Abstract
Nanoassemblies from amphiphilic block copolymers are promising nanomedicine platforms for cancer diagnosis and therapy due to their relatively small size, high loading capacity of drugs, controlled drug release, in vivo stability and prolonged blood circulation. Recent clinical trials with self-assembled polymeric micelles incorporating anticancer drugs have shown improved antitumor activity and decreased side effects encouraging the further development of nanoassemblies for drug delivery. This review summarizes recent approaches considering stimuli-responsive, multifunctionality and more advanced architectures, such as vesicles or worm-like micelles, for tumor-specific drug and gene delivery.
Introduction
Decades of cancer treatment research have led to a multimodal approach combining surgery, radiation and chemotherapy; however, advanced stage tumors are only slightly hindered with current therapies at the cost of considerable patient morbidity. Nanoparticle therapeutics have shown tremendous potential for achieving the so-far-elusive objective to specifically deliver drugs and genes in the body, and to increase the efficiency and reduce detrimental side effects by selectively targeting cancerous tissues [Citation1–3]. The tumor targeting in these nanomedicine technologies is based on the enhanced permeability and retention (EPR) effect [Citation4], i.e. the increased accumulation of macromolecules in tumor tissue, due to the high permeability of tumor blood vessels, and the retention of these macromolecules because of the impaired lymphatic drainage at the cancer site. Thus, the improved concentration ratios of target-to-non-target tissues and the increased drug residence at the target site, as well as the enhanced cellular uptake and intracellular stability, greatly emphasize the use of nanoparticulate delivery systems for cancer treatment.
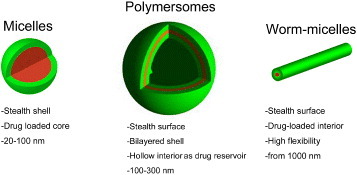
Highly ordered self-assembled polymer nanoparticles present several distinctive advantages for the development of multifunctional delivery systems, including the relative small size, the versatile capacity to integrate diagnostic and therapeutic functions within the constructs, and the ability to precisely control the interaction with the biological environment [Citation1–3, Citation5, Citation6]. The first nanoassemblies considered for tumor delivery in the late 1980s included drug-loaded polymeric micelles—a self-assembly of amphiphilic block copolymers consisting of (i) hydrophobic segments, forming the drug-loaded core and (ii) water-soluble segments, forming the biocompatible shell [Citation1]. Current clinical studies asses several micelles formulations, which incorporate such anticancer drugs as doxorubicin (Dox), paclitaxel, SN-38, cisplatin and DACHPt (activated oxaliplatin) (NK911, NK105, NK012, NC6001 and NC4016, respectively), and the first four drugs have advanced to phase II study [Citation7]. According to these clinical studies, polymeric micelles showed reduced side effects and high effectiveness to various intractable tumors including triple-negative breast cancers [Citation7]. Moreover, the progress of nanoassemblies into clinical evaluation has encouraged the further development of therapeutics based on polymeric nanoassemblies including micelles, worm-like micelles and vesicles (figure ). This review focuses on the recent advances in polymer nanoassemblies, including micelles and vesicles, with special focus on drug delivery and gene therapy. It provides a comprehensive perspective of recent breakthroughs and applications of nanoassemblies designed for improved tumor therapy.
Figure 1 Block copolymer nanoassemblies reported for tumor targeted drug and gene delivery.
Nanoassemblies design: Overcoming barriers
In biological environment, nanoassemblies are exposed to harsh conditions, and they should overcome certain biological barriers to reach the therapeutic targets [Citation5]. The knowledge gained through the extensive research on drug delivery systems during these years has helped to determine critical parameters for the design of self-assembled nanocarriers. As a result, high stability in physiological media, as well as stealth surface, charge and dimensions of the nanoassemblies have been identified as crucial factors for the design of long circulating drug carriers with the ability to extravasate and accumulate at tumor sites. Hydrophilic surfaces are essential to avoid the interaction with the plasma proteins and the recognition by the reticuloendothelial system (RES) providing stealth properties to the nanoassemblies. Accordingly, several hydrophilic backbones [Citation8, Citation9] have been used as building blocks to produce nanoassemblies with stealth properties and prolonged blood circulation. Among those, poly(ethylene glycol) (PEG) is most popular due to its linearity, lack of charge, immunogenicity, low polydispersity, and easy activation for conjugation [Citation10]. In addition, drug carriers should not be cationic but either neutral or slightly anionic to avoid a non-specific interaction with the luminal surface of blood vessels, which is highly negatively charged. Thus, cationic nanoassemblies are adsorbed on the vascular surface having a short in vivo half-life [Citation11]. Moreover, a relatively small size of the nanocarrier, in combination with a small polydispersity, is also key factor to achieve prolonged circulation. Current knowledge on the effect of the size of nanocarriers alleges that the nanoparticles should be in the range of 10–100 nm to avoid glomerular excretion and attain a deep tumor penetration [Citation3].
The efficiency of drug delivery through nanoassemblies can also be improved by adjusting their design to provide controlled drug release, identification of the targeted site, and specific cellular uptake. Thus, novel nanoassemblies have been considered, which have more advanced architectures and include stimuli-responsiveness, target recognition and imaging ability.
