salts stable down to low temperatures, regardless of the size and shape of the counter anions. Most BDT-TTP conductors have a β-type donor arrangement with almost uniform stacks. Introduction of appropriate substituents results in molecular packing that differs from the β-type. A vinylogous TTP, 2-(1,3-dithiol-2-ylidene)-5-(2-ethanediylidene-1,3-dithiole)-1,3,4,6-tetrathiapentalene (DTEDT) has yielded an organic superconductor (DTEDT)3Au(CN)2 as well as metallic radical cation salts, regardless of the counter anions. (Thio)pyran analogs of TTP, namely (T)PDT-TTP and its derivatives produce molecular conductors with novel molecular arrangements. A TTP analog with reduced π-electron system 2,5-bis(1,3-dithian-2-ylidene)-1,3,4,6-tetrathiapentalene (BDA-TTP) has afforded several organic superconductors. Highly conducting molecular metals with unusual oxidation states (+1, +5/3 and neutral) have been developed on the basis of 2,5-bis(1,3-dithiol-2-ylidene)-1,3,4,6-tetrathiapentalene (BDT-TTP) derivatives and analogous metal derivatives M(dt)2 (M = Ni, Au).
Introduction
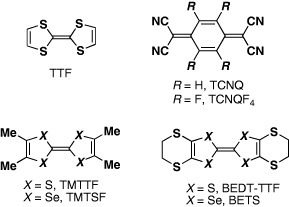
Since the publication of the report on the metallic conductivity in a tetrathiafulvalene-tetracyanoquinodimethane (TTF·TCNQ) complex [Citation1] and the subsequent discovery of the first organic superconductor (TMTSF)2PF6 (TMTSF=tetramethyltetraselenafulvalene) [Citation2], TTF and its derivatives have played a leading role in the development of new molecular metals and superconductors [Citation3–5]. To explore molecular organic superconductors, it is important to stabilize the metallic state down to low temperatures ( 4.2 K) by enhancing the dimensionality of electrical conductivity. In this way, the Peierls transition, which is observed in conventional one-dimensional (1D) conductors represented by TTF·TCNQ, can be suppressed. Since the discovery of 2D bis(ethelynedithio)-TTF (BEDT-TTF) metals and superconductors, substitution of ‘capped’ alkyl-dichalcogeno groups on TTF has been regarded as the most promising strategy for preparation of 2D metals [Citation6, Citation7]. However, ‘capped’ alkyl-dichalcogeno groups on π-electron frameworks other than TTF must be introduced. This limits the search for novel conducting components of metals stable down to low temperatures (scheme ).
Scheme 1 Molecular structures of TTF and TCNQ derivatives in this paper.
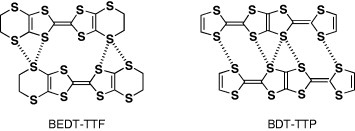
In this context, a new π-electron framework, having the ability to produce multidimensional metals without any substituent, is of significant interest. Thus, the introduction of substituent alkyl, benzo and/or methylthio groups, as well as unsubstitution, may also be effective in preparing 2D (or at least quasi-1D) metals for such π-electron systems, whereas the corresponding TTF derivatives usually yield completely 1D conductors. We have noticed a bis-fused TTF, 2,5-bis(1,3-dithiol-2-ylidene)-1,3,4,6-tetrathiapentalene (abbreviated as BDT-TTP or simply TTP) as a promising candidate for the construction of 2D metals. This molecule can also be designed by insertion of the 1,3,4,6-tetrathiapentalene moiety between two 1,3-dithiole rings of TTF. Because BDT-TTP has the so-called ladder-like array of sulfur atoms, as in BEDT-TTF, its cation radical salts are expected to form 2D conducting sheets thanks to the lateral intermolecular interactions based on sulfur–sulfur contacts incorporated in the BDT-TTP framework (scheme ). This article reviews the synthesis, structure and electrochemical properties of TTP donors, and the structure and properties of molecular conductors based on these TTP donors.
Scheme 2 Lateral interactions in BEDT-TTF and BDT-TTP conductors.
Synthesis, structure and properties of TTP molecules
Synthesis
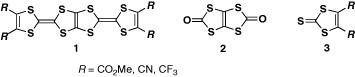
The first synthesis of BDT-TTP derivatives (1) (scheme ) was reported in 1980 [Citation8]. Schumaker and co-workers synthesized BDT-TTP derivatives with strongly electron-withdrawing substituents such as cyano, methoxycarbonyl or trifluoromethyl groups by means of a phosphite-mediated cross-coupling reaction between 1,3,4,6-tetrathiapentalene-2,5-dione (2) and corresponding 1,3-dithiole-2-thiones (3) (scheme ). However, this methodology has not been applied to the synthesis of the other BDT-TTP derivatives, probably because of difficulties in optimizing the reaction conditions for cross-coupling using 2. Furthermore, the details of the structure and properties of the products have not been reported, particularly the crystal structure, redox behavior and formation of conducting salts. The author and co-workers developed a versatile route for the synthesis of BDT-TTP derivatives early in 1990 as shown in scheme [Citation9, Citation10]. First, an unsymmetrical TTF moiety is constructed with protection groups for dithiolate. Then, the fused 1,3-dithiole ring is formed by deprotection of TTF dithiolate and successive reaction with triphosgene. Finally, the trialkylphosphite-mediated cross-coupling reaction between the resultant 1,3-dithiol-2-one fused with TTF (5) and 1,3-dithiole-2-thione derivatives afford the desired BDT-TTP derivatives. Unfortunately, the cross-coupling reaction in the final step does not proceed at all when unsubstituted or alkyl-substituted 3 is used. Thus, the unsubstituted derivative has been synthesized by demethoxycarbonylation of the bis(methoxycarbonyl) derivative using lithium bromide monohydrate in hexamethylphosphoric triamide (HMPA). In contrast, the tetramethyl derivative has not been synthesized thus far in spite of the expectation of as an analog of TMTSF.
Scheme 3 Molecular structures of 1–3.
Scheme 4 Synthesis of BDT-TTP derivatives.
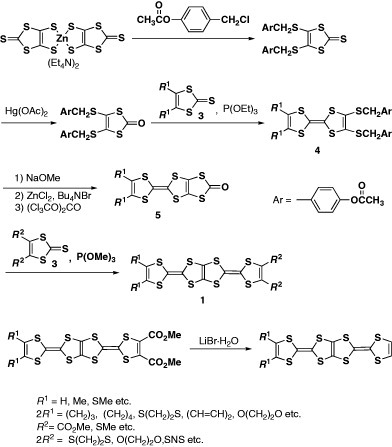
In a similar manner, selenium analogs ST-TTP and BDS-TTP (6) (scheme ) have been prepared in the cross-coupling reaction by the combination of 1,3-dithiole-2-thiones fused with TTF (or STF) (7) and 1,3-diselenol-2-ones (8) [Citation11]. More than 80 types of BDT-TTP derivatives and selenium analogs have been synthesized by this method to date [Citation9–41]. Yamada et al developed another synthetic strategy for 5 (scheme ) [Citation42]. One of the TTF frameworks is constructed via the Me3Al-promoted reaction (Me=methyl) of organotin compound 9 prepared from 2 with ester 1 0 in this procedure. However, P(OEt)3-mediated cross coupling is still required because of the extremely low solubility of 5 and 11 in common solvents.
Scheme 5 Synthesis of selenium analogs of BDT-TTP.
Scheme 6 Synthesis of 5 and11 via Me3 Al-promoted reaction.
Structure and electrochemical properties of BDT-TTP derivatives
Figure shows the molecular structure of neutral BDT-TTP determined by x-ray structure analysis [Citation10]. The BDT-TTP molecule adopts a chair conformation, in which the molecule is folded at the four sulfur atoms in the central tetrathiapentalene moiety. The dihedral angle is 27.17° (27.33°) between the two planes composed of S1-S2-C1-C2-S3-S4 (S5-S6-C7-C8-S7-S8) and S3-S4-C5-C6-S5-S6.
Figure 1 Top (a) and side (b) views of the molecular structure of BDT-TTP. (Reprinted with permission from [Citation10].)
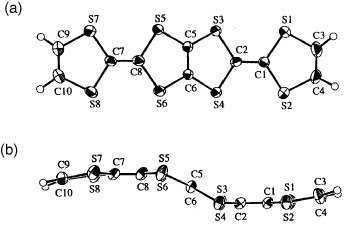
Table 1 summarizes the redox potentials of BDT-TTP derivatives measured by cyclic voltammetry. The cyclic voltammograms of most BDT-TTP derivatives exhibit four pairs of redox waves because they have four redox-active 1,3-dithiole rings. The first redox potential of BDT-TTP, E1=+0.44 V versus saturated calomel electrode (SCE) in benzonitrile, is higher than that of TTF (+0.35 V) by about 0.1 V in spite of the extension of π-conjugation. Instead, the E1 value of BDT-TTP is comparable to those of 4, 5-bis(methylthio)-TTF (12, 0.44 V) and 2-isopropylidene-1, 3-dithiolo[4,5-d]-TTF (13, 0.45 V) [Citation43] measured under identical conditions. In contrast, the tetrakis(methylthio) derivative (TTM-TTP) has an E1 value (+0.53 V) similar to the corresponding TTF TTM-TTF (+0.51 V). These results indicate that the positive charge formed by the first oxidation mainly distributes on one TTF moiety, while the other acts as a sulfur-based substituent. The ΔE (=E2−E1) values of BDT-TTP (0.18 V) and TTM-TTP (0.19 V) are smaller by 0.24 and 0.08 V compared with those of TTF (0.42 V) and TTM-TTF (0.27 V), respectively. Because such a small ΔE corresponds to a reduction of on-site Coulomb repulsion in the dicationic state of TTP derivatives, TTP conductors may be expected to show high conductivity.
Table 1 Redox potentialsa of BDT-TTP derivatives and the related compounds (scheme ).
Scheme 7 Molecular structures of the donors listed in table 1.
Structures and conducting properties of TTP conductors
Numerous molecular conductors based on TTP donors have been synthesized (see scheme ). Table 2 summarizes the molecular packing patterns and conducting properties of TTP-type conductors whose crystal structures have been determined. In this section, representative materials based on BDT-TTP derivatives are reported.
Scheme 8 Molecular structures of BDT-TTP derivatives listed in table 2.
Table 2 Molecular packing patterns and electrical properties of conducting salts based on BDT-TTP derivatives (scheme ); σrt refers to room-temperature conductivity.
(BDT-TTP)Ax and (ST-TTP)2A
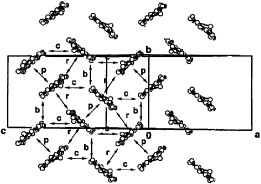
Several radical cation salts with various anions have been synthesized on the basis of BDT-TTP and its selenium analog ST-TTP. Most of these salts exhibit high room-temperature electrical conductivity σrt=1–1000 S cm−1 and show metal-like conducting behavior down to low temperatures (0.6–4.2 K) regardless of the size and shape of the counter anions [Citation11, Citation1Citation2, Citation44–51]. Figure shows the crystal structure of (BDT-TTP)2SbF6. BDT-TTP molecules form 2D conducting sheets, each of which is divided by an insulating layer composed of the anions, as expected. The packing pattern of the donors is classified as the β-type in BEDT-TTF conductors (figure ). The donors form a face-to-face stack with interplanar distances of 3.46 and 3.47 Å. The overlap mode of the donor molecules in the stack is the so-called ring-over-bond type. The slip distance is 1.6 Å along the long molecular axis in the stack. A similar molecular arrangement has also been found in most molecular conductors based on BDT-TTP and its selenium analogs ST-TTP and BDS-TTP with various anions (ClO4−, BF4−, ReO4−, PF6−, AsF6−, SbF6−, Re6S6Cl82−, Mo6Cl142−, etc.). Therefore, BDT-TTP is regarded as having the strong self-aggregating property of β-type molecular packing with little dimerization. This contrasts to the behavior of the array of donors in BEDT-TTF conductors, which strongly depends on both the shape and the size of the counter anions.
