Abstract
Large amounts of radioactive material were released from the Fukushima Daiichi nuclear plant in Japan, contaminating the local environment. During the early stages of such nuclear accidents, iodine I-131 (half-life 8.02 d) is usually detectable in the surrounding atmosphere and bodies of water. On the other hand, in the long-term, soil and water contamination by Cs-137, which has a half-life of 30.17 years, is a serious problem. In Japan, the government is planning and carrying out radioactive decontamination operations not only with public agencies but also non-governmental organizations, making radiation measurements within Japan. If caesium (also radiocaesium) could be detected by the naked eye then its environmental remediation would be facilitated. Supramolecular material approaches, such as host–guest chemistry, are useful in the design of high-resolution molecular sensors and can be used to convert molecular-recognition processes into optical signals. In this work, we have developed molecular materials (here, phenols) as an optical probe for caesium cation-containing particles with implementation based on simple spray-on reagents and a commonly available fluorescent lamp for naked-eye detection in the solid state. This chemical optical probe provides a higher spatial resolution than existing radioscopes and gamma-ray cameras.
1. Introduction
Caesium-133 is the heaviest stable alkali metal and it has had only a few significant applications, such as in atomic clocks [Citation1] or as gradient centrifugation reagents [Citation2]. More recently, however, caesium compounds as their brines have emerged as important materials for deep drilling operations due to the high densities of their solutions [Citation3] and their relatively low toxicity [Citation4]. Radiocaesium, on the other hand, including the two longer-lived isotopes of caesium Cs-134 (half-life 2.07 years) and Cs-137 (half-life 30.17 years), is a major source of environmental contamination in the vicinity of radiation leaks such as those which occurred at Chernobyl, Ukraine [Citation5], and more recently at Fukushima, Japan [Citation6, Citation7].Footnote4 During nuclear reactor breaches, iodine I-131 (half-life 8.02 d) is released to the atmosphere. Concurrently Cs-137 is released as a microparticulate aerosol leading to long-term soil and water contamination [Citation8].
In addition to the well-known nuclear reactor disasters, radiocaesium has been more gradually introduced into the biosphere through nuclear weapons testing over the last 60 years [Citation9]. Dual environmental hazards are presented by radiocaesium in that background radiation exposure is increased and, more significantly, it is known to be bioaccumulated, leading to a much greater risk of carcinogenesis in organisms [Citation10]. Although caesium is gradually eliminated from mammals when it is removed from dietary sources, levels of corporeal contamination may be maintained or increased in cases of chronic pollution surrounding nuclear disaster areas, where locally grown agricultural produce certainly contain significant amounts of radioactive contaminants. Uptake of radiocaesium by cultivated plants is therefore a significant source of risk to local populations [Citation11] since radiocaesium is usually simply left to decay, meaning that radiation leaks (and nuclear weapons tests) result in major legacies of environmental contamination [Citation12]. Materials that can be used to remove these toxic elements should be considered from the viewpoint of environmental protection.
Areas contaminated by accumulated Cs-134 and Cs-137 can be monitored from over-flying aircraft and, in Japan, both public agencies and non-governmental organizations undertake such measurements. Although the Japanese government is planning and implementing radioisotope decontamination operations, the serious problem of waste treatment remains. For instance, Toshiba [Citation13] and JAXA [Citation14] are developing a gamma ray camera to visualize radioactivity. Since caesium is released during reactor breaches in a particulate form it would be useful if these particulates could be detected within large volumes of contaminated ground soil in the solid state. For these reasons, it would be very useful to develop sensors for particles that contain caesium cations (Cs+) Footnote5 that could be used to screen large areas of contaminated land, especially in the vicinities of radiation leaks. This would facilitate identification of contaminated soil and the actual volume of soil requiring treatment could be reduced by a factor of more than 100 000 by visualization of Cs+-containing particles (see the supplementary information available at stacks.iop.org/STAM/14/015002/mmedia). Apart from measurement using specially calibrated radiation meters, there are no materials that can be applied to the rapid detection of microscopically distributed caesium cations using a simple procedure. Host–guest supramolecular materials promise simple detection of a variety of chemical species, especially specific ions [Citation15–Citation18]. Host–guest interactions may be converted into detectable signals such as electrical and optical outputs while, for instance, fluorescent molecular sensing materials provide greater spatial resolution [Citation19–Citation21].