Shape effect
Shape effects of the nanoassemblies have not been studied in detail in vivo, and most of the reported nanoassemblies for drug delivery are spherical. In nature, a number of viruses exhibit filamentous morphologies, such as H5N1 with 1 μm length [Citation12] and Ebola with length above 10 μm [Citation13]. This motivated the recent development and study of worm-micelles for drug delivery. Discher et al studied the shape effect of worm-micelles, also called filomicelles, from amphiphilic block copolymers, both in vitro and in vivo [Citation14, Citation15]. These worm-micelles were prepared from blends of degradable PEG-b-poly(lactic acid) (PEG-b-PLA) and the degradation of PLA by hydrolysis led to the self-shortening of worms and a clear transition toward spherical micelles. Moreover, the high flexibility of worm micelles allows them to penetrate nanoporous gels where 100 nm-sized vesicles cannot enter, suggesting that the tissue permeation of these nanostructures might be augmented in the in vivo situation. In in vitro flow experiments, cells take up the spherical and short worm-micelles faster than longer filaments because the latter are extended by the flow. The worm-micelles were shown to circulate in the blood stream for at least one week after intravenous injection, which is approximately ten times longer than their spherical counterparts. They also effectively delivered paclitaxel to solid tumors leading to remarkable antitumor activity. These findings not only suggest that it is not necessary for long-circulating nanocarriers to be spherical, but also reveal the strong effect of the nanoassemblies shape on their biological properties [Citation15].
Controlled drug delivery
A nanocarrier system incorporating stimuli-responsive property would be suitable to overcome some of the systemic and intracellular delivery barriers. Moreover, the selective release of the nanocarriers cargo should enhance the drug targeting and improve the efficiency of the delivered therapeutics. The stimuli used for controlling the drug release can be classified as endogenous and exogenous triggers. The former are intrinsic stimuli existing in the body given by the unique pathways of the nanoassemblies or the pathological characteristics of the malignancy, and the latter are external stimuli that selectively enhance the nanoassemblies release.
Endogenous triggers
Several endogenous stimuli have been used to design environment-responsive drug delivery systems. Accordingly, the selective drug release or destabilization of the nanoassemblies has been achieved using the differences in pH in the body, the intracellular reductive environment, and the glucose concentration as endogenous triggers.
Solid tumors and inflammatory tissues present mildly acidic conditions of approximately pH 6.8 [Citation16]. They provide a selective trigger for the drug release from nanoassemblies, since blood and normal tissues have a pH of 7.4. Moreover, because nanocarriers enter the cells via endocytosis and are localized in the endosomes or in the lysosomes [Citation17], the pH of endosomal and lysosomal compartments of cells (pH 5–6) is also a very useful stimulus.
Nanoassemblies from polyesters have shown enhanced drug release rate at low pH as the polyester hydrolysis increases with decreasing pH. In vivo studies with polymersomes composed of PEG-b-polyester demonstrated growth arrest and shrinkage of rapidly growing tumors [Citation18]. Although the rate of polyester hydrolysis increases at low pH, such system does not specifically release its cargo synchronously to pH changes. Alternatively, polymers that change polarity in response to pH will maintain structural integrity of the nanoassemblies and result in localized burst release. In this way, PEG-b-poly(L-histidine) (PEG-b-P(His)) was used to prepare pH-sensitive polymeric micelles incorporating Dox [Citation19]. These micelles showed an accelerated release of the drug when the pH decreased due to the ionization of the P(His) block forming the micelle core. PEG-b-poly(2-vinylpyridine) (PEG-b-P2VP) [Citation20] and poly(2-(methacryloyloxy)ethyl phosphorylcholine)-b-poly(2-(diisopropylamino)ethyl methacrylate) (PMPC-b-PDPA) [Citation21] have been shown to form pH-sensitive polymersomes and micelles.
Nanoassemblies having pH sensitivity can also be constructed by the use of an acid-labile bond between the drug and the carrier polymer, such as pH-sensitive Dox-loaded polymeric micelles prepared by chemically conjugating Dox to PEG-b-poly(aspartic acid) (PEG-b-P(Asp)) copolymers via an acid-labile hydrazone bond [Citation22]. These micelles specifically released Dox at endosomal-pH conditions (pH 5.0), whereas Dox was retained in the micelle core at physiological pH. They efficiently suppressed tumor growth in vivo, while the toxicity was negligible due to the minimal drug leakage [Citation23].
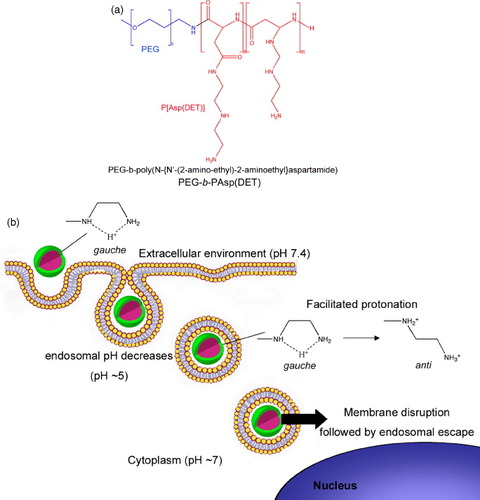
Synthetic self-assembled gene vectors based on cationic polymers can also use pH to enhance their transfection efficiency. The complete dissociation of the nanoassemblies to unimers at endocytic pH has been considered as a targeted strategy to deliver DNA [Citation24], though the endosomal escape of macromolecules is limited. Thus, the selective destabilization of endosomal membranes using pH-sensitive nanoassemblies is a potent strategy to increase the transfection efficiency and selectivity. Accordingly, Miyata et al reported polyion complex (PIC) micelles made from PEG-b-poly(N-{N′-(2-amino-ethyl)-2-aminoethyl}aspartamide) (PEG-b-P[Asp(DET)]) (figure (a)) [Citation25]. These micelles accomplished appreciably high in vitro and in vivo gene transfection due to the membrane destabilization at the acidic pH of late endosomal compartment, which corresponded to the protonation change of the ethylenediamine in the P[Asp(DET)] backbone (figure (b)) [Citation26]. Thus, after endocytosis of these polyplexes, the ethylenediamine unit in the block copolymer is expected to facilitate the efficient translocation of the micelle from the late endosomes toward the cytoplasm. Moreover, Lee et al prepared PIC micelles from PEG-b-poly[(N′-citraconyl-2-aminoethyl)aspartamide] (PEG-b-P(Asp(EDA-Cit))) with the ability to switch the charge from anionic to cationic at the endosomal pH due to the degradation of the citraconic amide side chain at pH 5.5 [Citation27, Citation28]. This strategy is very promising for loading a wide range of charged compounds, while controlling the stability, charge and biological fate of the nanoassemblies.