Figure 2 Crystal structure of (BDT-TTP)2SbF6 where dotted lines represent intermolecular S···S contacts. (Reprinted with permission from [Citation45].)
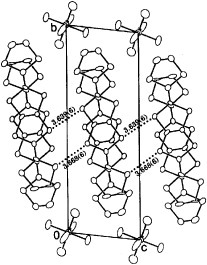
Figure 3 Donor sheet structure of (BDT-TTP)2SbF6. The intermolecular overlap integrals are a1=25.1, a2=25.3, c=0.8, p1=7.9 and p2=8.6× 10−3. (Reprinted with permission from [Citation45].)
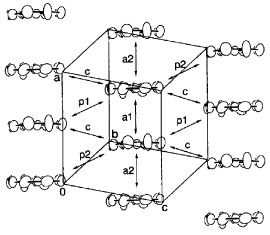
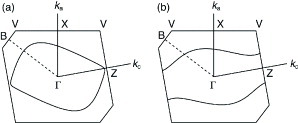
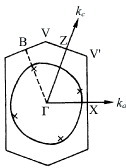
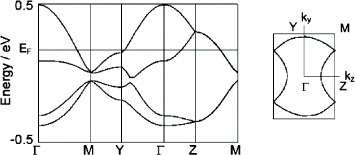
The overlap integrals of BDT-TTP salts were calculated by a molecular orbital method based on the extended Hückel approximation. The calculated intrastack overlap integrals between the highest occupied molecular orbitals (HOMOs) of (BDT-TTP)2SbF6 are almost identical (a1=25.1, a2=25.3× 10−3), which is consistent with the interplanar distances and the slip distances along the long molecular axis. Thus, the donors form an almost uniform stack in this salt, while β-BEDT-TTF salts have a strongly dimerized stack. On the other hand, the calculated lateral overlaps (Sl) are relatively large compared with the stacking ones (Ss). The ratio of the largest intrastack overlap to the largest lateral one (Ss/Sl) is 2.9, which corresponds to an intermediate value between a typical 2D metal, β-(BEDT-TTF)2I3 (Ss/Sl=1.9) [Citation72] and a typical quasi-1D metal, (TMTTF)2Br (Ss/Sl=6.6) [Citation73]. Thanks to such relatively large interstack interactions, the calculated Fermi surface is closed in the conducting layer (figure (a)). On the other hand, the Fermi surface, as deduced from the experimental reflection spectrum, is open in the kc direction, as shown in figure (b) [Citation74]. When the theoretical calculation is performed by excluding 3d orbitals of sulfur atoms, the shape of the calculated Fermi surface approaches that of the Fermi surface obtained from the reflection spectrum. However, the overlap integrals are underestimated by about 30%.
Figure 4 Fermi surface of (BDT-TTP)2SbF6 calculated from the transfer integrals obtained (a) theoretically and (b) from optical experiments.
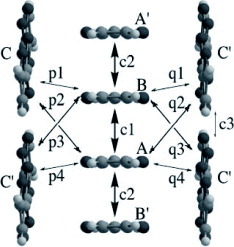
Three exceptional molecular packing patterns that differ from uniform β-type packing have been identified for BDT-TTP conductors. (BDT-TTP)2Cu(NCS)2 adopts θ-type arrangement of the donors, and exhibits semiconducting temperature dependence of resistivity. Yakushi and co-workers reported that this salt shows a phase transition accompanied with a change in the activation energy at 250 K, below which the donors are in a charge-ordering state as is often observed in θ-phase conductors [Citation75]. Kobayashi and co-workers reported that (BDT-TTP)3I has a unique 3D donor packing with six crystallographically independent donor molecules, as shown in figure [Citation50]. This salt shows semiconducting behavior with a small activation energy of 0.014 eV due to charge disproportionation. Kobayashi and co-workers also developed BDT-TTP conductors with divalent anions [MIICl4]2− (M=Co, Mn, Zn) [Citation51]. Figure shows the crystal structure of (BDT-TTP)3[MIICl4](EtOH)x (x ≈ 1.0). Three independent halves of BDT-TTP molecules (A, B, C) and half of a [MIICl4]2− anion are crystallographically independent. Comparison of the bond lengths indicates that the formal charge of molecule C is nearly 0 and those of molecules A and B are +1. Donors A and B form a column along the c-axis with a ring-over-bond overlap mode. In contrast, donor C forms a unique 1D ‘BDT-TTP tape’ along the c-axis and the molecular plane of donor C is approximately perpendicular to the planes of donors A and B. These salts exhibit a relatively high conductivity of σrt ≈ 5 S cm−1 and weak metal like behavior down to about 150 K. At low temperature, the resistivity gradually increases with decreasing temperature, however, the conductivity is high even at 30 K. Magnetic susceptibility measurement reveals temperature-independent metal-like behavior in the [ZnIICl4]2− salt.
Figure 5 Crystal structure of (BDT-TTP)3I. (Reprinted with permission from [Citation50].)
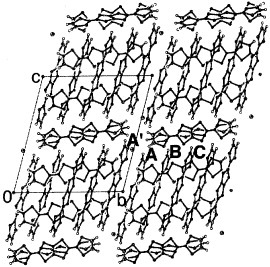
Figure 6 Donor-sheet structure of (BDT-TTP)3[MIICl4](EtOH)x (M=Co, Mn, Zn, x≈1.0). (Reprinted with permission from [Citation51].)
κ-Type salts based on TTP donors
Molecular packing that differs from the β-type can be realized if appropriate substituents are introduced into the BDT-TTP framework. Construction of a κ-type structure is of significant interest because many κ-type radical cation salts based on TTF derivatives show superconducting transitions [Citation4, Citation5, Citation7]. For TTF systems, the introduction of capped alkyl-dichalcogeno groups is indispensable for constructing κ-type conductors. The roles of the capped alkyl-dichalcogeno groups are as follows: (i) steric hindrance to avoid formation of stacking structure revealing in the formation of dimers and (ii) realization of 2D electronic structure through sulfur–sulfur lateral interaction. In fact, all TTF donors affording κ-type salts have one or two ethylenedithio, methylendithio or ethylenedioxy groups. In contrast, a ‘capped’ alkyl-dichalcogeno group is not needed for the BDT-TTP system, because it has enough sulfur atoms in the π-electron framework to realize lateral interactions. Thanks to the ability to form a 2D conduction layer, the introduction of a non-planar substituent such as a fused cyclohexene ring may be possible to construct a κ-type structure. In fact, 2-(4,5-cyclohexeno-1,3-dithiol-2-ylidene)-5-(1,3-dithiol-2-ylidene)-1,3,4,6-tetrathiapentanlene (CH-TTP) has afforded a κ-type salt with an I3− anion (figure ) [Citation52]. This salt exhibits high conductivity of σrt=38 S cm−1 and metal-like temperature dependence down to 1.4 K. Superconductivity has not been observed in spite of the κ-type molecular packing, probably because the upper band of this salt is not half-filled due to a nearly 3 : 1 donor : acceptor composition. Whereas all the κ-type superconductors discovered thus far have a 2:1 composition and a half-filled upper band as a result. The other BDT-TTP derivatives with a fused cyclohexene ring, CHEO-TTP [Citation25] and CHET-TS-TTP [Citation26], have also yielded κ-type conductors with ReO4− or SbF6− anion, both of which retain metallic conductivity down to 4.2 K.
Figure 7 Donor-sheet structure of (CH-TTP)(I3)0.31. The intermolecular overlap integrals are c=−4.32, p=22.2, r=7.61 and b=12.0× 10−3. (Reprinted with permission from [Citation52].)
In this regard, BDT-TTP derivatives fused with other six-membered rings are of interest to elucidate a correlation between molecular structure and the molecular array in the TTP conductors. BDT-TTP fused with dehydro-1,4-dioxane, in other words, ethylenedioxy-substituted BDT-TTP (EO-TTP), seems to afford the κ-type salts. However, x-ray structure analysis of (EO-TTP)2AsF6 revealed a β-type packing pattern with little dimerized stacking as observed in (BDT-TTP)2A [Citation53]. The torsion angle around the ethylene bridge in (EO-TTP)2AsF6 is 37°, which is about two-thirds of that in (CH-TTP)(I3)0.31 (57°). Considering a smaller dihedral angle, as well as fewer hydrogen atoms in the dehydrodioxane ring (four hydrogen atoms) than in the cyclohexene rings (eight hydrogen atoms), steric hindrance between the donors in (EO-TTP)2AsF6 should be smaller than that in (CH-TTP)(I3)0.31. Smaller steric repulsion may favor the formation of the stacking β-type structure. A tight-binding band calculation indicates that this salt has a closed Fermi surface similar to (BDT-TTP)2SbF6, although the 1D character is still strong owing to large overlaps in the stacking direction. EO-TTP salts with tetrahedral and octahedral anions including (EO-TTP)2AsF6 exhibit very high conductivity of σrt=380–1300 S cm−1 with a simple metallic temperature dependence down to 1.5–4.2 K. On the other hand, CPEO-TTP [Citation22] and EOET-TTP [Citation14], which have the additional substituent of an annelated cyclopentene ring or ethylenedithio group, have yielded κ-type salts with octahedral anions.
BTM-TTP conductors
The methylthio group seems to be more sterically hindered than the annelated cyclohexane group, being expected to afford κ-type or other type of packing of the donors. Bis(methylthio)-substituted TTP (BTM-TTP) has afforded two types of radical cation salts (needles and plates) with octahedral anions. x-ray structure analysis revealed that needle-like crystals of (BTM-TTP)2A (A=SbF6, TaF6) have the so-called β-type array of donors [Citation34, Citation54]. BTM-TTP molecules form a nearly uniform column in spite of the presence of bulky methylthio groups. Namely, both the interplanar distances and the slip distances are almost the same (1.6 Å) along the donor long axis. Calculation of the overlap integrals indicates that the donors are slightly dimerized in the stack. On the other hand, the interstack overlap integrals are 14–30% larger than the intrastack ones. A tight-binding calculation suggests that the calculated Fermi surface is quasi-1D.
The plate-like crystal of (BTM-TTP)2SbF6 adopts the so-called θ-type array (figure (a)) [Citation54]. The donors are uniformly stacked with an interplanar distance of 3.44 Å. The donors slip along both the short and long axes of the donor (figure (b)), in contrast to θ-BEDT-TTF salts, in which only slipping along the short axis is observed. The dihedral angle of the donors between the neighboring columns is large (130.7°), indicating smaller overlap integrals than those of the θ-salts with smaller dihedral angles. The calculated bandwidth (0.57 eV) is about half of that in β-(BTM-TTP)2SbF6. The calculated Fermi surface is closed, as is characteristic of a 2D metal.
Figure 8 (a) Conducting sheet structure of θ-(BTM-TTP)2SbF6 viewed along the donor long axis. The intermolecular overlaps between HOMOs are a=7.7, p=6.5 and q=3.6× 10−3. (b) The overlap mode of the donors in the stack. (Reprinted with permission from [Citation54].)
The β-type salts exhibit relatively high conductivity of σrt=30–120 S cm−1 and metallic temperature dependence down to 5 K. In contrast, θ-(BTM-TTP)2SbF6 exhibits relatively low conductivity of σrt=0.8 S cm−1 and semiconducting temperature dependence with an activation energy of 0.13 eV, probably because of narrow bandwidth and resultant large electron correlation in the quarter-filled band system.