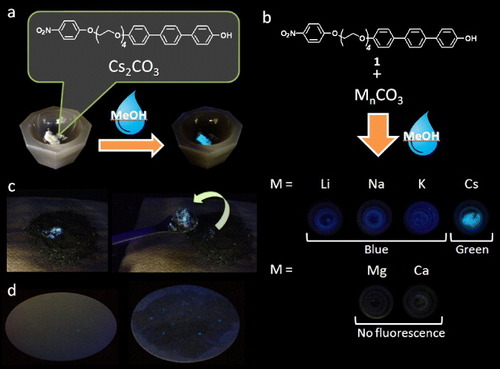
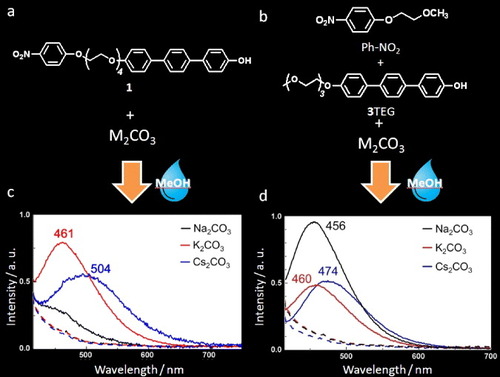
In this work, we have adopted the deprotonation of phenols as a probe reaction for detecting alkali metal cations. Phenols are a notable class of organic compound because of the acidity of their hydroxyl protons [Citation22]. Thus, deprotonation can occur by treatment with an inorganic base (e.g. Na2CO3, K2CO3) yielding a phenolate anion with associated countercation such as Na+ or K+. For substituted phenols, such as 4-hydroxy-1-biphenyl and 1-hydroxy-4,4’,4’’-terphenyl [HTP; 1-(4’-hydroxy phenyl)-4-phenylbenzene], substantial changes in fluorescence emission spectra are observed upon deprotonation [Citation23, Citation24], which are strongly affected by the nature of the substituent, solvent and deprotonating reagent. Furthermore, intermolecular processes such as fluorescence resonance energy transfer (FRET) [Citation25] or excimer emission [Citation26] can be designed into molecules to optimize or tune any sensing output signal. Using these features, we initially designed a class of HTP sensing elements and compounded one of them with an electron-accepting 4-nitrophenyl ether (4-NPE) group through a variable-length polyethylene glycol chain with the aim of selectively detecting alkali metal or alkali earth metal cations. Figure shows the chemical structure of 4-NPE-substituted HTP 1, which contains a tetraethylene glycol (TEG) linker separating the two aromatic moieties. Caesium cation-containing particles can be visualized after spraying with 1 in methanol. Significantly, because of our molecular design, the probe is selective for caesium cations making it useful for the detection of caesium in the environment. The probe is particularly suitable for detecting caesium salts in particulate form and allows them to be detected in the solid state with the naked eye.
Figure 1. Photographs of fluorescence changes of a mixture of 1 and various carbonate salts after addition of a drop of methanol. (a) Fluorescence change of a powdered mixture of 1 and Cs2CO3 under UV irradiation (365 nm) after addition of a drop of methanol. (b) Photographs of a mixture of 1 and various carbonate salts under UV irradiation (365 nm) after addition of a drop of methanol. (c) Photographs of 1 + Cs+ on dirt under room light (left) and under UV irradiation (365 nm, right) after spraying with methanol. (d) Photographs of 1 + Cs+ particles on filter paper (diameter 110 mm) under UV irradiation (365 nm) after spraying with methanol.