Figure 2 (a) Chemical structure of PEG-b-PAsp(DET) copolymer bearing an ethylenediamine unit at the side chain, which can form stable polyplexes with pH-sensitive protonation properties. (b) The protonation change of the ethylenediamine in the PAsp(DET) backbone of the polyplexes leads to the membrane destabilization at the acidic pH of late endosomal compartments facilitating the efficient translocation of the polyplexes to cytoplasm.
Instead of relying on pH changes to cleave acid-labile bonds or shift the polarity of the hydrophobic block, destabilization of nanoassemblies, by either oxidation or reduction in responsiveness, can result in the selective release of the encapsulated drugs. The redox triggering can occur at inflammatory and tumors sites since they present activated macrophages that release oxygen-reactive species. Moreover, the thiol-rich environment of the cytosol of cells offers a selective reductive stimulus for the controlled release of therapeutics. Hubbell et al prepared polymersomes using triblock copolymers consisting of PEG-b-poly(propylene sulfide)-b-PEG (PEG-b-PPS-b-PEG) [Citation29]. After exposing these vesicles to oxidative agents, the PPS block is oxidized to poly(propylene sulfoxide) and poly(propylene sulfone) leading to the hydrophilization of the originally hydrophobic block and the dissociation of the vesicles.
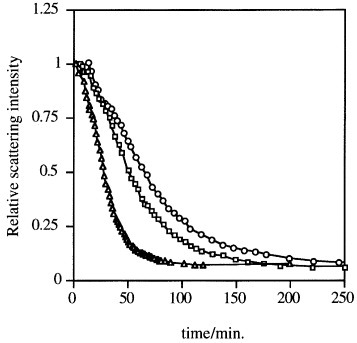
Disulfide bonds are another type of reduction-sensitive functional group that can be introduced to nanoassemblies since they can be selectively cleaved in the reductive intracellular environment. Accordingly, PIC micelles with disulfide-crosslinked cores selectively dissociated under reductive conditions, as found from the decrease in the static light scattering (SLS) intensity after the addition of dithiothreitol (DTT; figure ), and efficiently delivered the loaded pDNA [Citation30–33]. The intracellular glutathione, which concentration is 50 to 1000 times higher than extracellular glutathione, reductively cleaves the disulfide links leading to disruption of this system and enhancing the in vivo transfection efficiencies [Citation33].
Figure 3 PIC micelles sensitive to reductive conditions. Change in the relative scattering light intensity of disulfide-crosslinked PIC complex micelles after the addition of dithiothreitol (DTT) (○, 0.5 mM; ▵, 1.0 mM; □, 2.0 mM) in 10 mM PBS at pH 7.4. (Reprinted with permission from [Citation30] © 1999 American Chemical Society.)
Exogenous triggers
External triggers such as light, temperature and ultrasound can also be applied to achieve selective drug delivery through a destabilization of the carriers or an enhanced drug release. Poly(N-isopropylacrylamide) (PNIPAAm), has been widely studied as thermosensitive polymer for biomedical applications due to its sharp lower critical solution temperature (LCST) in water at approximately 32 °C [Citation34–36]. Moreover, the LCST of a thermosensitive polymer can be modulated by copolymerizing it with hydrophilic comonomers to increase LCST since PNIPAAm is in its precipitated form at body temperature. The copolymerization of NIPAAm with the hydrophilic dimethylacrylamide (DMAAm) resulted in a random copolymer (P(NIPAAm-co-DMAAm)) with an LCST slightly above body temperature (40 °C) [Citation36]. Thus, the release of Dox from P(NIPAAm-co-DMAAm)-b-PLA micelles was very slow at 37 °C, while the Dox release rate increased at 42.5 °C suggesting the potential of this system as temperature-sensitive nanoassembly. Another promising thermosensitive polymer is poly(2-isopropyl-2-oxazoline) (PiPrOx), with an LCST near physiological conditions [Citation37, Citation38]. Park et al prepared novel thermosensitive PIC micelles with a constant cloud-point temperature of approximately 32 °C via the complexation of a pair of oppositely charged block copolymers containing the thermosensitive PiPrOx segments, PiPrOx-b-P(Lys) and PiPrOx-b-P(Asp) [Citation39]. Since the LCST of PiPrOx can also be tuned by copolymerization [Citation40], these PiPrOx-PIC micelles have high potential as a size-regulated temperature-responsive nanocarrier for loading charged compounds.