TMEO-TTP and TMEO-ST-TTP salts
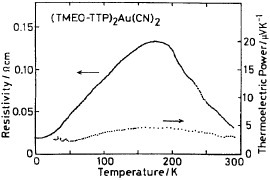
The BDT-TTP derivative with ethylenedioxy and two methylthio groups TMEO-TTP affords many metallic radical cation salts with octahedral and linear anions down to low temperatures (0.6–4.2 K), whereas the salts with tetrahedral anions are semiconductors [Citation14]. Among the metallic salts, the crystal structure of only (TMEO-TTP)2Au(CN)2 has been determined [Citation56]. This salt has the so-called β″-type array of donors similar to (BEDT-TTF)2ClO4(TCE) [Citation6] (see figure ). It shows a resistivity maximum around 180 K (figure ), while the other metallic TMEO-TTP salts exhibit simple metallic temperature dependence. The thermoelectric power and electron spin resonance (ESR) spectrum of this salt also exhibit anomalies around this temperature, indicating a metal-to-metal transition (see also figure ).
Figure 9 Donor-sheet structure of (TMEO-TTP)2Au(CN)2. The intermolecular overlap integrals are c=4.2, a1=18.3, a2=7.3, p1=−11.5 and p2=−9.7× 10−3. (Reprinted with permission from [Citation56].)
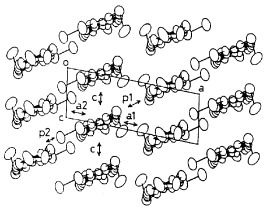
Figure 10 Electrical properties of (TMEO-TTP)2Au(CN)2. (Reprinted with permission from [Citation56].)
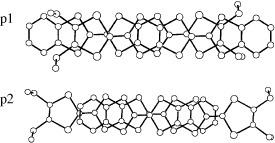
On the other hand, a selenium analog TMEO-ST-TTP forms a 1 : 1 salt with an Au(CN)2− anion [Citation59]. The Au(CN)2− anion is located in a cavity surrounded by four donor molecules in the ac-plane (see figure ). Although the TMEO-ST-TTP molecules seem at first glance to form a 2D network in the ac-plane, there is no significant lateral interaction along either a- or c-axis. The donors are strongly dimerized in the column, because one of the overlaps makes a large slip along both the long and short axes of the donor, as shown in figure . Thus, a tight-binding band calculation indicates that this is a 1D band insulator. In contrast to the Au(CN)2− anion, the octahedral anions (PF6−, AsF6− and TaF6−) have afforded conducting materials that retain metallic conductivity down to 4.2 K [Citation59, Citation60]. X-ray structure analysis revealed that these materials have β-type donor packing with the composition of (TMEO-ST-TTP)2A. In these salts, the overlap mode of the donors in the stack is the ring-over-bond type (figure ). The slip distances along the long molecular axis are about half of the 1,3-dithiole ring for p1 (1.6–1.7 Å), and one-and-a-half for p2 (4.9–5.0 Å). As a result, the donors are electronically dimerized along the stacking direction, although the interplanar distances are almost the same (3.50 and 3.52 Å for the PF6− salt). The ratio of the calculated overlap integrals (p1/p2) is 2.3 for the PF6− salt. A tight-binding band calculation suggests that the upper and lower bands are completely separated, which corresponds to strong dimerization along the stack, and results in the formation of an effectively half-filled band. The interstack overlaps are 10–20% of the larger intrastack overlap. Thus, the calculated Fermi surface is closed along the stacking direction, but open along the interstack direction as is observed in a quasi-1D system. However, the interstack interactions are sufficiently strong to suppress the metal-to-insulator transition at low temperature.
Figure 11 Crystal structure of (TMEO-ST-TTP)Au(CN)2. (Reprinted with permission from [Citation59].)
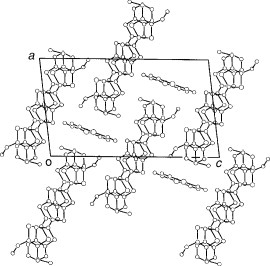
Figure 12 Overlap modes of donor molecules in (TMEO-ST-TTP)Au(CN)2. (Reprinted with permission from [Citation59].)
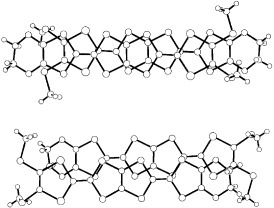
Figure 13 Overlap modes of donor molecules in (TMEO-ST-TTP)2AsF6. (Reprinted with permission from [Citation60].)
(BEDT-TTP)2I3 and (EP-TTP)2Au(CN)2
The title materials have the donor packing close to β-(BEDT-TTF)2I3 [Citation61, Citation62]. However, the degree of dimerization in the stack is much smaller than for BEDT-TTF salts, because the longer π-electron framework of BDT-TTP is less sensitive to overlap integral than TTF systems. A tight-binding band calculation of (BEDT-TTP)2I3 suggests the existence of a 2D Fermi surface as shown in figure , which is also supported by angle-dependent magnetoresistance [Citation76]. On the other hand, the calculated Fermi surface of (EP-TTP)2Au(CN)2 is quasi-1D, because the lateral interaction is smaller than that of (BEDT-TTP)2I3 owing to steric hindrance by the projected propylenedithio group along the lateral direction. Both the salts exhibit high conductivity of σrt=600–900 S cm−1 and retain metallic conductivity down to 0.5–0.6 K. The temperature dependence of resistivity approximately obeys the T2 law. Such simple metallic behavior indicates that the metallic states of these salts are significantly stabilized. As a result, they show no superconductivity in spite of being isostructural with the ambient-pressure superconductor β-(BEDT-TTF)2I3.
Figure 14 The Fermi surface of (BEDT-TTP)2I3.
Conducting materials based on TMET-TTP and its analogs
A BDT-TTP derivative with both methylthio and ethylenedithio groups TMET-TTP and its selenium analogs have afforded θ-type salts regardless of the counter anions [Citation19, Citation20, Citation35, Citation37, Citation63]. It is noteworthy that even TCNQ has yielded the θ-type conductors [Citation20]. In these materials, the donors make pseudo-stacks, in which they slip along both the short and long axes of the donor as observed for θ-(BTM-TTP)2SbF6 [Citation54]. The dihedral angles between molecular planes of adjacent stacks are as large as 118–128°. Most of the θ-type salts based on TMET-TTP-type donors have uniform stacks, whereas the donors form dimerized stacks in (TMES-TTP)4I3. The calculated Fermi surface is 2D and is similar to that of θ-type BEDT-TTF salts. These θ-type salts show a metal-to-insulator transition at low temperatures or exhibit semiconducting behavior at and below room temperature. Destabilization of the metallic state in TMET-TTP conductors is probably due to the narrow bandwidth and resultant strong electron correlation derived from large dihedral angles between molecular planes, as pointed out in θ-BEDT-TTF salts by H Mori et al [Citation77].
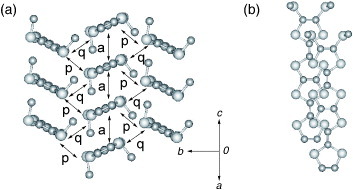
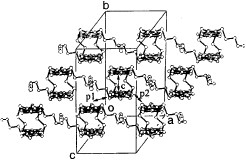
The BDT-TTP derivatives with long alkyl chains have been investigated by T Mori and co-workers to change the ratio of donor to anion in TMET-TTP salts, most of which have the composition (TMET-TTP)4A. Among them, the crystal structures of (C2TET-TTP)2ClO4, (C2TET-TS-TTP)2A (A=PF6, ClO4, BF4) and (C4TET-TTP)I3 have been determined [Citation28, Citation29]. The radical cation salts based on C2TET-TTP and C2TET-TS-TTP are isostructural. They have β-type donor packing contrary to the initial expectation of the θ-type array, although the ratio of the donor to anion (2 : 1) has indeed changed from 4 : 1. Surprisingly, the donors form a uniform stack in spite of the presence of sterically hindered ethylthio groups, whereas most donors with large substituents are dimerized to avoid steric hindrance as is observed in CH-TTP (κ-type) and TMEO-ST-TTP (dimerized β-type) salts, with the exception of TMET-TTP (θ-type) and BTM-TTP (both β- and θ-types). In the C2TET-TTP salts (figure ), large steric hindrance in the stack derived from the ethyl chain is reduced by a larger slip along the donor long axis (4.7 Å) than that of (BDT-TTP)2ClO4 (1.6 Å), which also has a uniform β-type donor array. Whereas the donors form a pseudo stack so that steric hindrance is avoidable by a slip along the donor short axis in the θ-type conductors. All the C2TET-(TS)-TTP salts with tetrahedral and octahedral anions exhibit metallic conductivity down to 1.5 K owing to the uniform stacks of a relatively large bandwidth. On the other hand, two donor molecules and an anion are alternatively stacked in (C4TET-TTP)I3, in which the donors are stacked in a head-to-tail manner (figure ). The dimer is surrounded by four I3− anions in the ac-plane, and the interdimer overlaps p1 and p2 are only −0.6 and 0.03×10−3, respectively. Therefore, the bandwidth of this material is expected to be very small. It shows low conductivity of 0.025 S cm−1 at room temperature, and has a large activation energy of 0.23 V, which corresponds with a small bandwidth and a 1 : 1 composition.
Figure 15 Donor sheet structure of (C2TET-TTP)2ClO4. The intermolecular overlap integrals are b=−20.2, p1=7.0, p2=2.1, p3=−0.9 and p4=9.6× 10−3. (Reprinted with permission from [Citation28].)
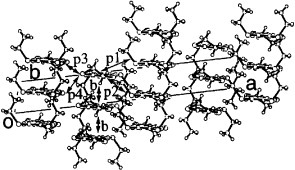
Figure 16 Donor sheet structure of (C4TET-TTP)I3. The intermolecular overlap integrals are c=−16.6, p1=−0.6 and p2=0.03× 10−3. (Reprinted with permission from [Citation28].)
(TTM-TTP)Ax
Tetrakis(thiomethyl) derivative TTM-TTP has afforded three types of I3− salts: (TTM-TTP)2I3, (TTM-TTP)I3 and (TTM-TTP)(I3)5/3 [Citation64, Citation65]. Among them, (TTM-TTP)2I3 is a low-conductive (σrt=0.03 S cm−1) semiconductor with a trimerized donor stack [Citation64]. The crystal includes a neutral donor molecule in the anion layer. (TTM-TTP)I3 exhibits very high conductivity of σrt=700 S cm−1, in spite of the fully oxidized +1 state of the donor. It shows metal-like temperature dependence down to TMI=160 K (figure ) [Citation64]. TTM-TTP molecules form uniform columns with a relatively short interplanar distance of 3.45 Å (figure ). The arrangement of the donors may be classified as β-type. However, effective lateral interaction is inhibited, because the methylthio groups protrude from the long molecular axis. A tight-binding band calculation suggests that (TTM-TTP)I3 has a large bandwidth of W=1 eV. The nature of the insulating state of (TTM-TTP)I3 has been investigated, but has not get been elucidated [Citation76–81]. On the other hand, (TTM-TTP)(I3)5/3 also has uniform donor stacks similar to (TTM-TTP)I3, and it exhibits metallic conductivity down to TMI=20 K (see also figure ). The donor molecule in this material has an anomalously large positive charge of +5/3. The metallic conductive behavior in spite of large positive charges of +1 and +5/3 in TTM-TTP conductors is due to the small on-site Coulomb energy as well as the large bandwidth derived from uniform stacks [Citation65]. The AuI2− and MX4− (M=Fe, Ga,X=Cl, Br) anions also yield 1:1 salts; however, anions that are shorter than I3− make dimerized columns [Citation66–68]. As a result, they exhibit the semiconducting behavior characteristic of band insulators. In contrast, (TTM-TTP)FeBr1.8Cl2.2 exhibits metallic behavior similar to (TTM-TTP)I3, because it occasionally forms uniform donor columns [Citation68].