2. Experimental section
2.1. Materials
Solvents and materials were purchased from Tokyo Chemical Industry Co Ltd (Tokyo, Japan) and Wako Pure Chemical Co (Osaka, Japan), and were used without further purification. 1H and 13C nuclear magnetic resonance (NMR) spectra were measured at 298 K from CDCl3 or d6-dimethyl sulfoxide (DMSO) solutions of the samples using a JEOL model AL300BX (300 MHz) spectrometer with tetramethylsilane (Me4Si) as an internal standard. Chemical shifts (δ) and coupling constants (J) are given in parts per million (ppm) and hertz (Hz), respectively. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) measurements were carried out using a Shimadzu-Kratos AXIMA + mass spectrometer with dithranol as a matrix. High-resolution mass spectra (HRMS, ESI-positive) were acquired with JMS-MS700 (JEOL), and elemental analysis was performed with a PE 2400II (PerkinElmer) system.
2.2. Measurements
Electronic absorption spectra were measured from tetrahydrofuran solutions of the samples using a Shimadzu model UV-3600 UV/Vis/NIR spectrophotometer. Fourier transform infrared (FTIR) spectra were measured from KBr pellets using a Nicolet model 760X spectrometer. Fluorescence spectra were measured from solid samples using an Otsuka Electronics, model MCPD-7000 spectrometer. Fluorescence lifetimes were measured using a Horiba model FluoroCube UltraFast-3000U setup.
2.3. Synthesis
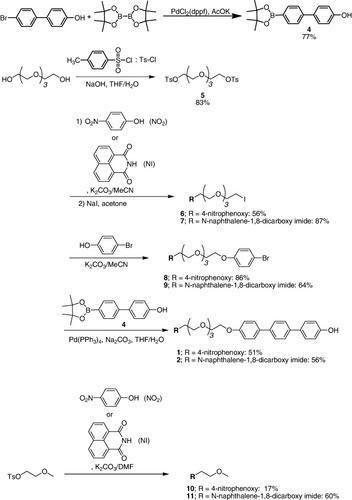
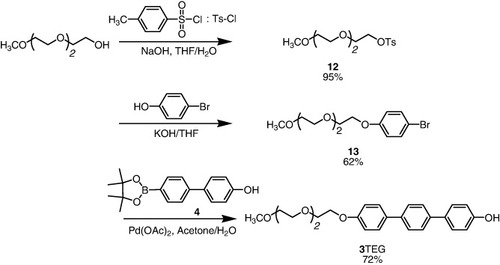
Synthetic routes of 1 and 2 are shown in scheme . Simple substituted derivatives are designated 3TEG (TEG = 2-{2-[2-(methoxyethoxy)ethoxy]ethoxy}) (scheme ).
Scheme 1. Synthesis of the receptors 1 and 2.
Scheme 2. Synthesis of the receptors 3TEG.
2.3.1. 4-Hydroxy-4’-(4,4’,5,5’-tetramethyl-1,3,2-dioxaborolan-2-yl) biphenyl (4).
4'-Bromo-[1,1'-biphenyl]-4-ol (0.498 g, 2 mmol), potassium acetate (KOAc, 0.588 g, 6 mmol), PdCl2(dppf) (dppf = 1,1'-bis(diphenylphosphino)ferrocene; 0.043 g, 0.06 mmol), and bis(2,2,3,3-tetramethyl-2,3-butanedionato)diboron (0.761 g, 3.0 mmol) were dissolved in anhydrous DMSO (12 ml). The mixture was heated at 80 °C for 4 h with stirring under N2. The mixture was cooled to room temperature and the solid filtered off and washed with dichloromethane. After evaporation of the solvents under reduced pressure, the residue was purified by silica gel column chromatography (AcOEt/hexane = 1/2, where AcOEt stands for ethyl acetate), to give 0.351 g of 4 as a white powder in 59% yield. 1H-NMR (300 MHz, CDCl3, 25 °C): δ = 1.36 (s, 12H), 5.16 (s, 1H), 6.91 (d, 2H, J = 9.0), 7.41–7.63 (m, 4H), 7.86 (d, 2H, J = 9.0) ppm. MALDI-TOF (dithranol): m/z = 296.1 [M]+.