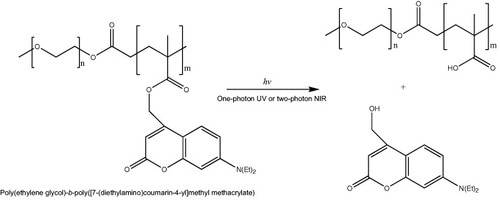
An attractive feature of light-responsive polymeric assemblies is that the drug release can be induced at a specific time at the site of light exposure. Light in the ultraviolet (UV), visible (VIS) or near-infrared (NIR) region has been applied as the trigger [Citation41]; however, since NIR light penetrates deeper into tissues and induces minimal damage to healthy cells, it is of particular interest for biomedical applications [Citation42]. Jiang et al reported a PEG-b-poly(2-nitrobenzyl methacrylate) system [Citation43] where the cleavage of 2-nitrobenzyl moieties occurred by photolysis either via one photon UV (365 nm) or two-photon NIR (700 nm) excitation. The formation of carboxylic acid after irradiation shifted the hydrophilic/hydrophobic balance and resulted in the break up of the micelles, or the swelling of the micelle core when it was crosslinked with a diamine. Nevertheless, the sensitivity of this system was low because of inefficient two-photon absorption. Recently, NIR photosensitive polymeric micelles were prepared with block copolymers bearing coumarin chromophores, namely, [7-(diethylamino)coumarin-4-yl]methyl, having a large two-photon absorption cross section (figure ). The photolysis of [7-(diethylamino)coumarin-4-yl]methyl esters by one-photon UV or two-photon NIR irradiation leads to the release of 7-diethylamino-4-(hydroxymethyl)coumarin, and the conversion of the ester groups to carboxylic acid shifting the hydrophobic backbone to hydrophilic poly(methacrylic acid) (PMA) (figure ). Thus, the disruption under irradiation of polymeric micelles prepared with this block copolymer released both preloaded nile red and photocleaved coumarin molecules from the hydrophobic micelle core [Citation44].
Figure 4 Photosensitive block copolymer for nanoassemblies. Chemical structure of PEG-b-poly([7-(diethylamino)coumarin-4-yl]methyl methacrylate) and photolysis reaction after UV or NIR irradiation.
Ultrasound can also be used to release the drug from nanoassemblies while precisely controlling the irradiation position inside the body. Ultrasound has been used as a non-invasive trigger for the in vitro and in vivo release of drugs from PEG-b-poly(propylene oxide) (PPO)-b-PEG (poloxamer) micelles [Citation45–47] implying that ultrasound could be focused on a localized tumor and the anti-cancer agent can be released from the micelles and delivered directly to the malignancy.
Active targeted nanoassemblies
The controlled interaction of the nanoassemblies with the pathological tissues is a key point in the design of efficient drug delivery systems. Given the PEG shielding, the activity of most long circulating nanocarriers decreases due to their poor cellular uptake, which is often a disadvantage for exerting drug efficacy compared to free drugs that rapidly move into the interior of the cells. Therefore, it is likely that if a drug carrier is actively transported then the specificity and bioavailability of those drug delivery systems should exceed those of a drug delivery system that just take advantage of the EPR effect to target the tumor.
A wide variety of targeting molecules have been assessed, with varying degrees of success, for their potential application in cancer therapy, including humanized antibodies and single-chain Fv generated from murine hybridoma or phage display, minibodies, aptamers and peptides [Citation48]. Several tumor-specific antibodies have been used to modify PEG-based assemblies in order to recognize tumor-associated antigens and increase the exposure of the nanoassemblies to the malignant cells [Citation49, Citation50]. Nevertheless, the antibody-targeted carrier systems still have several limitations regarding problems with immunogenicity, stability and storage, and expensive and time-consuming procedures.
Aptamers [Citation51] have been recently considered as targeting moieties that counterpart the potential of antibodies for diagnostic and therapeutic applications [Citation52]. Aptamers are RNA or DNA oligonucleotides that fold by intramolecular interaction into three-dimensional conformations with the ability to specifically bind targeted proteins. The small size, lack of immunogenicity, and ease of isolation of aptamers have resulted in their rapid progress into clinical trials [Citation53]. These molecules have been considered for targeted delivery of Dox-loaded polymeric micelles to prostate cancer presenting selective tumor accumulation and uptake by the cancer cells [Citation54, Citation55]. Biotinylated nanoassemblies have also been shown to attach to cells and to surfaces coated with the biotin receptor avidin. In this way, they efficiently deliver Dox from pH sensitive micelles, which consist of PEG-b-P(His) and Biotin-P(His)-b-PEG-b-PLA and show improved activity compared to the non-targeted micelles [Citation56]. Stable worm micelles have also been targeted using end-biotinylated block copolymers to mediate high-affinity binding to both avidin-bearing surfaces and biotin-specific receptors on smooth muscle cells [Citation57].
An alternative to cell-specific cancer targeting strategies is the use of HIV-derived transactivator of transcription (TAT) peptide, which has a strong capability to translocate nanoparticles into cells and to enhance the cellular uptake of the nanoassemblies. Accordingly, Lee et al reported polymeric micelles from TAT-P(His)-b-PEG-b-PLA for the delivery of Dox. The surface of these micelles can hide TAT during circulation at pH 7.4; it can also expose TAT at the slightly acidic tumor extracellular pH to enhance internalization of the micelles. This pathway increased intracellular concentrations of Dox and the drug potency in various cell lines including Dox-resistant cells in vitro and in vivo [Citation58].