Figure 17 Temperature dependence of resistivity of (TTM-TTP)I3 and (TTM-TTP)(I3)5/3.
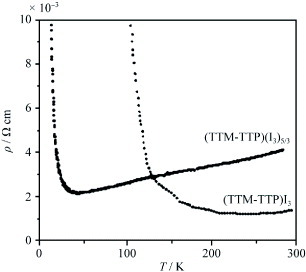
Figure 18 Stacking structure of (TTM-TTP)I3. (Reprinted with permission from [Citation64].)
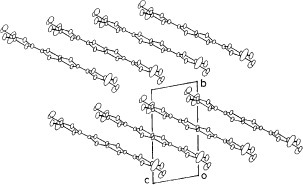
(CPTM-TTP)4A(A = PF6, AsF6, SbF6)
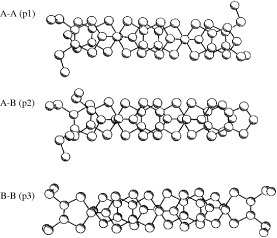
The title materials are isostructural and form molecular packing similar to β-type salts [Citation17]. However, they have a tetramerized column in which two crystallographically independent donors A and B are stacked as AABB as shown in figure . Therefore, the donor sheet structure of these salts is closer to that of λ–(BETS)2GaCl4, an ambient-pressure superconductor with Tc=8 K [Citation82], rather than to that of β-type BEDT-TTF salts. The donor molecules are stacked in a head-to-tail manner in A–A (p1) and B–B (p3), and in a head-to-head manner in A–B (p2). The interplanar distances are 3.58, 3.61 and 3.40 Å for p1, p2 and p3, respectively (for the AsF6− salt). All the overlap modes are the ring-over-bond type: however, the slip distance along the long molecular axis (D) of p3 (4.73 Å) is much larger than those of p1 (1.55 Å) and p2 (1.68 Å). Such a large D value of p3 does not affect the overlap integral very much in this case, because BDT-TTP is about twice as long as TTF. Thus, the overlap of p3 (18.1× 10−3 for the AsF6− salt) is only about 20% smaller than those of the other intrastack interactions. On the other hand, there are relatively large interstack interactions (q2 and q4). As a result, the Fermi surface calculated by the tight-binding method is a typical eclipse characteristic of 2D metals. Among these salts, the PF6− and AsF6− salts exhibited metallic conductivity down to 4.2 K, although maximum resistivity was observed around 80 K. In contrast, an increase in resistivity below 50 K has been observed for the SbF6− salt. Although the low-temperature behavior of the SbF6− salt is not clear, it is plausible that the largest anion of SbF6− among those three salts induces a strong electron correlation.
Figure 19 Overlap modes of donor molecules in (CPTM-TTP)4AsF6.
TTP analogs including non-TTF moiety
Various TTP donors possessing non-TTF moieties have been synthesized (see scheme ) [Citation83–125]. Molecular packing patterns and electrical properties of molecular conductors based on these TTP analogs are summarized in table 3.
Table 3 Molecular packing patterns and electrical properties of conducting salts based on TTP donors containing non-TTF moieties (scheme ).
Scheme 9 Molecular structures of analogous TTP derivatives.
Scheme 10 Molecular structures of analogous TTP derivatives listed in table 3.
Radical cation salts based on vinylogous TTP donors, DTEDT and DSEDS
The BDT-TTP molecule has a stripe-like structure, and all sulfur atoms have the same phase in the HOMO (figure ). On the contrary, vinylogous TTP donor 2-(1,3-dithiol-2-ylidene)-5-(2-ethanediylidene-1,3-dithiole)-1,3,4,6-tetrathiapentalene (DTEDT) has a crooked structure, and the π-orbitals of sulfur atoms at the terminal of the vinylogous 1,3-dithiole ring in the HOMO are out of phase with those of the other sulfur atoms (see also figure ). Thus, DTEDT conductors are expected to show both sterically and electronically less effective lateral interactions than BDT-TTP salts. However, similar to BDT-TTP, DTEDT has afforded many metallic radical cation salts regardless of the size and shape of the counter anions. Furthermore, (DTEDT)3Au(CN)2 has exhibited a superconducting transition at 4 K under ambient pressure (figure ) [Citation83, Citation84]. The donors form conducting sheets parallel to the ac-plane as shown in figure . The array of the donors is close to that of most BDT-TTP salts of the β-type with an almost uniform stack (figure ). In contrast to most β-type salts based on unsymmetrical donors, DTEDT molecules are arranged in such a way that the stacking and lateral directions are parallel in (DTEDT)3Au(CN)2. Because DTEDT has a crooked structure and the phase of the sulfur atoms in the HOMO is reversed at the terminal vinylogous 1,3-dithiole ring, such a parallel array results in the most effective intermolecular interaction both sterically and electronically. On the other hand, a selenium analog of DTEDT, DSEDS, has afforded a metallic TaF6− salt, in which the array of donors is identical to the average structure of (DTEDT)3Au(CN)2 [Citation86]. Therefore, DTEDT may have a strong tendency to form the β-type donor arrangement similar to BDT-TTP. A tight-binding band calculation suggests that (DTEDT)3Au(CN)2 has a closed Fermi surface, although the one-dimensional character along the stacking direction is stronger because the stacks are almost uniform.
Figure 20 HOMO of DTEDT (left) and BDT-TTP (right).
Figure 21 Temperature dependence of resistivity of (DTEDT)3Au(CN)2. (Reprinted with permission from [Citation83].)
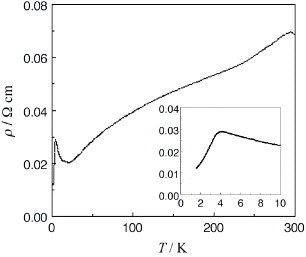
Figure 22 Crystal structure of (DTEDT)3Au(CN)2. (Reprinted with permission from [Citation83].)
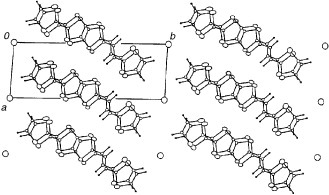
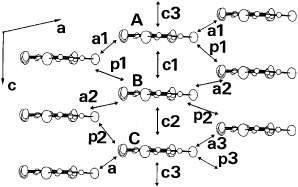
Figure 23 Donor-sheet structure of (DTEDT)3Au(CN)2. The intermolecular overlap integrals are c1=−19.2, c2=−19.9, c3=−10.8, a1=−1.5, a2=−1.3, a3=−0.7, p1=−3.8, p2=−3.4 and p3=−6.9× 10−3.
Radical cation salts based on (thio)pyran analogs
Three-dimensional (3D) organic conductors are of significant interest from the viewpoint of the search for molecular superconductors with higher critical temperatures (Tc), because M3C60 (M=alkali metal) in which C60 molecules are arranged in a 3D fashion show superconductivity with the highest Tc ∼ 40 K among molecular materials [Citation144]. It has also been pointed out that an increase of 3D character through an insulating anion layer results in higher Tc in BEDT-TTF superconductors [Citation145, Citation146]. However, a 3D Fermi surface has not yet been observed in solids based exclusively on organic molecules, but it has been theoretically suggested by a tight-binding band calculation in a metal dithiolene system, MexH4−xN[Pd(dmise)2]2 (x=2, 3) [Citation147]. TTP donors containing the pyran-4-ylidene moiety, 2-(1,3-dithiol-2-ylidene)-5-(pyran-4-ylidene)-1,3,4,6-tetrathiapentalene (PDT-TTP) [Citation89], and its thiopyran analog (TPDT-TTP) [Citation90], are potentially interesting for the preparation of 3D conductors, because a significant intermolecular overlap along the long molecular axis is expected through chalcogen atoms in the (thio)pyran ring, in addition to the π–π stacking and lateral chalcogen–chalcogen interactions.
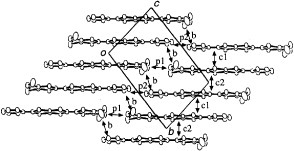
The ethylenedithio derivative of PDT-TTP (ET-PDT) has yielded a 4 : 1 salt with the PF6− anion (ET-PDT)4PF6(cn) (cn=1-chloronaphthalene) [Citation89]. The ET-PDT molecules form conducting sheets, each being divided by insulating layers composed of the anions and solvent molecules. The array of the donors is classified as the so-called λ-type (figure ). Thus, two crystallographically independent ET-PDT molecules A and B form a face-to-face stack with a four-folded period AABB similar to (CPTM-TTP)4A (A=PF6, AsF6, SbF6) [Citation17]. The molecules are stacked in a head-to-tail manner for molecules AA′ and BB′ and in a head-to-head manner for AB (figure ). This salt has an almost uniform stack from the viewpoint of interplanar distances (3.60–3.62 Å). However, the head-to-tail overlap between the unsymmetrical π-electron frameworks of PDT-TTP prevents effective intrastack overlaps, because there is little consistency in the phases of the molecular coefficients between the outer 1,3-dithiole ring and the pyran ring. The calculated overlap integrals of the head-to-tail stack, p1 (8.6× 10−3) and p3 (4.9× 10−3), are only about 30–50% of the head-to-head overlap p2 (−16.4× 10−3), indicating that the ET-PDT molecules are strongly tetramerized along the stacking direction. Such difference in the manner of overlap does not affect the overlap integrals in the (CPTM-TTP)4A (19.7–21.3× 10−3 for A=PF6), which has a symmetrical π-electron framework of BDT-TTP. On the other hand, relatively large interstack overlaps a1 (4.4× 10−3) and c2 (−4.5× 10−3) are coupled with the largest intrastack overlap integral p2 (figure ). Thus, the overlap along the c-direction is much larger than that along the a-direction. As a result, the calculated Fermi surface is open along ka.
Figure 24 Donor-sheet structure of (ET-PDT)4PF6 (cn) (Reprinted with permission from [Citation89].)
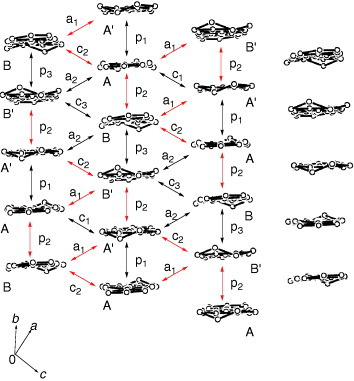
Figure 25 Overlap modes of the donor molecules in (ET-PDT)4PF6(cn). (Reprinted with permission from [Citation89].)
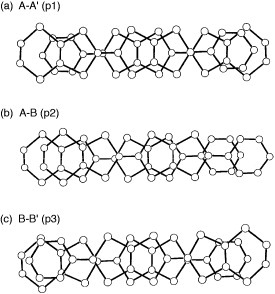
Figure 26 A schematic drawing of overlaps between the donor molecules in (ET-PDT)4PF6(cn); bars and broken lines denote the donor molecules projected along the long molecular axis and relatively large intra- and interstack interactions, respectively.
