Abstract
We report on the characterization, assembly and electroanalytical performance of Prussian blue/polypyrrole (PBPPy) composite nanoparticles synthesized by the reverse micelle method. Scanning electron microscopy suggests the formation of nanosized PBPPy particles with diameters between 40 and 50 nm. Optical absorption confirms that the particles are composed of Prussian blue (PB) and polypyrrole. PB and PBPPy nanoparticles were anchored onto the surface of cysteine-modified Au electrodes. Cyclic voltammetry experiments show that PB- or PBPPy-modified electrodes exhibit intrinsic electrochemical properties and a high electrocatalytic activity towards H2O2. PBPPy-modified electrodes exhibit a higher sensitivity to H2O2 than PB-modified electrodes. A linear calibration curve in the concentration range 0.99 μM–8.26 mM H2O2 is constructed with a detection limit of 0.23 μM at a signal-to-noise ratio of 3. Excellent stability is observed for PBPPy-composite-nanoparticle-modified electrodes even in a pH 6 phosphate buffer solution with a high H2O2 concentration (0.99 mM). Glutaraldehyde and were also employed to immobilize glucose oxidase for the development of PBPPy-based biosensors. The results show that PBPPy composite nanoparticles can be used to develop oxidase-based biosensors.
Introduction
Prussian blue (PB) is well known for its attractive photophysical, magnetic, electrochromic and electrochemical properties and has been widely used over the past few years [Citation1, Citation2]. In particular, PB has been recognized as a promising low-potential transducer for H2O2 reduction. The study of the electrocatalytic properties of PB and the development of PB-based chemical or biological sensors have attracted the most attention [Citation3, Citation4].
Nanomaterials exhibit unique optical, electronic and catalytic properties, and have promising applications in various areas [Citation5, Citation6]. Owing to their large surface-to-volume ratio and increased surface activity, nanosized PB particles have received increasing attention in the field of electroanalysis and biosensors [Citation7, Citation8]. Qian and Yang coated PB onto SiO2 nanoparticles and carbon nanotubes by electrostatic interaction [Citation9]. Then, they assembled PB-modified nanomaterials onto a poly(diallyldimethyl ammonium chloride)-modified glass carbon electrode. Their results show that PB on the surface of nanomaterials is stable and maintains its intrinsic electrochemical properties and high electrocatalytic activity towards H2O2. Xian et al developed a novel modified electrode using highly dispersed PB nanoparticles protected by poly(vinylpyrrolidone) [Citation10]. The electrocatalytic reduction of hemoglobin was observed at a PVP-protected PB-nanoparticle-modified electrode, which enabled the development of a stable electrochemical sensor for hemoglobin. Liu et al have immobilized, by the self-assembly method, PB particles with a size of about 50 nm on a gold electrode, using cysteine as a bridge between the gold surface and the nanoparticles [Citation11]. PB nanoparticles were prepared by mixing FeCl3 with K3FeCN6 in the presence of H2O2. Multilayer films of PB nanoparticles protected by poly(diallyldimethylammonium chloride) and glucose oxidase have been fabricated for glucose biosensing [Citation12]. Multiplayer films can also be constructed by the electrostatic interaction between negatively charged PB nanoparticles and positively charged poly(allylaminehydrochloride) and they show a high catalytical activity for H2O2 [Citation1] However, according to common opinion, PB films are not very stable in neutral and alkaline solutions. They are also easily destroyed by the catalytic reduction of H2O2 owing to the formation of OH− ions, which can disrupt the Fe–CN bond in PB. This instability of PB films makes them unsuitable for the development PB-based electrodes or biosensors.
The use of micellar water droplets as a novel environment for nanoparticle synthesis has attracted much interest for its potential advantages over the traditional nanoparticle synthesis methods [Citation14–16]. The size of micelle cores is expressed by the molar ratio of water to surfactant. The reaction is restricted in the water cores, and the diameter of the obtained product particles can be controlled by adjusting the size of the water core. Various nanoparticles have been synthesized in water cores of reverse micelles in organic solvents [Citation17–19].