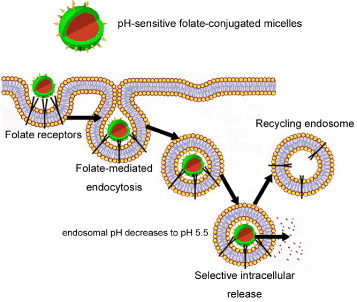
The use of the existing endocytosis pathways for specific drug delivery is a practical strategy, and the conjugation of normally endocytosed ligands to nanocarriers frequently improves their uptake and accumulation. Among these ligands, folic acid has been intensively investigated as a means for tumor-specific delivery of a broad range of experimental therapies including several conceptually new treatments [Citation59]. The folate receptor (FR) is able to bind and transport a wide variety and a broad size range of chemical conjugates of folic acid, antifolate drugs and immunological agents. This property generates an immense interest in pH-sensitive nanoassemblies that would selectively target FR-positive malignancies (figure ). Furthermore, several malignant tumors are known to overexpress FR [Citation60–63] and significant correlations have been made between the FR expression level and the grade and differentiation status of the tumor; the highest expression of FR is associated with poorly differentiated and more aggressive tumors [Citation64]. Accordingly, Bae et al were able to selectively target pharyngeal cancer cells in vitro and in vivo with folate-conjugated pH-sensitive micelles incorporating Dox [Citation65]. Moreover, Dox-loaded pH-sensitive micelles prepared from folate-PEG-b-P(His) were able to overcome multidrug resistance in MCF-7/DoxR solid tumors both in vitro and in vivo [Citation66].
Figure 5 Folate targeted nanoassemblies. Enhanced cellular uptake by folate-mediated endocytosis of folate-conjugated nanoassemblies. The plasma membrane envelops the nanoassembly/folate receptor complex to form an endosome. As the lumen of the maturing endosome acidifies up to pH 5.5, the receptor changes the conformation and releases the conjugate.
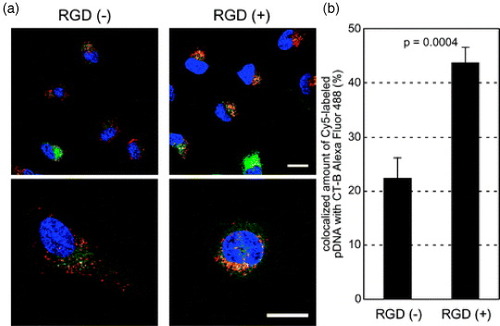
The tumor endothelial cells overexpress several cell-surface molecules promoting cell invasion and proliferation during tumor vascular remodeling and angiogenesis that can be used as pilot molecules for nanoassemblies [Citation67]. One such molecule is the αvβ3 integrin, which plays a key role in endothelial cell survival during angiogenesis [Citation68]. In fact, αvβ3 was recently used as an endothelial cell target in several nanoassemblies approaches such as tumor targeted paclitaxel-loaded nanoparticles [Citation69] and non-viral gene delivery [Citation70, Citation71]. Moreover, Oba et al recently reported that cRGD functionalized PIC micelles were distributed in the perinuclear region preferentially through caveolae-mediated endocytosis (figure (a)). This doubled the signal of the micelles in the caveosomes (figure (b)), which may be a desirable pathway for avoiding the lysosomal degradation of delivered genes, and enhanced the transfection efficiency of the nanoassemblies [Citation72].
Figure 6 Intracellular distribution of cRGD-conjugated disulfide-crosslinked PIC micelles. Polyplex micelles loading Cy5-labeled pDNA (red) and CT-B Alexa Fluor 488 conjugate, a marker for the lipid rafts and the caveosomes (green), were incubated with HeLa cells for 1 h, washed and reincubated for 11 h. The cell nuclei were stained with Hoechst 33342 (blue). (a) CLSM images of RGD (−) micelles (left) and RGD (+) micelles (right). The scale bars represent 20 μm. (b) Quantification of Cy5-labeled pDNA colocalized with CT-B in the inner-cytoplasm. Error bars represent SEM (n=10). (Reprinted with permission from [Citation72] ©2008 The American Society for Pharmacology and Experimental Therapeutics.)
Another interesting approach considered is the functionalization of polymeric vesicles with the adhesive properties of leukocytes, i.e. leuko-polymersomes. Thus, functionalizing the terminal groups on the outer shell of vesicles via avidin–biotin chemistry, Hammer et al attached adhesion ligands, selectins and integrins, mimicking the two critical adhesion pathways that leukocytes utilize to achieve adhesion in the fast fluid flow of blood vessels [Citation73]. These leuko-polymersomes achieved specific adhesion at hydrodynamic flow rates at which leukocytes adhere. Given the close relationship between tumors and inflammatory processes, such vesicles might be very useful not only for monitoring or treating inflammation, but also for cancer application [Citation74].
Despite various remarkable results, the cell targeting strategies have found impediments to be translated to in vivo conditions due to complications from opsonization by serum components and competitive interactions with other cells. Though promising, targeted carriers must contact the desired target either by convection or diffusion before their pilot molecules can increase the chances of cellular uptake and localized release. Therefore, future design of nanoassemblies with targeting ligands should thoroughly consider the benefits from active targeting versus passive targeting via the EPR effect.
Imaging, diagnosis and therapy: theranostic nanoassemblies
Theranostic nanoassemblies should be able to diagnose and deliver targeted therapy and monitor the response to the therapy [Citation75]. This integration of diagnostic imaging capacity with therapeutic involvement is critical for addressing the challenges of cancer alterations and heterogeneities. As a platform technology, nanoassemblies have the advantage of being able to combine targeting to multiple tumor markers and delivery of multiple agents, simultaneously.