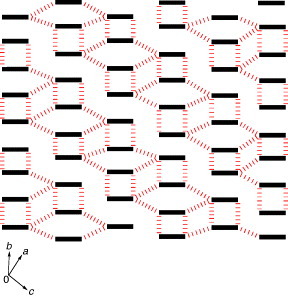
Radical cation salts of bis(methylthio)-derivative TM-TPDS have quite a different donor packing from that of (ET-PDT)4PF6(cn). The crystal structure of (TM-TPDS)2AsF6 is shown in figure . This salt does not form a 2D conducting sheet in contrast to the typical TTP conductors. Thus, the TM-TPDS molecules are arranged in a ‘windmill’ manner in the bc-plane, whereas they form a conduction column along the a-axis [Citation92]. The anions are located in a cavity formed in the center of the windmill and are surrounded by hydrogen atoms of the methylthio groups and of the thiopyran rings in the donors. The unsymmetrical TM-TPDS molecules are stacked in a head-to-tail manner along the a-axis and are slightly dimerized. Several sulfur–sulfur contacts are shorter than the sum of van der Waals radii between the central tetrathiapentalene moiety and the thiopyran ring or methylthio groups. The calculated overlap integrals between the columns are as large as ∼10% of the intracolumnar overlaps, thanks to the relatively large molecular orbital coefficient of the sulfur atom in the thiopyran ring and to the short sulfur–sulfur contacts. However, the calculated Fermi surface is closed only along the ka direction because intrastack interactions are still dominant in this material. (TM-TPDS)2AsF6 has high conductivity of σrt=240 S cm−1 and metal-like conducting behavior down to 100 K. At lower temperatures, the resistivity gradually increased. This metal-to-semiconductor transition should not be a Peierls transition derived from highly 1D character, because no obvious decrease of magnetic susceptibility has been observed below TMI.
Figure 27 Crystal structure of (TM-TPDS)2AsF6 projected onto the bc-plane. Dotted lines show the intermolecular S···S contacts.
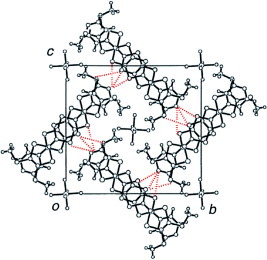
Exchange of sulfur (or oxygen) atoms with selenium atoms on tetrathiapentalene, methylthio and (thio)pyran-4-ylidene moieties is expected to form a windmill structure with a higher 3D character. For this reason, bis(methylseleno) derivatives were investigated. SM-PDT has yielded single crystals of PF6− and AsF6− salts suitable for x-ray structure analysis and electrical conductivity measurement [Citation93]. Unexpectedly, these salts adopt a fully oxidized state with a composition of (SM-PDT)A(PhCl)x (A=PF6 and AsF6), in which the arrangement of donors is quite different from that of (TM-TPDS)2AsF6. Thus, the donor columns are completely divided by the counter anions and by disordered solvent molecules along the a-axis (figure ), indicating the absence of lateral interaction. Instead, the donor columns are bridged by short selenium–selenium contacts within the sum of the van der Waals radii to form a 2D sheet-like structure. The Se···Se contacts between selenomethyl groups may be more preferable in this salt than the edge-to-side interactions through the tetrathiapentalene moiety, as observed in (TM-TPDS)2AsF6. The donors are dimerized in the column with slip distances along the donor long axis of 1.4 and 4.7 Å. On the other hand, the interstack overlap integrals through the selenomethyl groups are ∼1/4 of the intrastack ones (figure ), and they are significantly larger than the longitudinal interaction. Calculation suggests that this salt is a band insulator because of its fully oxidized state and dimerized stack. However, it has relatively high conductivity of σrt=4.8 S cm−1. Considering the high conductivity (in spite of being a 1 : 1 salt) and the relatively large edge-to-edge interaction, the 2 : 1 salts with the same donor array are expected to exhibit metallic conductivity down to low temperature.
Figure 28 Crystal structure of (SM-PDT)PF6(PhCl)x viewed along the c-axis. Disordered solvent molecules are located in the cavity.
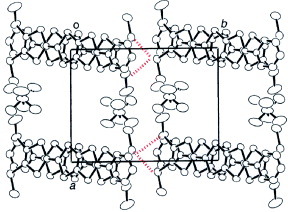
Figure 29 Donor column structure viewed along the a-axis and definition of the overlap integrals of (SM-PDT)PF6(PhCl)x. The intermolecular overlap integrals are c1=6.3, c2=8.1, p1=−2.3, p2=0.00 and b=−0.8× 10−3. (Reprinted with permission from [Citation93].)
TTP donors with reduced π-electron system
TTP-type donors have yielded many molecular metals stable down to low temperatures, but only one superconductor. This is probably due to excessive stabilization of the metallic state by the large bandwidth as well as reduced on-site Coulomb repulsion [Citation148].
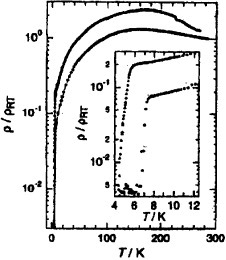
Yamada et al have developed molecular conductors based on BDH-TTP and 2,5-bis(1,3-dithian-2-ylidene)-1,3,4,6-tetrathiapentalene (BDA-TTP) [Citation97, Citation98]Footnote1 whose π-electron systems have been reduced by the exchange of double bonds in the outer 1,3-dithiole rings of BDT-TTP with a saturated ethylene or propylene bridge. Cyclic voltammetry revealed that the ΔE value becomes larger than that in BDT-TTP, suggesting an increase of the on-site Coulomb repulsion by reduction of the π-electron system. Among the conducting materials based on BDH-TTP and BDA-TTP, most BDH-TTP conductors are metallic down to low temperatures. X-ray structure analysis of (BDH-TTP)2PF6 reveals that the donors adopt the κ-type arrangement [Citation97]. In this material, intradimer overlap is much smaller than those of κ-type BEDT-TTF superconductors due to steric hindrance at the terminal sp3 carbons. In contrast, interdimer interaction should be larger than in BEDT-TTF salts, because all the rings are five-membered. As a result, the split of the HOMO bands characteristic of κ-type conductors should not occur in this system. Thus, the metallic state of this material is stabilized considerably by the realization of a quarter-filled band instead of the half-filled one as observed in κ-type BEDT-TTF superconductors [Citation5]. On the other hand, BDA-TTP conductors have a strong tendency to take β-type molecular packing. In these materials, intrastack interaction is smaller compared with that of typical β-type conductors and the degree of dimerization is very large, because the six-membered rings at the terminal of BDA-TTP largely protrude from the molecular plane as shown in figure . It is noteworthy that BDA-TTP affords several superconductors with various anions of octahedral (PF6−, AsF6−, SbF6−), tetrahedral (FeCl4−, GaCl4−) and linear (I3−) shapes (figure ) [Citation98, Citation129–131].
Figure 30 Molecular structure of BDA-TTP in (BDA-TTP)2SbF6; the dihedral angles φ1 and φ2 are 47.9 and 33.2°, respectively. (Reprinted with permission from [Citation98].)
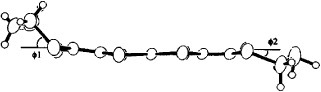
Figure 31 Temperature dependence of normalized resistivity in BDA-TTP superconductors. (Reprinted with permission from [Citation98].)
The author and co-workers have developed many reduced TTP derivatives, in which one of the 1,3-dithiole rings is removed [Citation43, Citation100–103]. An ethylenedithio derivative, MeDTET yields many metallic radical cation salts stable down to 0.6–1.5 K regardless of the counter anions. Among metallic MeDTET salts, x-ray structure analysis of the PF6− salt revealed that this salt has the (MeDTET)3PF6(TCE)x composition as well as κ-type packing of donors [Citation134]. No observation of superconductivity, even though it is a κ-type salt, is probably owing to reduced on-site Coulomb repulsion derived from a partially filled band rather than a half-filled band. A cyclopentane analog CPDTET also yields κ-type salts, (CPDT-TTF)2(AsF6)0.72 and (CPDT-TTF)2(SbF6)0.88 [Citation135], in which two donor molecules are strongly dimerized with each other in a ‘head-to-head’ manner (figure ). However, the transport properties are very different between these two salts. (CPDT-TTF)2(AsF6)0.72 exhibits metallic temperature dependence down to 4.2 K. In contrast, (CPDT-TTF)2(SbF6)0.88 shows a metal-to-semiconductor transition at 200 K, and it retains metallic behavior down to low temperatures under a high pressure of 5.6 kbar. The different transport properties are probably due to different occupancies of the anion sites. The band structure of the SbF6− salt is very close to a half-filled band, leading to a metal–semiconductor transition due to strong electron correlation. In contrast, the band filling of the AsF6− salt is far from half-filled because there are more anion defects than in the SbF6- salt, which hampers the semiconducting transition. On the other hand, a selenium analog CPDT-STF forms a TCNQ complex with 2D β-type donor packing. (CPDT-STF)(TCNQ) exhibits metallic conductivity down to low temperatures [Citation102]. A dimethyl-substituted derivative, MeDTDM, yields β-type salts. They show metallic conductivity down to 20 K, below which the resistivity increases [Citation101].
Figure 32 Overlap mode in a dimer of (CPDT-TTF)2(AsF6)0.72.
Nishikawa et al investigated TTP analogs, MeDH-TTP and MeDA-TTP, with more reduced π-conjugation [Citation103, Citation104]. MeDA-TTP salts with octahedral and tetrahedral anions are isostructural and have β-type donor packing similar to that in BDA-TTP superconductors [Citation104]. They exhibit metallic behavior down to 165–205 K, whereas the ClO4− salt is a semiconductor with a small activation energy of 0.028 eV. (MeDH-TTP)2AsF6 crystallizes in a κ-type structure [Citation103]. A tight-binding calculation suggests the dimerization gap in this salt is much smaller than in typical κ-type BEDT-TTF conductors, leading to an almost 3/4-filled band (figure ). This salt shows semiconducting behavior below room temperature with a small activation energy of 0.032 eV. ESR measurements suggest an antiferromagnetic transition at 40 K [Citation149]. A detailed study of the dependence of resistivity on T-P revealed simple metallic behavior above Pc=2.4 GPa [Citation150], which is much larger than the critical pressure for a typical Mott transition in κ-(BEDT-TTF)2Cu[N(CN)2]Cl (Pc=0.03 GPa) [Citation151] and is similar to that observed in the charge-ordered insulator (DIDCNQ)2Ag (Pc=2.2 GPa) [Citation152]. Considering this salt has a quarter-filled band and a large Pc, the most plausible origin of the insulating state for (MeDH-TTP)2AsF6 is the charge-ordering state [Citation150].
Figure 33 Band structure and Fermi surface of (MeDH-TTP)2AsF6. (Reprinted with permission from [Citation149].)
Higher homologs of TTP
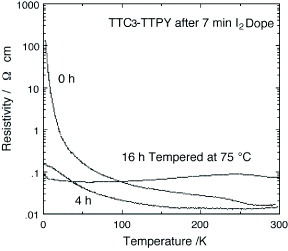
The higher homologs of TTP donors are also of interest, not only as a promising component for molecular metals but also as a candidate for molecular electronics. A tris-fused TTF, 2,2′-bi(1″,3″-dithiol-2″-ylidene-1,3,4,6-tetrathiapentalene-5-ylidene) (BDT-TTPY, scheme ), has been synthesized by the P(OEt)3-mediated coupling reaction of 1,3-dithiol-2-one fused with TTFs (5) [Citation107, Citation108]. The cyclic voltammograms of BDT-TTPY derivatives measured in benzonitrile-carbon disulfide are composed of three pairs of redox waves, each corresponding to a two-electron transfer [Citation107]. The solubility of BDT-TTPYs in common organic solvents is extremely low compared with the corresponding BDT-TTP derivatives. Therefore, the usual electrochemical oxidation technique cannot be adopted for the preparation of conducting materials. Instead, chemical doping of the BDT-TTPY suspension with appropriate oxidants such as iodine or NO2X (X=BF4, SbF6) has been carried out. Among the conducting materials obtained, several iodine complexes have high conductivity of σrt=0.7–16 S cm−1 on compressed pellet samples [Citation107, Citation108], and several have very small activation energies of 0.017–0.033 eV. Therefore, these materials are expected to exhibit metallic conductivity if conductivity measurements are performed on single crystals. A crystal of TTC3-TTPY doped with iodine vapor exhibits metal-like conductivity down to 1.3 K, as shown in figure [Citation110].