In this work, we synthesized PB and Prussian blue/ polypyrrole (PBPPy) nanoparticles in reverse microemulsion. The stability of the PBPPy-modified electrode is apparently enhanced. Cyclic voltammetry was employed to study the electrochemical properties and high electrocatalytic activity towards H2O2. Glutaraldehyde and were also employed to immobilize glucose oxidase for the development of PBPPy-based biosensors.
Materials and methods
Reagents
117, isooctane, sodium bis (2-ethylhexyl) sulfosuccinate (AOT), cysteine and glucose oxidase (EC 1.1.3.4, type II, 15 500 U g−1) were purchased from Fluka, Fisher, Acros Organics, BBI and Sigma, respectively. Pyrrole and glutaraldehyde were obtained from SCRC (Shanghai, China). They were used as received. All the other chemicals were of analytical grade and used without further purification. All the solutions were prepared using Milli-Q water. The supporting electrolyte is a pH 6.0 phosphate buffer solution (PBS) containing 0.01 M Na2HPO4, 0.01 M KH2PO4 and 0.1 M KCl. All the H2O2 or glucose solutions were prepared with the above-mentioned PBS solution. All experiments were carried out at room temperature.
Preparation of PB and PBPPy nanoparticles in reverse microemulsion
Prussian blue nanoparticles were prepared by the reverse micelle method. The desired amount of AOT was dissolved in isooctane ([AOT]=0.5 M) to obtain 2 ml of solution. Then 50 μl of aqueous solution with 1 M KCl and 0.1 M HCl was added to AOT solution to form the microemulsion. The solution was stirred vigorously to ensure complete dissolution of the reactants. In turn, 40 μl of 0.1 M FeCl3, the desired amount of water and 40 μl 0.1 M K4Fe(CN)6 were added to the reverse microemulsion solution. The water-to-surfactant molar ratio (w) was 12.22 in the microemulsion unless mentioned otherwise. After the mixing, the solution turned blue immediately, indicating the formation of PB. The above solution was kept overnight to complete the reaction of FeCl3 and K4Fe(CN)6.
The formation of PBPPy composite nanoparticles in the reverse microemulsion could be illustrated similarly as above, except that 50 or 100 μl of pyrrole was added before K4Fe(CN)6. The water-to-surfactant molar ratio was 12.22 in the microemulsion unless mentioned otherwise. Pyrrole monomers in isooctane approached the oil/water interface and were oxidized by FeCl3 in the reverse microemulsion to form polypyrrole. The solution turned completely black after 2 h, indicating the formation of PPy. Vigorous stirring was continued overnight. Then, when K4Fe(CN)6 was added, PPy acted as a steric stabilization substance for the nucleation and growth of PB. After the reaction was complete, PBPPy composite nanoparticles were formed, and the solution turned dark blue.
To separate PB or PBPPy nanoparticles from the solution, centrifugation at 14 000 rpm for 10 min was performed. The resultant precipitate was centrifuged and washed with ethanol, water and acetone in this sequence several times and finally dispersed in 1 ml of water. The obtained PB or PBPPy colloid solution was stable and transparent even after storing at 4 °C for one month. The PBPPy prepared with 50 μl of pyrrole was denoted PBPPyI and that with 100 μl of pyrrole PBPPyII.
Anchoring PB or PBPPy nanoparticles onto cysteine-modified Au electrode
The Au disk electrode (diameter 2 mm) was cleaned with piranha solution (H2O2:H2SO4=1:3) for 20 min followed by a thorough rinse with pure water.
Cysteine was assembled on a Au electrode prior to the deposition of nanoparticles. The Au electrode was dipped in 2 mM cysteine solution overnight, followed by rinsing twice with pure water for 1 min. Then a drop (2 μl) of PB or PBPPy colloid solution was cast on the surface of the cysteine-modified Au electrode. A small bottle tightly covered the electrode to serve as a closed evaporation chamber. After 12 h, the nanoparticle-modified electrode was rinsed twice with pure water.