Real-time diagnosis, therapy monitoring and feedback on efficiency are the essential components of theranostic nanoassemblies, and the recent advances on their design have brought these goals closer. In this way, several approaches combining tumor targeting with magnetic resonance imaging (MRI) and drug delivery have been reported. Accordingly, theranostic micelles showed an increased and αvβ6-dependent cell targeting in H2009 lung cancer cells. These micelles were functionalized with a lung cancer-targeting peptide (LCP) and loaded with superparamagnetic iron oxide (SPIO) and Dox for MRI and therapy, respectively. T2-weighted MRI images showed clear contrast in the H2009 cells incubated with LCP-encoded micelles while enhancing the cytotoxicity. The integrated diagnostic and therapeutic design of multifunctional nanomedicine potentially allows for image-guided, target-specific treatment of lung cancer [Citation76]. Also, theranostic cRGD-functionalized polymeric micelles were loaded with Dox and SPIO nanoparticles. In vitro and in vivo MRI and cytotoxicity studies demonstrated the ultrasensitive MRI imaging and αvβ3-specific cytotoxic response of these theranostic micelles [Citation77].
Not only MRI has been used as the imaging modality of theranostic nanodevices, quantum dots (QD)-based theranostic nanoassemblies were recently reported. The high surface-to-volume ratio of QDs enables the construction of smart multifunctional assemblies, where the QDs serve not only as an imaging agent but also a nanoscaffold. The QD–aptamer–Dox system (QD–Apt(Dox)) directed to the prostate cancer sensed the drug release based on a bi-FRET design. In the drug-loading state, both QD and Dox were not fluorescent because the QD fluorescence was quenched by the Dox, while the Dox fluorescence was quenched by the aptamer. On the other hand, in the drug-release state, the Dox was released from the QD–Apt complex, thus turning both QD and Dox fluorescence back ON. This multifunctional QD was demonstrated to enhance the therapeutic specificity against the targeted prostate cancer cells (LNCaP) compared to non-specific cells (PC3) in vitro [Citation78].
Although in vivo data of theranostic nanoassemblies are limited, the results obtained so far suggest the high potential and feasibility of this approach. The emergence of theranostic nanoassemblies will lead to a new conception of cancer treatments where therapies can be personalized to individual patients and real-time adjustment for improved efficiency.
Conclusion
Nanoassemblies are potent platforms for the development of cancer-specific drug delivery systems. Several characteristics of the nanoassemblies have been identified as crucial for their targeting success and already become imperative for the proper nanostructure design. Yet, the current methods to assess the optimal properties of the nanoassemblies are inadequate or inefficient mainly because of the dynamism of the biological environment. Thus, the success of cancer-targeted nanoassemblies must be evaluated experimentally for every particular case. A deeper understanding in cancer biology, nanobiology and polymer chemistry will catalyze optimization pathways to achieve more efficient anti-tumor systems. Gaining insights to the differences between normal and pathological tissues will also lead to the development of new selective triggers, targeted therapies and a highly promising role for stimuli-responsive nanoassemblies. Moreover, since most of the therapeutic targets are located intracellularly, the controlled release and tissue targeting of the nanoassemblies should evolve to specific drug delivery to subcellular targets. This approach may enhance the drug function, overcome drug resistance and lead to unprecedented therapeutic effects.
References
- KataokaKHaradaANagasakiY 2001 Adv. Drug Deliv. Rev. 47 113 http://dx.doi.org/10.1016/S0169-409X(00)00124-1
- DuncanR 2003 Nat. Rev. Drug Discov. 2 347 http://dx.doi.org/10.1038/nrd1088
- SenterP DKopecekJ 2004 Mol. Pharm. 1 395 http://dx.doi.org/10.1021/mp040010x
- MatsumuraYMaedaH 1986 Cancer Res. 46 6387