Figure 34 Temperature dependence of resistivity of TTC3-TTPY after 7 min iodine doping followed by annealing at 75 °C. (Reprinted with permission from [Citation110].)
Analogous TTPY donors containing a π-electron framework other than TTF have also been synthesized. In particular, extended TTPYs, in which an aromatic ring, such as benzene, thiophene or furan, is inserted into the central TTF moiety, exhibit higher solubility in common solvents [Citation11Citation2]. As a result, CT complexes have been formed by the usual mixing technique in organic solvent. The TCNQF4 complexes based on 14–16 exhibit relatively high conductivity of σrt=10−1–100 S cm−1 on compressed pellets. Higher homologues of TTPY possessing up to seven donor units have also been synthesized thanks to increased solubility using a thiophene-inserted TTF unit [Citation113].
Analogous metal complexes of BDT-TTPY, M (dt)>2: highly conducting single-component molecular conductors
The derivatives of metal bis(TTF-dithiolato) (M(dt)2), where dt is 2-(1,3-dithiol-2-ylidene)-1,3-dithiole-4,5-dithiolate, designed by exchange of the central double bond of BDT-TTPY with a metal ion, have received significant attention as candidates for single-component molecular metals, because both the donor (TTF) and acceptor (metal bis(dithiolato)) moieties are present in the molecule. Schumaker and Engler have reported, the synthesis of metal complexes with TTF-dithiolato ligands M(dt)2 as analogs of BDT-TTPY [Citation8]. Unfortunately, most anionic M(dt)2 complexes are readily oxidized to insoluble neutral complexes due to the presence of strongly electron-donating TTF moieties. The first synthesis and characterization of anionic M(dt)2 complexes have been reported independently by the groups of Underhill (M=Hg) and Matsubayashi (M=Au) [Citation114, Citation115]. Several M(dt)2 derivatives have been prepared since then: however, the synthesis of single crystals of neutral complexes remains difficult.
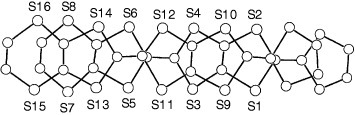
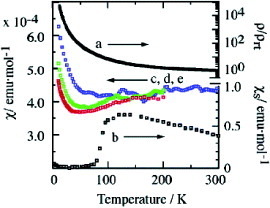
Kobayashi and co-workers have succeeded in growing single crystals of various M(dt)2 derivatives using an electrocrystallization technique and have studied their structures and physical properties [Citation119–123, Citation138–141]Footnote2. Among them, Ni(tmdt)2 exhibits very high conductivity of σrt=400 S cm−1 on a single crystal. The temperature dependence of the resistivity and magnetic susceptibility reveals metallic conducting behavior down to 0.6 K [Citation120]. Figure shows the crystal structure of Ni(tmdt)2. The packing pattern of the molecules viewed along the long molecular axis resembles the so-called β′-type as observed in BEDT-TTF conductors. On the other hand, half of the molecules are overlapped in the stack: namely, one molecule is stacked to extend over two molecules. A tight-binding band calculation, assuming a HOMO-LUMO (lowest unoccupied molecular orbital) gap of 0.1 eV, suggests that this material has a 3D Fermi surface. This result is consistent with the Fermi surface experimentally obtained by measuring the de Haas–van Alphen effect [Citation151]. A gold complex, Au(tmdt)2, is isostructural with Ni(tmdt)2. The ESR, nuclear magnetic resonance, and magnetic susceptibility measurements revealed an antiferromagnetic transition at 110 K in this complex (figure ) [Citation139]. Electrical conductivity of single-crystal Au(tmdt)2 revealed metallic behavior down to low temperatures, indicating coexistence of itinerant electrons and localized magnetically ordered spins. Thus, Au(tmdt)2 is a new antiferromagnetic molecular metal with a critical temperature TN above 100 K.
Figure 35 Crystal structure of Ni(tmdt)2 viewed along the molecular (a) short and (b) long axis. (Reprinted with permission from ref. [Citation137].)
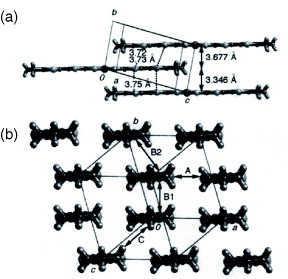
Figure 36 Resistivities and susceptibilities of Au(tmdt)2: (a), resistivity of compacted pellet (ρrt=0.067Ω cm); (b), spin susceptibility χs by ESR; (c)–(e), magnetic susceptibilities by SQUID at 30 kOe (blue, <300 K), 10 kOe (red, <200 K) and 3 kOe (green, <200 K). (Reprinted with permission from [Citation13Citation9].)
Summary and outlook
In summary, BDT-TTP derivatives and their analogs have yielded numerous radical cation salts and charge-transfer complexes that retains metallic conductivity down to low temperatures (4.2 K). Over 100 TTP-type molecular metals stable down to liquid helium temperatures have been obtained thus far. Stabilization of the metallic state in TTP conductors is mainly achieved by increasing dimensionality, thanks to lateral interaction through chalcogen atoms in the tetrathiapentalene moiety. It is noteworthy that a TTP vinylog, DTEDT, has yielded many molecular conductors exhibiting metallic conductivity down to 4.2 K regardless of the counter anions, although its half-unit of vinylogous TTF itself has never yielded any metallic material down to low temperatures. Furthermore, highly conducting metals with unusual oxidization states (+1, +5/3 and neutral) have also been developed, thanks to the long molecular structure with large π-electron conjugation of TTP-type molecules. The fusion of two (or more) donor units, in other words, the insertion of the tetrathiapentalene moiety into π-electron donors, is a reliable guiding principle to design stable molecular metals. However, the metallic state is too stabilized to cause superconducting transitions in many cases, with the exception of BDA-TTP systems. Furthermore, various chemical modifications of TTP molecules are important to control the bandwidth, band-filling and on-site Coulomb repulsion in their conducting materials and to develop new superconductors with higher Tc and novel molecular metals with multifunctionality, such as magnetic molecular metals or photoinduced highly conducting materials.
Acknowledgments
The author is grateful to Dr T Shirahata and Professor H Nishikawa for valuable discussions. This work was partially supported by Grants-in-Aid for Scientific Research (Nos. 15073216, 18GS0208 and 20110006) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, from the Japan Society for the Promotion of Science, and from the Ehime University COE incubation program.
Notes
For a overview of the reduced TTP conductors see [Citation136].
For a overview of molecular conductors based on M(dt)2 derivatives see [Citation137].
References
- FerrarisJCowanD OWalatkaJrVPerlsteinJ H 1973 J. Am. Chem. Soc. 95 948 http://dx.doi.org/10.1021/ja00784a066
- JéromeDMazaudARibaultMBechgaardK 1980 J. Phys. Lett. 41 L95 http://dx.doi.org/10.1051/jphyslet:0198000410409500
- BechgaardKCarneiroKRasmussenF BOlsenMRindorfGJacobsenC SPedersenH JScottJ C 1981 J. Am. Chem. Soc. 103 2440 http://dx.doi.org/10.1021/ja00399a065
- 2004 TTF Chemistry—Fundamentals and Applications of Tetrathiafulvalene, JYamada TSugimoto Kodansha-Springer
- IshiguroTYamajiKSaitoG 1998 Organic Superconductors 2nd edn Springer Series Solid-State Science, vol 88, Berlin Springer
- SaitoGEnokiTToriumiKInokuchiH 1982 Solid State Commun. 42 557 http://dx.doi.org/10.1016/0038-1098(82)90607-X
- WilliamsJ MFerraroJ RThornR JCarlsonK DGeiserUWangH HKiniA MWhangboM-H 1992 Organic Superconductors New Jersey Prentice-Hall
- SchumakerR REnglerE M 1980 J. Am. Chem. Soc. 102 6652 http://dx.doi.org/10.1021/ja00541a086
- MisakiYNishikawaHKawakamiKKoyanagiSYamabeTShiroM 1992 Chem. Lett. 21 2321 http://dx.doi.org/10.1246/cl.1992.2321
- MisakiYMatsuiTKawakamiKNishikawaHYamabeTShiroM 1993 Chem. Lett. 22 1337 http://dx.doi.org/10.1246/cl.1993.1337
- MisakiYKochiTYamabeTMoriT 1998 Adv. Mater. 10 588 http://dx.doi.org/10.1002/(SICI)1521-4095(199805)10:8<588::AID-ADMA588>3.0.CO;2-U
- MisakiYKochiTTaniguchiMYamabeTTanakaKTakimiyaKMorikamiAOtsuboTMoriT 1999 Synth. Met. 102 1675 http://dx.doi.org/10.1016/S0379-6779(98)00574-8
- MisakiYNishikawaHYamabeTMoriTInokuchiHMoriHTanakaS 1993 Chem. Lett. 22 729 http://dx.doi.org/10.1246/cl.1993.729