2.3.2. Tetraethylene glycol bis(4-toluenesulfonate) [Citation38] (5).
Sodium hydroxide (1.06 g, 26 mmol) dissolved in water (10 ml) and tetraethylene glycol (1.71 g, 8.8 mmol) in tetrahydrofuran (THF, 10 ml) were placed in a flask and the mixture was cooled on an ice bath with magnetic stirring. To the mixture was added dropwise p-toluenesulfonyl chloride (3.50 g, 17 mmol) in THF (10 ml) over 1 h with continuous stirring and cooling of the mixture below 5 °C. The solution was stirred at 0–5 °C for an additional 4 h, and then poured into ice water (50 ml). The mixture was extracted twice with dichloromethane. The combined organic extracts were washed with saturated aqueous sodium chloride solution, and then dried over anhydrous Na2SO4. After evaporation of the solvent, the resulting crude product was purified by chromatography on silica gel with ethyl acetate, giving the product as a colorless oil in an 83% yield. 1H-NMR (300 MHz, CDCl3, 25 °C): δ = 2.44 (s, 6H), 3.55–3.58 (m, 8H), 3.67 (t, 4H, J = 4.6), 4.15 (t, 4H, J = 4.6), 7.33 (d, 4H, J = 8.1), 7.79 (d, 4H, J = 8.4) ppm. MALDI-TOF (dithranol): m/z = 525.15 [M + Na]+, 541.16 [M + K]+.
2.3.3. 11-Iodo-3,6,9-trioxaundecyl-(4-nitrophenyl)ether (6).
Potassium carbonate (0.691 g, 5.0 mmol) and potassium iodide (0.002 g, 0.1 mmol) were added to a CH3CN (20 ml) solution of a mixture of 4-nitrophenol (0.347 g, 2.5 mmol) and 5 (5.02 g, 10 mmol), and the resulting suspension was refluxed for 13 h under N2. The reaction mixture was allowed to cool to room temperature and the solvents evaporated under reduced pressure. The residue was dissolved in CH2Cl2, washed with water and dried over anhydrous Na2SO4. After evaporation of the solvent under reduced pressure, the residue was dissolved in acetone (50 ml), NaI (15.0 g, 100 mmol) was added, and the mixture was heated at reflux for 24 h. The reaction mixture was allowed to cool to room temperature and then the solvent was removed under reduced pressure. The residue was taken into CH2Cl2 and the solution was washed with water, dried over anhydrous Na2SO4, and then the solvent was removed under reduced pressure. The crude product was subjected to column chromatography (SiO2, AcOEt/hexane: 1/1 v/v) which yielded the product as a colorless oil (0.60 g) in a 56% yield. 1H-NMR (300 MHz, CDCl3, 25 °C): δ = 3.25 (t, 2H, J = 7.0), 3.77–3.66 (m, 10H), 3.91–3.88 (m, 2H), 4.23 (t, 2H, J = 3.0), 6.98 (d, 2H, J = 9.2), 8.19 (d, 2H, J = 9.2) ppm. 13C-NMR (75 MHz, CDCl3, 25 °C): δ = 2.92, 68.14, 69.34, 70.15, 70.57, 70.65, 70.87, 71.88, 114.52, 125.83, 141.50, 163.78 ppm. MALDI-TOF (dithranol): m/z = 448.16 [M + Na]+, 464.11 [M + K]+. HRMS (ESI) m/z calculated for C14H20INNaO6 [M + Na]+: 448.0225, found 448.0233.
2.3.4. N-(11-Iodo-3,6,9-trioxaundecyl)naphthalene-1,8-di-carboxylic acid imide (7).
1H-NMR (300 MHz, CDCl3, 25 °C): δ = 3.22–3.17 (m, 2H), 3.73–3.53 (m, 16H), 3.85–3.81 (m, 2H), 4.47–4.42 (m, 2H), 7.75 (t, 2H, J = 7.7), 8.22 (d, 2H, J = 8.3), 8.60 (d, 2H, J = 7.3) ppm. 13C-NMR (75 MHz, CDCl3, 25 °C): δ = 2.92, 39.04, 67.88, 70.06, 70.11, 70.48, 70.58, 71.83, 122.50, 126.85, 128.08, 131.18, 131.46, 133.90, 164.11 ppm. MALDI-TOF (dithranol): m/z = 484.36 [M + H]+, 506.37 [M + Na]+, 522.26 [M + K]+.