- DavisM EChenZ GShinD M 2008 Nat. Rev. Drug Discov. 7 771 http://dx.doi.org/10.1038/nrd2614
- NishiyamaNKataokaK 2006 Pharmacol. Ther. 112 630 http://dx.doi.org/10.1016/j.pharmthera.2006.05.006
- MatsumuraY 2008 Japan . J. Clin. Oncol. 38 793 http://dx.doi.org/10.1093/jjco/hyn116
- OwensD EPeppasN A 2006 Int. J. Pharm. 307 93 http://dx.doi.org/10.1016/j.ijpharm.2005.10.010
- HeMZhaoZYinLTangCYiC 2009 Int. J. Pharm. 373 165 http://dx.doi.org/10.1016/j.ijpharm.2009.02.012
- LeeJ HLeeH BAndradeJ D 1995 Prog. Polym. Sci. 20 1043 http://dx.doi.org/10.1016/0079-6700(95)00011-4
- PapisovMBogdanovASchafferBNossiffNShenTWeisslederR 1993 J. Magn. Magn. Mater. 122 383 http://dx.doi.org/10.1016/0304-8853(93)91115-N
- ShortridgeK F et al 1998 Virology 252 331 http://dx.doi.org/10.1006/viro.1998.9488
- BrayMGeisbertT W 2005 Int. J. Biochem. Cell Biol. 37 1560 http://dx.doi.org/10.1016/j.biocel.2005.02.018
- KimYDalhaimerPChristianD ADischerD E 2005 Nanotechnology 16 S1 http://dx.doi.org/10.1088/0957-4484/16/3/001
- GengYDalhaimerPCaiSTsaiRTewariMMinkoTDischerD E 2007 Nat. Nanotech. 2 249 http://dx.doi.org/10.1038/nnano.2007.70
- EnginKLeeperD BCaterJ RThistlethwaiteA JTupchongLMcFarlaneJ D 1995 Int. J. Hypertherm. 11 211 http://dx.doi.org/10.3109/02656739509022457
- SavicRLuoLEisenbergAMaysingerD 2003 Science 300 615 http://dx.doi.org/10.1126/science.1078192
- AhmedFPakunluR ISrinivasGBrannanABatesFKleinM LMinkoTDischerD E 2006 Mol. Pharm. 3 340 http://dx.doi.org/10.1021/mp050103u
- LeeE SShinH JNaKBaeY H 2003 J. Control. Release 90 363 http://dx.doi.org/10.1016/S0168-3659(03)00205-0
- BorchertULipprandtUBilangMKimpflerARankAPeschka-SussRSchubertRLindnerPForsterS 2006 Langmuir 22 5843 http://dx.doi.org/10.1021/la060227t
- GiacomelliCMenL LBorsalR 2006 Biomacromolecules 7 817 http://dx.doi.org/10.1021/bm0508921
- BaeYFukushimaSHaradaAKataokaK 2003 Angew. Chem. Int. Ed. Engl. 42 4640 http://dx.doi.org/10.1002/anie.200250653
- BaeYNishiyamaNFukushimaSKoyamaHYasuhoriMKataokaK 2005 Bioconjug. Chem. 16 122 http://dx.doi.org/10.1021/bc0498166
- LomasHCantonIMacNeilSDuJArmesS PRyanA JLewisA LBattagliaG 2008 Adv. Mater. 19 4238 http://dx.doi.org/10.1002/adma.200700941
- AkagiDObaMKoyamaHNishiyamaNFukushimaSMiyataTNagawaHKataokaK 2007 Gene Ther. 14 1029 http://dx.doi.org/10.1038/sj.gt.3302945
- MiyataKObaMNakanishiMFukushimaSYamasakiYKoyamaHNishiyamaNKataokaK 2008 J. Am. Chem. Soc. 130 16287 http://dx.doi.org/10.1021/ja804561g
- LeeYFukushimaSBaeYHikiSIshiiTKataokaK 2007 J. Am. Chem. Soc. 129 5362 http://dx.doi.org/10.1021/ja071090b
- LeeYIshiiTCabralHKimH JSeoJ-HNishiyamaNOshimaHOsadaKKataokaK 2009 Angew. Chem. Int. Ed. Engl. 48 5309 http://dx.doi.org/10.1002/anie.200900064
- NapoliAValentiniMTirelliNMullerMHubbellJ A 2004 Nat. Mater. 3 183 http://dx.doi.org/10.1038/nmat1081
- KakizawaYHaradaAKataokaK 1999 J. Am. Chem. Soc. 121 11247 http://dx.doi.org/10.1021/ja993057y
- MiyataKKakizawaYNishiyamaNHaradaAYamasakiYKoyamaHKataokaK 2004 J. Am. Chem. Soc. 126 2355 http://dx.doi.org/10.1021/ja0379666
- MatsumotoSChristieR JNishiyamaNMiyataKIshiiAObaMKoyamaHYamasakiYKataokaK 2009 Biomacromolecules 10 119 http://dx.doi.org/10.1021/bm800985e
- MiyataKKakizawaYNishiyamaNYamasakiYWatanabeTKoharaMKataokaK 2004 J. Control. Release 109 15 http://dx.doi.org/10.1016/j.jconrel.2005.09.043
- SchildH G 1992 Prog. Polym. Sci. 17 163 http://dx.doi.org/10.1016/0079-6700(92)90023-R
- ChungJ EYokoyamaMOkanoT 2000 J. Control. Release 65 93 http://dx.doi.org/10.1016/S0168-3659(99)00242-4
- KohoriFSakaiKAoyagiTYokoyamaMYamatoMSakuraiYOkanoT 1999 Colloids Surf. B: Biointerface 16 195 http://dx.doi.org/10.1016/S0927-7765(99)00070-3
- UyamaHKobayashiS 1992 Chem. Lett. 1643
- ParkJ-SAkiyamaYWinnikF MandK 2007 Macromolecules 37 6786 http://dx.doi.org/10.1021/ma049677n
- ParkJ-SAkiyamaYYamasakiYKataokaK 2007 Langmuir 23 138 http://dx.doi.org/10.1021/la061431j
- ParkJ-SKataokaK 2007 Macromolecules 40 3599 http://dx.doi.org/10.1021/ma0701181
- RijckenC J FSogaOHenninkW Evan NostrumC F 2007 J. Control. Release 120 131 http://dx.doi.org/10.1016/j.jconrel.2007.03.023