For the preparation of a glucose biosensor, 10 mg of glucose oxidase was dissolved in 1 ml of water; then 1 μl of glucose oxidase solution was added onto the PBPPyII anchored electrode and dried. One microliter of 0.1% glutaraldehyde was dropped onto the surface of the cysteine–PBPPyII–glucose oxidase electrode, and the electrode was dried at room temperature. The obtained Au–cysteine–PBPPyII–(glucose oxidase+ glutaraldehyde) electrode was named biosensor I.
The biosensor II of the Au–cysteine–PBPPyII–glucose oxidase–Nafion electrode was fabricated by dropping 2 μl of Nafion solution (1 wt.%, diluted with ethanol) on the electrode after 1 μl of glucose oxidase solution was added onto the PBPPyII anchored electrode and dried. Finally, the obtained biosensor was dried at room temperature and rinsed twice with pure water before use.
Characterization
The morphology of PB and PBPPy nanoparticles was studied by scanning electron microscopy (SEM, S-4800 UHR FE-SEM). Fourier transform infrared (FTIR) spectra were recorded using a NEXUS 870 (Nicolet), and UV-visible absorbance was measured using a Thermo Electron Evolution 600 spectrometer.
Electrochemical experiments were conducted using an Autolab PGSTAT 30 System (Ecochemie, The Netherlands) in a three-electrode configuration. A nanoparticle-modified Au electrode with a diameter of 2 mm was used as the working electrode, a platinum wire as the auxiliary electrode and a Ag/AgCl electrode as the reference electrode. The supporting electrolyte is 10 mM PBS (pH 6.0) with 0.1 M KCl prepared under vigorous stirring. All experiments were carried out at room temperature. Amperometric measurements were performed at an applied potential while the H2O2 or glucose solution was being added to 10 ml of PBS under continuous stirring.
Results and discussion
For the preparation of PB nanoparticles, Fe3+ was firstly entrapped in micellar water droplets enveloped by AOT with negative charges in isooctane. Further adsorption of Fe(CN)64− resulted in the formation of PB nanoparticles in micellar water droplets. The formation of PBPPy composite nanoparticles in the reverse microemulsion could be illustrated similarly to that of PB, except that pyrrole was added before K4Fe(CN)6. Pyrrole monomers in isooctane approach the oil/water interface. The polymerization of pyrrole into polypyrrole, doping with chloride ions and the reduction of Fe3+ to Fe2+ occur instantaneously and simultaneously, as inferred from the appearance of the black solution. It is possible that only some of the Fe3+ was reduced to Fe2+ because Fe3+ at the center of the water core is not easily available to the pyrrole monomer in the oil phase. When K4Fe(CN)6 was added, polypyrrole acted as a steric stabilization substance for the nucleation and growth of PB and Prussian white. After the reaction was complete, the solution turned dark blue, indicating the formation of PBPPy composite nanoparticles. Then, Prussian white was oxidized to PB rapidly by atmospheric oxygen [Citation20].
To record SEM images, the colloid solution containing PB or PBPPyII nanoparticles was dropped onto a piece of Au-covered glass slide fixed to sample stage with conductive Ag paste. The images of PB and PBPPyII are shown in figure . Most nanoparticles are nearly spherical. The diameters of PBPPyII (40–50 nm) are larger than those of PB (15–25 nm) owing to the polymerization of pyrrole occurring at the oil/water interface. No distinguishable hollow or core–shell structures are observed because of insufficient SEM resolution. Some aggregated nanoparticles are observed. The mentioned samples were not pre-coated with a carbon or Au film for SEM. The observation of clear SEM images suggests that the obtained PB or PBPPyII nanoparticles are conducting.
Figure 1 SEM images of PB (a) and PBPPyII (b) with w=12.2.