- MisakiYNishikawaHKawakamiKYamabeTMoriTMoriHTanakaS 1993 Chem. Lett. 22 2073 http://dx.doi.org/10.1246/cl.1993.2073
- MisakiYKawakamiKMatsuiTYamabeTShiroM 1994 J. Chem. Soc. Chem. Commun. 459
- MoriTMisakiYKawakamiKYamabeTMoriHTanakaS 1995 Synth. Met. 70 875 http://dx.doi.org/10.1016/0379-6779(94)02686-S
- MisakiYKawakamiKFujiwaraHMiuraTKochiTTaniguchiMYamabeTMoriTMoriHTanakaS 1997 Mol. Cryst. Liq. Cryst. 296 77 http://dx.doi.org/10.1080/10587259708032315
- AshizawaMAragakiMMoriTMisakiYYamabeT 1997 Chem. Lett. 26 649 http://dx.doi.org/10.1246/cl.1997.649
- MoriTAshizawaMAragakiMMurataKMisakiYTanakaK 1998 Chem. Lett. 27 253 http://dx.doi.org/10.1246/cl.1998.253
- AragakiMHoshinoHMoriTMisakiYTanakaKMoriHTanakaS 2000 Adv. Mater. 12 983 http://dx.doi.org/10.1002/1521-4095(200006)12:13<983::AID-ADMA983>3.0.CO;2-7
- MoriTKawamotoTMisakiYTanakaKMoriHTanakaS 1998 Bull. Chem. Soc. Japan 71 1321 http://dx.doi.org/10.1246/bcsj.71.1321
- MisakiYTanakaKTaniguchiMYamabeTMoriT 1999 Adv. Mater. 11 25 http://dx.doi.org/10.1002/(SICI)1521-4095(199901)11:1<25::AID-ADMA25>3.0.CO;2-E
- TaniguchiMMisakiYTanakaKYamabeTMoriT 1999 Synth. Met. 102 1721 http://dx.doi.org/10.1016/S0379-6779(98)00895-9
- MisakiY et al 1999 Synth. Met. 102 1781 http://dx.doi.org/10.1016/S0379-6779(98)00320-8
- TaniguchiMMiuraTMisakiYTanakaKYamabeTMoriT 2002 Mol. Cryst. Liq. Cryst. 380 151 http://dx.doi.org/10.1080/713738710
- TakahashiKTanakaKTaniguchiMMisakiYTanakaKMoriT 2001 Synth. Met. 120 955 http://dx.doi.org/10.1016/S0379-6779(00)00641-X
- MisakiYNakayashikiTYoshidaSTanakaSMoriT 2003 Synth. Met. 135–136 669 http://dx.doi.org/10.1016/S0379-6779(02)00771-3
- KimuraSKuraiHMoriTMoriHTanakaS 2001 Bull. Chem. Soc. Japan 74 59 http://dx.doi.org/10.1246/bcsj.74.59
- AragakiMKimuraSKatsuharaMKuraiHMoriT 2001 Bull. Chem. Soc. Japan 74 833 http://dx.doi.org/10.1246/bcsj.74.833
- KimuraSHanazatoSKuraiHMoriTMisakiYTanakaK 2001 Tetrahedron Lett. 42 5729 http://dx.doi.org/10.1016/S0040-4039(01)01072-3
- TanakaKTaniguchiMMisakiYYamabeTMoriT 1999 Synth. Met. 102 1720 http://dx.doi.org/10.1016/S0379-6779(98)00893-5
- PapavassilliouG CMisakiYTakahashiKYamadaJMousdisG AShirahataTIseT 2001 Z. Naturforsch. B56 297
- IshizuKWatanabeMTanahashiTMisakiYAshizawaMMoriT 2008 J. Phys.: Conf. Ser. 132 012021 http://dx.doi.org/10.1088/1742-6596/132/1/012021
- NodaMMisakiYTanakaK 2006 Curr. Appl. Phys. 6 943 http://dx.doi.org/10.1016/j.cap.2005.01.052
- AshizawaMNiiHMoriTMisakiYTanakaKTakimiyaKOtsuboT 2003 Bull. Chem. Soc. Japan 76 2091 http://dx.doi.org/10.1246/bcsj.76.2091
- BandoYAshizawaMKawamotoTMoriT 2008 Bull. Chem. Soc. Japan 81 947 http://dx.doi.org/10.1246/bcsj.81.947
- BandoYMastuzawaTTakashitaNKawamotoTMoriTYamadaJ 2005 Bull. Chem. Soc. Japan 78 1442 http://dx.doi.org/10.1246/bcsj.78.1442
- AshizawaM et al 2004 Bull. Chem. Soc. Japan 77 1449 http://dx.doi.org/10.1246/bcsj.77.1449
- FujiwaraHLeeH-JCuiH-BKobayashiHFujiwaraEKobayashiA 2003 Chem. Lett. 32 482 http://dx.doi.org/10.1246/cl.2003.482
- FujiwaraHLeeH-JCuiH-BKobayashiHFujiwaraEKobayashiA 2004 Adv. Mater. 16 1765 http://dx.doi.org/10.1002/adma.200400148
- FujiwaraEAonumaSFujiwaraHSugimotoTMisakiY 2008 Chem. Lett. 37 84 http://dx.doi.org/10.1246/cl.2008.84
- YamadaJSatokiSMishimaSArakiNTakahashiKMasudaNNishimotoYTakasakiSAnzaiH 1996 J. Org. Chem. 61 3987 http://dx.doi.org/10.1021/jo952255e
- MisakiYNishikawaHFujiwaraHKawakamiKYamabeTYamochiHSaitoG 1992 J. Chem. Soc. Chem. Commun. 1408
- MoriTMisakiYFujiwaraHYamabeT 1994 Bull. Chem. Soc. Japan 67 2685 http://dx.doi.org/10.1246/bcsj.67.2685
- MisakiYFujiwaraHYamabeTMoriTMoriHTanakaS 1994 Chem. Lett. 23 1653 http://dx.doi.org/10.1246/cl.1994.1653
- DeluzetABatailPMisakiYAuban-SenzierPCanadellE 2000 Adv. Mater. 12 436 http://dx.doi.org/10.1002/(SICI)1521-4095(200003)12:6<436::AID-ADMA436>3.0.CO;2-G
- PerruchasSBoubekeurKCanadellEMisakiYAuban-SenzierPPasquierCBatailP 2008 J. Am. Chem. Soc. 130 3335 http://dx.doi.org/10.1021/ja074774s
- CuiHOtsukaTKobayashiATakedaNIshikawaMMisakiYKobayashiH 2003 Inorg. Chem. 42 6114 http://dx.doi.org/10.1021/ic034459g
- OtsukaTCuiHKobayashiAMisakiYKobayashiH 2002 J. Solid State Chem. 168 444 http://dx.doi.org/10.1006/jssc.2002.9727
- ChuiH-BOtsukaTKobayashiAMisakiYKobayashiH 2003 Bull. Chem. Soc. Japan 76 97 http://dx.doi.org/10.1246/bcsj.76.97
- KusamotoTFujiwaraEKobayashiAChuiH-BOtsuka OkanoYFujiwaraHKobayashiH 2006 Bull. Chem. Soc. Japan 79 527 http://dx.doi.org/10.1246/bcsj.79.527
- MisakiYMiuraTTaniguchiMFujiwaraHYamabeTMoriTMoriHTanakaS 1997 Adv. Mater. 9 714 http://dx.doi.org/10.1002/adma.19970090906
- MisakiYTanakaKTaniguchiMYamabeTMoriT 1999 Chem. Lett. 28 1249 http://dx.doi.org/10.1246/cl.1999.1249
- NodaMYasudaMNakanoYFuenoHItoATanakaKFujiwaraHSugimotoTMisakiY 2008 Chem. Lett. 37 396 http://dx.doi.org/10.1246/cl.2008.396
- MoriTMurataKMisakiYTanakaK 1998 Chem. Lett. 27 435 http://dx.doi.org/10.1246/cl.1998.435
- MoriTInokuchiHMisakiYNishikawaHYamabeTMoriHTanakaS 1993 Chem. Lett. 22 2085 http://dx.doi.org/10.1246/cl.1993.2085
- MoriTMisakiYNishikawaHYamabeTMoriHTanakaS 1995 Synth. Met. 70 873 http://dx.doi.org/10.1016/0379-6779(94)02685-R
- MisakiYNishikawaHYamabeTMoriTInokuchiH 1994 Bull. Chem. Soc. Japan 67 2368 http://dx.doi.org/10.1246/bcsj.67.2368
- MisakiYTaniguchiMTanakaKTakimiyaKMorikamiAOtsuboTMoriT 2002 J. Solid State Chem. 168 608 http://dx.doi.org/10.1006/jssc.2002.9758
- MisakiYTaniguchiMTanakaKTakimiyaKMorikamiAOtsuboTMoriT 1999 Chem. Lett. 28 859 http://dx.doi.org/10.1246/cl.1999.859
- MoriTMisakiYYamabeTMoriHTanakaS 1995 Chem. Lett. 24 549 http://dx.doi.org/10.1246/cl.1995.549
- MoriTMisakiYYamabeT 1994 Bull. Chem. Soc. Japan 67 3187 http://dx.doi.org/10.1246/bcsj.67.3187
- MoriTInokuchiHMisakiYNishikawaHYamabeTMoriHTanakaS 1993 Chem. Lett. 22 733 http://dx.doi.org/10.1246/cl.1993.733
- MoriTInokuchiHMisakiYYamabeTMoriHTanakaS 1994 Bull. Chem. Soc. Japan 67 661 http://dx.doi.org/10.1246/bcsj.67.661
- MoriTMisakiYYamabeT 1997 Bull. Chem. Soc. Japan 70 1809 http://dx.doi.org/10.1246/bcsj.70.1809
- KawamotoTAragakiMMoriTMisakiYYamabeT 1998 J. Mater. Chem. 8 285 http://dx.doi.org/10.1039/a703200e
- KatsuharaMAragakiMMoriTMisakiYTanakaK 2000 Chem. Mater. 12 3186 http://dx.doi.org/10.1021/cm000404q
- KatsuharaMAragakiMKimuraSMoriTMisakiYTanakaK 2001 J. Mater. Chem. 11 2125 http://dx.doi.org/10.1039/b101542g
- MisakiYNishikawaHYamabeTMoriTMoriHTanakaS 1995 Synth. Met. 70 1153 http://dx.doi.org/10.1016/0379-6779(94)02797-3
- KawamotoTAshizawaMMoriTYamauraJKatoRMisakiYTanakaK 2002 Bull. Chem. Soc. Japan 75 435 http://dx.doi.org/10.1246/bcsj.75.435
- OstuboSCuiH-BLeeH-JFujiwaraHTakahashiKOkanoYKobayashiH 2006 Chem. Lett. 35 130 http://dx.doi.org/10.1246/cl.2006.130
- MoriTKobayashiASasakiYKobayashiHSaitoGInokuchiH 1984 Bull. Chem. Soc. Japan 57 627 http://dx.doi.org/10.1246/bcsj.57.627
- MoriTKobayashiASasakiYKobayashiHSaitoGInokuchiH 1984 Chem. Lett. 13 957 http://dx.doi.org/10.1246/cl.1984.957
- OuyangJYakushiKMisakiYTanakaK 1998 J. Phys. Soc. Japan 67 3191 http://dx.doi.org/10.1143/JPSJ.67.3191
- OuyangJYakushiKMisakiYTanakaK 2001 Phys. Rev. B 63 054301 http://dx.doi.org/10.1103/PhysRevB.63.054301
- MoriTOnoSMisakiYYamabeTMoriHTanakaS 1995 Synth. Met. 85 1571 http://dx.doi.org/10.1016/S0379-6779(97)80353-0
- MoriHTanakaSMoriT 1998 Phys. Rev. B 57 12023 http://dx.doi.org/10.1103/PhysRevB.57.12023
- MoriTKawamotoTYamauraJEnokiTMisakiYYamabeTMoriHTanakaS 1997 Phys. Rev. Lett. 79 1702 http://dx.doi.org/10.1103/PhysRevLett.79.1702
- MaesatoMSasoYKagoshimaSMoriTKawamotoTMisakiYYamabeT 1999 Synth. Met. 103 2109 http://dx.doi.org/10.1016/S0379-6779(98)00470-6
- FujimuraNNambaAKambeTNogamiYOshimaKMoriTKawamotoTMisakiYYamabeT 1999 Synth. Met. 103 2111 http://dx.doi.org/10.1016/S0379-6779(98)00473-1
- OnukiMHirakiKTakahashiTJinnoDKawamotoTMoriTTanakaKMisakiY 2001 J. Phys. Chem. Solid 62 405 http://dx.doi.org/10.1016/S0022-3697(00)00176-1
- KobayashiHUdagawaTTomitaHBunKNaitoTKobayashiA 1993 Chem. Lett. 22 1559 http://dx.doi.org/10.1246/cl.1993.1559
- MisakiYHiguchiNFujiwaraHYamabeTMoriTMoriHTanakaS 1995 Angew. Chem. Int. Edn Engl. 34 1222 http://dx.doi.org/10.1002/anie.199512221