- WeisslederR 2001 Nat. Biotech. 19 327 http://dx.doi.org/10.1038/86684
- JiangJTongXMorrisDZhaoY 2006 Macromolecules 39 4633 http://dx.doi.org/10.1021/ma060142z
- BabinJPelletierMLepageMAllardC FMorrisDZhaoY 2009 Angew. Chem. Int. Ed. Engl. 121 3379 http://dx.doi.org/10.1002/ange.200900255
- BatrakovaE VKabanovA V 2008 J. Control. Release 130 98 http://dx.doi.org/10.1016/j.jconrel.2008.04.013
- PittW GHusseiniG AStaplesB J 2004 Expert Opin. Drug Deliv. 1 37 http://dx.doi.org/10.1517/17425247.1.1.37
- HusseiniG AMyrupG DPittW GChristensenD ARapoportN Y 2000 J. Control. Release 69 43 http://dx.doi.org/10.1016/S0168-3659(00)00278-9
- AllenT M 2002 Nat. Rev. Cancer 2 750 http://dx.doi.org/10.1038/nrc903
- TorchilinV PLevchenkoT SLukyanovA NKhawB AKlibanovA LRammohanRSamokhinG PWhitemanK R 2001 Biochim. Biophys. Acta. 1511 397 http://dx.doi.org/10.1016/S0005-2728(01)00165-7
- TorchilinV PLukyanovA NGaoZPapahadjopoulos-SternbergB 2003 Proc. Natl Acad. Sci. USA 100 6039 http://dx.doi.org/10.1073/pnas.0931428100
- EllingtonA DSzostakJ W 1990 Nature 346 818 http://dx.doi.org/10.1038/346818a0
- BrodyE NGoldL 2000 J. Biotechnol. 74 5
- VinoresS A 2003 Curr. Opin. Mol. Ther. 5 673
- FarokhzadO CJonSKhademhosseiniATranT-N TLaVanD ALangerR 2004 Cancer Res. 64 7668 http://dx.doi.org/10.1158/0008-5472.CAN-04-2550
- FarokhzadO CChengJTeplyB ASherifiAJonSKantoffP WRichieJ PLangerR 2006 Proc. Natl. Acad. Sci. USA 103 6315 http://dx.doi.org/10.1073/pnas.0601755103
- LeeE SNaKBaeY H 2005 Nano Lett. 5 325 http://dx.doi.org/10.1021/nl0479987
- DalhaimerPEnglerA JParthasarathyRDischerD E 2004 Biomacromolecules 5 1714 http://dx.doi.org/10.1021/bm049884v
- LeeE SGaoZKimDParkKKwonI CBaeY H 2008 J. Control. Release 129 228 http://dx.doi.org/10.1016/j.jconrel.2008.04.024
- LeamonC PLowP S 2001 Drug Discov. Today 6 44 http://dx.doi.org/10.1016/S1359-6446(00)01594-4
- ToffoliGCernigoiCRussoAGalloABagnoliMBoiocchiM 1997 Int. J. Cancer 74 193 http://dx.doi.org/10.1002/(SICI)1097-0215(19970422)74:2<193::AID-IJC10>3.0.CO;2-F
- RossJ FChaudhuriP KRatnamM 1994 Cancer 73 2432 http://dx.doi.org/10.1002/1097-0142(19940501)73:9<2432::AID-CNCR2820730929>3.0.CO;2-S
- BuenoRAppasaniKMercerHLesterSSugarbakerD 2001 J. Thoracic Cardiovasc. Surg. 121 225 http://dx.doi.org/10.1067/mtc.2001.111176
- EvansC OYoungA NBrownM RBratD JParksJ SNeishA S 2001 J. Clin. Endocrinol. Metab. 86 3097 http://dx.doi.org/10.1210/jc.86.7.3097
- ReddyJ AAllagaddaV MLeamonC P 2005 Curr. Pharm. Biotech. 6 131 http://dx.doi.org/10.2174/1389201053642376
- BaeYNishiyamaNKataokaK 2007 Bioconjug. Chem. 18 1131 http://dx.doi.org/10.1021/bc060401p
- LeeE SNaKBaeY H 2005 J. Control. Release 103 405 http://dx.doi.org/10.1016/j.jconrel.2004.12.018
- YancopoulosG DKlagsbrunMFolkmanJ 1998 Cell 93 661 http://dx.doi.org/10.1016/S0092-8674(00)81426-9
- BrooksP CMontgomeryA MRosenfeldMReisfeldR AHuTKlierGChereshD A 1994 Cell 79 1157 http://dx.doi.org/10.1016/0092-8674(94)90007-8
- DanhierFVromanBLecouturierNCrokartNPourcelleVFreichelsHJérômeCMarchand-BrynaertJFeronOPréatV 2009 J. Control. Release 140 166 http://dx.doi.org/10.1016/j.jconrel.2009.08.011
- WickhamT J 2003 Nat. Med. 9 135 http://dx.doi.org/10.1038/nm0103-135
- HoodJ DBednarskiMFraustoRGuccioneSReisfeldR AXiangRChereshD A 2002 Science 296 2404 http://dx.doi.org/10.1126/science.1070200
- ObaMAoyagiKMiyataKMatsumotoYItakaKNishiyamaNYamasakiYKoyamaHKataokaK 2008 Mol. Pharm. 5 1080 http://dx.doi.org/10.1021/mp800070s
- HammerD ARobbinsG PHaunJ BLinJ JQiWSmithL AGhotoghchianP PTherienM JBatesF S 2008 Faraday Discuss. 139 129 http://dx.doi.org/10.1039/b717821b
- CoussensL MWerbZ 2002 Nature 420 860 http://dx.doi.org/10.1038/nature01322
- SummerBGaoJ 2008 Nanomedicine 3 137 http://dx.doi.org/10.2217/17435889.3.2.137
- GuthiJ SYangS-GHuangGLiSKhemtongCKessingerC WPeytonMMinnaJ DBrownK CGaoJ 2009 Mol. Pharm. at press
- NasongklaNBeyERenJAiHKhemtongCGuthiJ SChinS-FSherryA DBoothmanD AGaoJ 2006 Nano Lett. 6 2427 http://dx.doi.org/10.1021/nl061412u
- BagalkotVZhangLLevy-NissenbaumEJonSKantoffP WLangerRFarokhzadO C 2007 Nano Lett. 7 3065 http://dx.doi.org/10.1021/nl071546n