Figure (a) shows the UV-visible spectra of PB and PBPPyII colloid solutions. The characteristic absorption bands of Prussian blue at 680 nm are observed for both PB and PBPPyII samples, which are attributed to the charge transfer from Fe2+ to Fe3+ in Prussian blue. Compared with that of the PB sample, the absorbance of the PBPPyII sample at 680 nm is lower, suggesting that polypyrrole within the nanostructure of PBPPyII affects the absorbance of PB. During the preparation of PBPPyII nanoparticles, polypyrrole is obtained firstly by the polymerization of a pyrrole monomer with the help of FeCl3, and then PB is deposited into the framework of polypyrrole nanoparticles. The absorption results can be interpreted to mean that most of the polypyrrole is interlaced with PB.
Figure 2 UV-visible spectra (a) of PB (1) and PBPPyII (2) colloid solutions. FTIR spectra (b) of PB (1) and PBPPyII (2) nanoparticle samples.
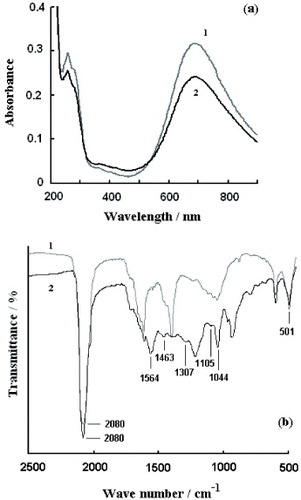
The molecular structure of the resulting PB and PBPPyII is characterized by FTIR spectroscopy and is shown in figure (b). The peak at 2080 cm−1 is the characteristic CN stretching absorption band of Prussian blue and its analogues, and the absorption bands at 501 cm−1 are due to the formation of M–CN–M’ structures (M=metal); they indicate the presence of PB [Citation20]. No obvious difference is found between the FTIR spectra of the PB nanoparticles prepared by the reverse micelle method and conventional methods [Citation20]. Curve 2 in figure (b) has characteristic absorption bands of polypyrrole. The peaks at 1564 and 1463 cm−1 correspond to the fundamental vibration of the pyrrole ring. The peaks at 1307, 1105, 1044 can be assigned to the in-plane vibration.
Figure shows the cyclic voltammograms of the PB- and PBPPy-modified electrodes in PBS without (1) and with (2) H2O2. The observed peaks are attributed to the conversion from Prussian white to Prussian blue. The sharp peak indicates a regular inorganic polycrystalline structure [Citation21]. The waveform with an ΔEp of approximately 93 mV reveals a rapid electrochemistry of the immobilized redox active PB. Cysteine provides a favorable microenvironment for the electron exchange between nanoparticles and the electrode. The electron transfer mechanisms for a modified electrode in the presence of potassium electrolyte may be formulated as
Figure 3 Cyclic voltammograms of Au–cysteine–PB (a), Au–cysteine–PBPPyI (b) and Au–cysteine–PBPPyII (c) electrodes in PBS (10 mM, pH 6.0) with 0.1 M KCl without (1) and with (2) 0.99 mM H2O2. Scan rate: 100 mV s−1.
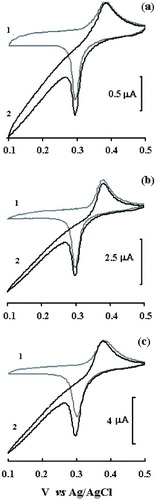
The transfer of electrons is compensated for by the entrapment of cations from supporting electrolytes to maintain electroneutrality. As shown in figure , the reduction peak is sharper than the oxidation peak, which is similar to that in a previous report [Citation1]. The presence of 0.1 M KCl in the supporting solution prevents the release of K+ from K4Fe4II[FeII(CN)6]3. Therefore, the reduction of Fe4III[FeII(CN)6]3 is easier than the oxidation of K4Fe4II[FeII(CN)6]3. The redox process is the four-electron oxidation of K4Fe4II[FeII(CN)6]3 to Fe4III[FeII(CN)6]3 rather than that of Fe2II[FeII(CN)6] to Fe4III[FeII(CN)6]3. In the presence of H2O2, the current due to Prussian blue reduction starts increasing (with respect to that obtained in buffer solution) whereas in the reverse scan, oxidation current decreases, as expected in the case of a mediated electrochemical reduction. This result demonstrates that PB has an electrocatalytic activity towards the reduction of H2O2. This further confirms that PB or PBPPy is assembled onto the cysteine-modified Au surface and still keeps its electrochemical properties. As shown in figure , the shapes of the voltammograms in the presence and absence of polypyrrole are the same, but the PBPPy-composite-nanoparticle-modified electrode exhibits better electrocatalytic activity toward H2O2. The former result means that the existence of conductive polypyrrole does not interfere with the electron transfer between PB and the electrode. The latter result means that polypyrrole in PBPPy induces the immobilization of PB and also provides a favorable microenvironment for the electrocatalytic activity of PB dispersed in PBPPy.