- MisakiYHiguchiNFujiwaraHOhtaTYamabeTMoriTMoriHTanakaS 1996 Mol. Cryst. Liq. Cryst. 284 27 http://dx.doi.org/10.1080/10587259608037908
- MisakiYOhtaTHiguchiNFujiwaraHYamabeTMoriTMoriHTanakaS 1995 J. Mater. Chem. 5 1571 http://dx.doi.org/10.1039/jm9950501571
- FujiwaraHMisakiYYamabeTMoriTMoriHTanakaS 2000 J. Mater. Chem. 10 1565 http://dx.doi.org/10.1039/b002790l
- MisakiYSasakiTOhtaTFujiwaraHYamabeT 1996 Adv. Mater. 8 804 http://dx.doi.org/10.1002/adma.19960081006
- MisakiYKaibukiTTakahashiKTaniokaHTanakaK 2002 Mol. Cryst. Liq. Cryst. 380 151 http://dx.doi.org/10.1080/713738695
- MisakiYFujiwaraHMaruyamaTTaniguchiMYamabeTMoriTMoriHTanakaS 1999 Chem. Mater. 11 2360 http://dx.doi.org/10.1021/cm981035p
- MisakiYFujiwaraHYamabeT 1996 J. Org. Chem. 61 3650 http://dx.doi.org/10.1021/jo9516244
- KaibukiTMisakiYTanakaKTakimiyaKMorikamiAOtsuboT 1999 Synth. Met. 102 1621 http://dx.doi.org/10.1016/S0379-6779(98)00469-X
- MisakiYKaibukiTTaniguchiMTanakaKKawamotoTMoriTNakamuraT 2000 Chem. Lett. 29 1274 http://dx.doi.org/10.1246/cl.2000.1274
- TakahashiKNakayashikiTTaniguchiMMisakiYTanakaK 2001 Chem. Lett. 30 162 http://dx.doi.org/10.1246/cl.2001.162
- IwamotoSWatanabeSFuenoHTanakaKMisakiY 2008 Chem. Lett. 37 82 http://dx.doi.org/10.1246/cl.2008.82
- MisakiYNatsumeYTakahashiKFuenoHTanakaK 2003 Synth. Met. 135–136 671 http://dx.doi.org/10.1016/S0379-6779(02)00773-7
- YamadaJMishimaSAnzaiHTamuraMNIshioYKajitaKSatoTNishikawaHIkemotoIKIkuchiK 1996 Chem. Commun. 2517
- YamadaJWatanabeMAnzaiHNishikawaHIkemotoIKikuchiK 1999 Angew. Chem., Int. Edn Engl. 38 810 http://dx.doi.org/10.1002/(SICI)1521-3773(19990315)38:6<810::AID-ANIE810>3.0.CO;2-I
- YamadaJWatanabeMAkutsuHNkakatsujiSNishikawaHIkemotoIKikuchiK 2001 J. Am. Chem. Soc. 123 4174 http://dx.doi.org/10.1021/ja002290p
- YamadaJWatanabeMToitaTAkustuHNakatsujiSNishikawaHIkemotoIKIkuchiK 2002 Chem. Commun. 1118
- MisakiYNishikawaHKawakamiKKoyanagiSYamabeTShiroM 1993 Chem. Lett. 22 445 http://dx.doi.org/10.1246/cl.1993.445
- MisakiYTaniguchiMMiuraTFujiwaraHYamabeTKawamotoTMoriT 1997 Adv. Mater. 9 633 http://dx.doi.org/10.1002/adma.19970090808
- TaniguchiMMisakiYYamabeTTanakaKMurataKMoriT 1999 Solid State Commun. 111 559 http://dx.doi.org/10.1016/S0038-1098(99)00247-1
- NishikawaHIsakaTKodamaTIkemotoIKikuchiKYamadaJMisakiY 2001 Synth. Met. 120 903 http://dx.doi.org/10.1016/S0379-6779(00)00957-7
- NishikawaHSekiyaHWatanabeDKodamaTKikuchiKIkemotoIYamadaJ 2005 Synth. Met. 154 277 http://dx.doi.org/10.1016/j.synthmet.2005.07.070
- MisakiYTakahashiKWatanabeSFuenoHTanakaK 2003 Synth. Met. 137 937 http://dx.doi.org/10.1016/S0379-6779(02)01191-8
- MisakiYYamanakaTMurakamiYFuenoHTanakaK 2008 J. Phys.: Conf. Ser. 132 012022 http://dx.doi.org/10.1088/1742-6596/132/1/012022
- NishikawaHKawauchiSMisakiYYamabeT 1996 Chem. Lett. 25 43 http://dx.doi.org/10.1246/cl.1996.43
- MisakiYKawakamiKHiguchiNNishikawaHMiuraTYamabeT 1996 Mol. Cryst. Liq. Cryst. 284 337 http://dx.doi.org/10.1080/10587259608037936
- SakoKOkuHMisakiYTanakaKTatemitsuH 2003 Synth. Met. 137 901 http://dx.doi.org/10.1016/S0379-6779(02)01133-5
- AshizawaMKimuraSMoriTMisakiYTanakaK 2004 Synth. Met. 141 307 http://dx.doi.org/10.1016/S0379-6779(03)00416-8
- FujiwaraHNishikawaTMisakiYYamabeT 1999 Synth. Met. 102 1737 http://dx.doi.org/10.1016/S0379-6779(98)00942-4
- TakahashiKTaniokaHFuenoHMisakiYTanakaK 2002 Chem. Lett. 31 1002 http://dx.doi.org/10.1246/cl.2002.1002
- MisakiYKuboAMatsudaWFuenoHTanakaK 2006 Curr. Appl. Phys. 6 939 http://dx.doi.org/10.1016/j.cap.2005.01.043
- NavorN LRobertsonNWallaceEKilburnJ DUnderhillA EBartlettP NWebsterM 1996 J. Chem. Soc., Dalton Trans. 823
- NakanoMKurodaAMatsubayashiG 1997 Inorg. Chim. Acta 254 189 http://dx.doi.org/10.1016/S0020-1693(96)05145-6
- UnderhillA EWebsterMSvenstrupNBecherJ 1996 Chem. Commun. 1363
- MisakiYTaniYTaniguchiMMaitaniTTanakaKBechgaardK 2000 Mol. Cryst. Liq. Cryst. 343 59 http://dx.doi.org/10.1080/10587250008023503
- KumasakiMTanakaHKobayashiA 1998 J. Mater. Chem. 8 301 http://dx.doi.org/10.1039/a704518b
- FujiwaraEKobayashiAFujiwaraHKobayashiH 2004 Inorg. Chem. 43 1122 http://dx.doi.org/10.1021/ic034670s
- TanakaHOkanoYKobayashiHSuzukiWKobayashiA 2001 Science 291 285 http://dx.doi.org/10.1126/science.291.5502.285
- SuzukiWFujiwaraEKobayashiAHasegawaEMiyamotoTKobayashiH 2002 Chem. Lett. 31 936 http://dx.doi.org/10.1246/cl.2002.936
- SasaMFujiwaraEKobayashiAIshibashiKKerakuraKOkanoYFujiwaraHKobayashiH 2005 J. Mater. Chem. 15 155 http://dx.doi.org/10.1039/b413597k
- FujiwaraEYamamotoKShimamuraMZhouBKobayashiATakahashiKOkanoYCuiHKobayashiH 2007 Chem. Mater. 19 553 http://dx.doi.org/10.1021/cm062131q
- MisakiYTaniYTakahashiKTanakaK 2002 Mol. Cryst. Liq. Cryst. 379 71 http://dx.doi.org/10.1080/713738644
- KuboKNakanoAYamamotoH MKatoR 2008 J. Am. Chem. Soc. 128 12358 http://dx.doi.org/10.1021/ja0638004
- TakahashiKNakayashikiTMisakiYTanakaK 2002 Mol. Cryst. Liq. Cryst. 376 107 http://dx.doi.org/10.1080/10587250210766
- ShevyakovaIBuravovLTkachevaVZorinaLKhasanovSSimonovSYamadaJCanadellEShibaevaRYagubskiiE 2004 Adv. Funct. Mater. 14 660 http://dx.doi.org/10.1002/adfm.200305170
- YamadaJ 2004 J. Physique IV 114 439 http://dx.doi.org/10.1051/jp4:2004114103
- YamadaJToitaTAkustuHNakatsujiSNishikawaHIkemotoIKIkuchiKChoiE SGrafDBrooksJ S 2003 Chem. Commun. 2230
- ChoiE SGrafDBrooksJ SYamadaJ AKikuchiKTokumotoM 2004 Phys. Rev. B 70 024517. http://dx.doi.org/10.1103/PhysRevB.70.024517
- YamadaJFujimotoKAkustuHNakatsujiSMiyazakiAAimatsuMKuboSEnokiTKikuchiK 2006 Chem. Commun. 1331
- SetifiF et al 2002 Inorg. Chem. 41 3761 http://dx.doi.org/10.1021/ic0255961
- SetifiFGolhenSOuahabLMiyazakiAOkabeKEnokiTToitaTYamadaJ 2002 Inorg. Chem. 41 3786. http://dx.doi.org/10.1021/ic025553k
- MisakiYNishikawaHYamabeTMoriTMoriHTanakaS 1993 Chem. Lett. 22 1341 http://dx.doi.org/10.1246/cl.1993.1341
- FujiwaraHMisakiYTaniguchiMYamabeTMoriTMoriHTanakaS 1998 J. Mater. Chem. 8 1711 http://dx.doi.org/10.1039/a801375f
- YamadaJAkustuHNishikawaHKikuchiK 2004 Chem. Rev. 104 5057 http://dx.doi.org/10.1021/cr0306687
- KobayashiAFujiwaraEKobayashiH 2004 Chem. Rev. 104 5243 http://dx.doi.org/10.1021/cr030656l
- KobayashiATanakaHKumasakiMToriiHNarymbetovBAdachiT 1999 J. Am. Chem. Soc. 121 10763 http://dx.doi.org/10.1021/ja9921017
- SuzukiWFujiwaraEKobayashiAFujishiroYNishiboriETakataMSakataMFujiwaraHKobayashiH 2003 J. Am. Chem. Soc. 125 1486 http://dx.doi.org/10.1021/ja0292243
- YamamotoKFujiwaraEKobayashiAFujishiroYNishiboriESakataMTakataMTanakaHOkanoYKobayashiH 2005 Chem. Lett. 34 1090 http://dx.doi.org/10.1246/cl.2005.1090
- TanakaHOkanoYKobayashiHSuzukiWKobayashiA 2002 J. Am. Chem. Soc. 124 10002 http://dx.doi.org/10.1021/ja027024l
- FujiwaraE et al 2008 Inorg. Chem. 47 863 http://dx.doi.org/10.1021/ic701100r
- SuzukiWFujiwaraEKobayashiAFujishiroYNishiboriETakataMSakataMOkanoYKobayashiH 2003 Chem. Lett. 32 1106 http://dx.doi.org/10.1246/cl.2003.1106
- HaddenR C 1992 Acc. Chem. Res. 25 127 http://dx.doi.org/10.1021/ar00015a005
- YamochiHKomatsuTMatsukawaNSaitoGMoriTKusunokiMSakaguchiK 1993 J. Am. Chem. Soc. 115 11319 http://dx.doi.org/10.1021/ja00077a034
- NakamuraTKomatsuTSaitoGOsadaTKagoshimaSMiuraNKatoKMaruyamaYOshimaK 1993 J. Phys. Soc. Japan 62 4373 http://dx.doi.org/10.1143/JPSJ.62.4373
- SatoAKobayashiHNaitoTSakaiFKobayashiA 1997 Inorg. Chem. 36 5262 http://dx.doi.org/10.1021/ic970507j
- KanodaK 1997 Hyperfine Interact. 104 235 http://dx.doi.org/10.1023/A:1012696314318
- NishikawaH et al 2006 Chem. Lett. 35 912 http://dx.doi.org/10.1246/cl.2006.912
- YasuzukaSKobayashiKNishikawaHYoshinoHMurataK 2006 J. Phys. Soc. Japan 75 83710 http://dx.doi.org/10.1143/JPSJ.75.083710
- LimelettePWzietekPFlorensSGeorgesACostiT APasquierCJéromeDBatailP 2003 Phys. Rev. Lett. 91 16401 http://dx.doi.org/10.1103/PhysRevLett.91.016401
- ItouTKanodaKMurataKMatsumotoTTakahashiT 2004 Phys. Rev. Lett. 93 216408 http://dx.doi.org/10.1103/PhysRevLett.93.216408
- TanakaHTokumotoMIshibashiSGrafDChoiE SBrooksJ SYasuzukaSOkanoYKobayashiHKobayashiA 2004 J. Am. Chem. Soc. 126 10518 http://dx.doi.org/10.1021/ja046895n