The synthesis reaction of PB or PBPPy nanoparticles is restricted to the water cores, and the diameter of the obtained product particles can be controlled by adjusting the size of the water core. The size of the micelle cores is characterized by the molar ratio of water to surfactant, w=[H2O]/[AOT]). The microemulsions with w=8.0, 10.0, 12.2, 15.0 and 20.0 were prepared with the same parameters as that with w=12.2. The obtained PBPPy nanoparticles were immobilized onto the cysteine-modified electrode. The highest electrocatalytic activity toward H2O2 was observed for w=12.2. The specific surface area and catalytical efficiency increase with decreasing nanoparticle size, but the number of obtained nanoparticles decreases owing to centrifugation. This is why the best performance was achieved for an intermediate ratio. Note also that the microemulsion solution became turbid for w=20.0.
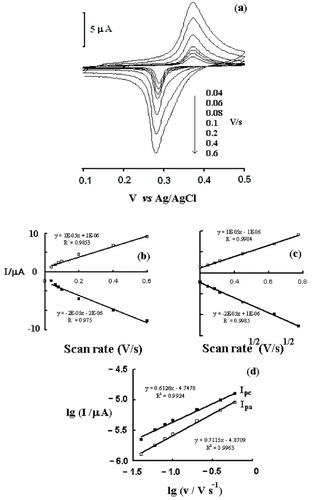
The effect of potential scan rate (v) on oxidation peak current (Ipa) and reduction peak current (Ipc) has been investigated in the range of 40–600 mV s−1 (figure ). A linear relationship is observed between the peak current and the square root of the scan rate for the redox reaction between PB and Prussian white (figure (c)). This linearity could indicate that the diffusion of K+ within PB or PBPPy is rate-limiting, since the concentration of K+ in the bulk solution is 0.1 M and the observed redox peaks involve K+ ion diffusion from the support electrolyte to the electrode surface (during the reduction step) or from the electrode to the solution (during the oxidation step) to ensure electroneutrality [Citation22]. Figure (d) shows the linear relation between log Ip and log v. The slope of log Ipa versus log v is 0.71 and that of log Ipc versus log v is 0.61. For an ideal diffusion-controlled reaction, the slope is 0.5; for ideal thin-layer electrochemistry, the slope is 1 [Citation12]. Here, the slopes are 0.71 and 0.61, indicating that both diffusion control and surface control exist in the reaction. The plots of anodic and cathodic peak currents versus scan rate are approximately linear from 40 to 600 mV s−1 suggesting facile charge transfer kinetics in this sweep rate range (figure (b)). The shift or separation of anodic and cathodic peak potentials is negligible, also suggesting facile charge transfer kinetics in this sweep rate range.
Figure 4 (a) Cyclic voltammograms of the Au–cysteine–PBPPyII in PBS at different scan rates. (b) Dependences of anodic and cathodic peak currents on scan rate. (c) Dependences of anodic and cathodic peak currents on the square root of the scan rate (v1/2). (d) Linear relationship between log Ip and log v.
The current response of the cysteine–PBPPyII modified Au electrode as a function of the applied potential for 0.99 mM H2O2 was studied. The reduction current response to H2O2 increases rapidly with the decrease in applied potential from 400 to 100 mV, then a slow enhancement can be observed from 100 to −200 mV (not shown). Once the applied potential becomes −200 mV or more negative, PB becomes unstable and the interfering effect of oxygen increases. Therefore, −100 mV (versus Ag/AgCl) was chosen as the optimal working potential.
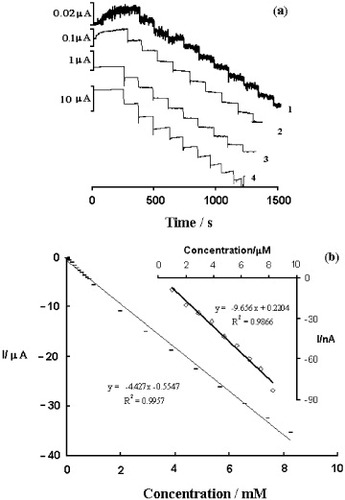
When held at a constant potential of −100 mV, the Au–cysteine–PBPPyII electrode showed a cathodic current response to added H2O2. Repeated additions of H2O2 solution resulted in a stepwise increase in cathodic current (figure (a)). The electrode shows a very fast response within several seconds. This must be related to the immobilization of PBPPy nanoparticles on the surface of the electrode and to the fast diffusion of H2O2 into a reaction layer of PBPPyII. As shown in figure (b), the dependence of catalytic current on H2O2 concentration is linear from 0.99 μM to 8.26 mM, with a sensitivity of 4.427 μA mM−1 and a detection limit of 0.23 μM at a signal-to-noise ratio of 3. The electrocatalytic reduction of H2O2 at the PB-modified electrode involves at least three steps: (i) diffusion of H2O2 into the reaction layer of PB, (ii) the chemical oxidation of a reduced form of PB (i.e., Prussian white) by H2O2 and (iii) the electrochemical reduction of the oxidized form of PB formed in a preceding step. The wide linear dependence of reduction current on the H2O2 concentration should be due to the synchronization between the mentioned three steps. The reversibility of the Au–cysteine–PBPPyII electrode was investigated at a H2O2 concentration of 3.5 μM. The mean current was –34.9 nA with a ripple of 5.8%. The Au–cysteine–PBPPyII electrode shows no decrease in the extent of its original response after five tests.
Figure 5 (a) Curve of typical amperometric response curve of the Au–cysteine–PBPPyII electrode to successive addition of 0.1 (1), 1 (2), 10 (3) and 100 (4) mM H2O2 in PBS (10 mM, pH 6.0) with 0.1 M KCl at −100 mV versus Ag/AgCl. (b) H2O2 calibration curve obtained with Au–cysteine–PBPPyII electrode in PBS (10 mM, pH 6.0) with 0.1 M KCl.
According to common opinion, PB films are not very stable in neutral and alkaline solutions; they are also easily destroyed during the catalytic reduction of H2O2 owing to the formation of OH− ions, which can disrupt the Fe–CN bond in PB [Citation2]. The stability of the PBPPyII-modified electrode depends on the nature of the supporting electrolyte, the number of voltammetric cycles and the potential range. Our results suggest that KCl is the most suitable supporting electrolyte. The cyclic voltammograms of the Au–cysteine–PBPPyII electrodes were carried out in pH 6 blank PBS or PBS containing 0.099 and 0.99 mM H2O2. The fifth and 100th cycles of the Au–cysteine–PBPPyII-modified Au electrode were almost identical (not shown) indicating the excellent stability of the PBPPyII modified electrode. On the one hand, the peak current of the cyclic voltammogram does not decrease in pH 6 PBS, which indicates the good stability of the PBPPyII-modified electrode in nearly neutral solutions. On the other hand, the peak current of the cyclic voltammogram remains almost unchanged even in pH 6 PBS with a high H2O2 concentration, which indicates that the obtained PBPPyII-modified electrode could resist H2O2 to some extent. The good stability can be attributed to two aspects: (i) polypyrrole in composite nanoparticles prevents PB from being decomposed by OH− ions formed during the catalytic reduction of H2O2; (ii) PBPPy nanoparticles could be firmly attached to the cysteine-modified Au electrode, and cysteine is favorable for the electron transfer between Au electrodes and anchored PBPPy nanoparticles. In fact, PB nanoparticles could be anchored onto the cysteine-modified electrodes and PBPPyII directly onto the unmodified blank electrodes, but both of them give no stable or reproducible signal to H2O2.
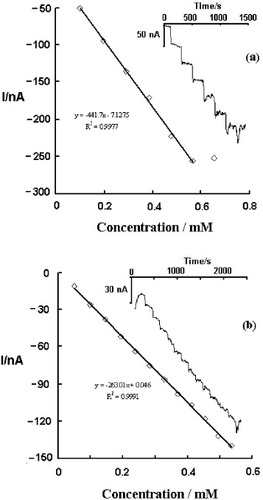
Glutaraldehyde and were used to immobilize glucose oxidase for developing possible biosensor applications of PBPPy nanoparticles. The inset of figure shows a current–time plot of the enzyme electrode during successive step changes in glucose concentration. When an aliquot of glucose is added to the buffer solution, reductive current increases steeply to reach a stable value. The response times (reaching 90% of the maximum response) are less than 1 min for biosensor I and 1.5 min for biosensor II, which indicates fast diffusion. By contrast, the diffusion hindrance of biosensor II is larger than that of biosensor I, while the response sensitivity of biosensor I (442 nA mM−1) is larger than that of biosensor II (263 nA mM−1). However, biosensor II shows a wider linear range than biosensor I. As shown in figure , the linear range of biosensor I for glucose concentration extends between 99 μM and 0.566 mM with a correlation coefficient of 0.9977 and a detection limit of 0.23 μM at a signal-to-noise ratio of 3. The corresponding range for biosensor II is 49.5 μM–0.536 mM and the correlation coefficient is 0.9991. No decrease in current was observed after biosensor I was tested 8 times continuously in PBS with 250 μM glucose. However, only a 61.2% current response was retained for biosensor II. The observed current decrease for biosensor II should be due to the leakage of enzymes under
. For biosensor I, the glutaraldehyde crosslinks the enzymes and cysteine by covalent bonds to form a stable construction. These results show that PBPPy composite nanoparticles could effectively catalyze glucose oxidation and could be used in oxidase-based biosensors. Various immobilization strategies are being studied for fabricating stable and sensitive biosensors.
Figure 6 (a) Calibration curve of biosensor I. The inset shows the typical current–time response of biosensor I upon successive addition of 10 mM glucose in PBS (10 mM, pH 6.0) with 0.1 M KCl at −100 mV versus Ag/AgCl. (b) Calibration curve of biosensor II. The inset shows typical current–time response of biosensor II upon successive addition of 5 mM glucose in PBS (10 mM, pH 6.0) with 0.1 M KCl at −100 mV versus Ag/AgCl.
Conclusions
In this study, PBPPy composite nanoparticles were synthesized by the reverse micelle method in isooctane/AOT microemulsion. Excellent stability is observed for PBPPy-composite-nanoparticle-modified electrodes even in pH 6 PBS with a high H2O2 concentration. On the one hand, PPy in composite nanoparticles prevents PB from being decomposed by OH− ions formed during the catalytic reduction of H2O2. On the other hand, PBPPy nanoparticles could be stably absorbed on a cysteine-modified Au electrode, and cysteine is favorable for electron transfer between Au electrode and anchored PBPPy nanoparticles. The PB- or PBPPy-modified electrode exhibits its intrinsic electrochemical properties and high electrocatalytic activity towards H2O2. Glutaraldehyde and were applied to immobilize glucose oxidase to study possible applications of PBPPy nanoparticles to biosensor development. The results show that PBPPy composite nanoparticles could be used in oxidase-based biosensors. Further research will be focused on optimizing the diameter of PBPPy composite nanoparticles and studying various immobilization strategies for fabricating highly stable biosensors with a higher sensitivity and a wider linear range.
References
- KaryakinA A 2001 Electroanalysis 13 813 http://dx.doi.org/10.1002/1521-4109(200106)13:10<813::AID-ELAN813>3.0.CO;2-Z
- RicciFPalleschiG 2005 Biosens. Bioelectron. 21 389 http://dx.doi.org/10.1016/j.bios.2004.12.001
- ZhangXWangJOgorevcBSpichigerU E 1999 Electroanalysis 11 945 http://dx.doi.org/10.1002/(SICI)1521-4109(199909)11:13<945::AID-ELAN945>3.0.CO;2-7
- RicciFAmineATutaC SCiucuA ALucarelliFPalleschiGMosconeD 2003 Anal. Chim. Acta 485 111 http://dx.doi.org/10.1016/S0003-2670(03)00403-3
- AbdolmajidB MMohammadR GRassoulDTaherehsadatRSiavashRSaeedR-ZParvizN 2009 Mater. Sci. Eng. C 29 187 http://dx.doi.org/10.1016/j.msec.2008.06.016
- NoponenVBhatSSievänenEKolehmainenE 2008 Mater. Sci. Eng. C 28 1144 http://dx.doi.org/10.1016/j.msec.2007.10.001
- BustosEManríquezJOrozcoGGodínezL A 2005 Langmuir 21 3013 http://dx.doi.org/10.1021/la047478r
- OhnukiHSaikiTKusakariAIchiharaMIzumiM 2008 Thin Solid Films 516 8860 http://dx.doi.org/10.1016/j.tsf.2007.11.059
- QianLYangX 2006 Colloids Surf. A 78 123 http://dx.doi.org/10.1016/j.colsurfa.2005.12.008
- XianYZhouYXianYZhouLWangHJinL 2005 Anal. Chim. Acta 546 139 http://dx.doi.org/10.1016/j.aca.2005.04.090
- LiuS-QXuJ-JChenH-Y 2002 Electrochem. Commun. 4 421 http://dx.doi.org/10.1016/S1388-2481(02)00336-3
- ZhaoWXuJ-JShiC-GChenH-Y 2005 Langmuir 21 9630 http://dx.doi.org/10.1021/la051370+
- FioritoP AGoncalesV RPonzioE ATorresiS I C 2005 Chem. Commun. 3 366 http://dx.doi.org/10.1039/b412583e
- CarvalhoC M LCabralJ M S 2000 Biochimie 82 1063 http://dx.doi.org/10.1016/S0300-9084(00)01187-1
- HouGZhuLChenDJiangM 2007 Macromolecules 40 2134 http://dx.doi.org/10.1021/ma062373b
- SetuaPChakrabortyASethDBhattaMSatyamP VSarkarN 2007 J. Phys. Chem. C 111 3901 http://dx.doi.org/10.1021/jp067475i
- LiZZhangJMuTDuJLiuZHanBChenJ 2004 Colloids Surf. A 243 63 http://dx.doi.org/10.1016/j.colsurfa.2004.05.010
- JangJYoonH 2005 Langmuir 21 11484 http://dx.doi.org/10.1021/la051447u
- ZhangWQiaoXChenJ 2006 Chem. Phys. 330 495 http://dx.doi.org/10.1016/j.chemphys.2006.09.029
- RegueraE 1999 Trans. Metal Chem. 24 648 http://dx.doi.org/10.1023/A:1006942415737
- KaryakinA AKaryakinaE E 1999 Sensors Actuators B 57 268 http://dx.doi.org/10.1016/S0925-4005(99)00154-9
- MosconeDD’OttaviDCompagnoneDPalleschiG 2001 Anal. Chem. 73 2529 http://dx.doi.org/10.1021/ac001245x
- IvamaV MSerranoS H P 2003 J. Braz. Chem. Soc. 14 551 http://dx.doi.org/10.1590/S0103-50532003000400010