2.3.5. 11-(4-Bromophenoxy)-3,6,9-trioxaundecyl-(4-nitrophenyl)ether (8).
K2CO3 (0.349 g, 2.5 mmol) was added to a solution of a mixture of 4-bromophenol (0.128 g, 1.3 mmol) and 6 (0.305 g, 0.63 mmol) in CH3CN (10 ml), and the resulting suspension was refluxed for 15 h under N2. The reaction mixture was allowed to cool to room temperature and solvent removed under reduced pressure. The residue was dissolved in ethyl acetate and the solution was washed with water, dried over anhydrous Na2SO4, and then solvent was removed under reduced pressure. The crude product was subjected to column chromatography (SiO2, AcOEt/hexane 1/1 v/v) yielding the product as a white solid (0.213 g, 64%). 1H-NMR (300 MHz, CDCl3, 25 °C): δ = 3.74–3.65 (m, 8H), 3.89–3.82 (m, 4H), 4.13–4.07 (m, 2H), 4.20 (t, 2H, J = 1.9), 6.78 (d, 2H, J = 9.0), 6.96 (d, 2H, J = 9.3), 7.33 (d, 2H, J = 8.7), 8.18 (t, 2H, J = 4.6) ppm. 13C-NMR (75 MHz, CDCl3, 25 °C): δ = 67.62, 68.14, 69.35, 69.62, 70.62, 70.64, 70.81, 70.88, 113.00, 114.53, 116.37, 125.84, 132.19, 141.53, 157.83, 163.79 ppm. MALDI-TOF (dithranol): m/z = 492.02 [M + Na]+. HRMS (ESI) m/z calculated for C20H24BrNNaO7 [M + Na]+: 492.0624, found 492.0634.
2.3.6. N-[11-(4-Bromophenoxy)-3,6,9-trioxaundecyl]naphthalene-1,8-dicarboxylic acid imide (9).
1H-NMR (300 MHz, CDCl3, 25 °C): δ = 3.63–3.61 (m, 6H), 3.71–3.86 (m, 2H), 3.84–3.76 (m, 4H), 4.15–4.02 (m, 2H,), 4.44 (t, 2H, J = 6.0), 6.76 (d, 2H, J = 4.6), 7.32 (d, 2H, J = 7.5), 7.74 (t, 2H, J = 8.0), 8.20 (d, 2H, J = 7.3), 8.59 (d, 2H, J = 6.0) ppm. 13C-NMR (75 MHz, CDCl3, 25 °C): δ = 39.07, 67.56, 67.89, 69.52, 70.12, 70.59, 70.74, 112.89, 116.36, 122.56, 126.88, 128.14, 131.22, 131.52, 132.12, 133.94, 157.83, 164.18 ppm. MALDI-TOF (dithranol): m/z = 528.12 [M + H]+. HRMS (ESI) m/z calculated for C26H26BrNNaO6 [M + Na]+: 550.0833, found 550.0841.
2.3.7. 11-(4’’-Hydroxy-1’’-phenyl-1’,4’-phenyl-1,4-phenoxy)-3,6,9-trioxaundecyl-(4-nitrophenyl)ether (1).
4 (0250 g, 0.53 mmol) and 8 (0.250 g, 0.50 mmol) were dissolved in THF (16 ml). 2.0 M aqueous Na2CO3 (16 ml) was added and the solution was purged with N2 gas. Tetrakis(triphenylphosphine) palladium(0) (3 mg, 0.0031 mmol) was added and the mixture was heated at 80 °C for 24 h with vigorous stirring under N2. After cooling to room temperature, solvents were removed under reduced pressure and the residue dissolved in ethyl acetate, followed by washing with water and drying of the organic phase over anhydrous Na2SO4. The solution was filtered and solvent removed under reduced pressure. The crude product was purified by column chromatography (silica gel) using THF/hexane = 9/1 as the eluent to yield 0.152 g (51%) as an orange solid. 1H-NMR (300 MHz, d6-DMSO, 25 °C): δ = 3.58–3.56 (m, 8H), 3.78–3.76 (m, 4H), 4.12–4.10 (m, 2H), 4.24–4.21 (m, 2H), 6.85 (d, 2H, J = 8.6), 7.01 (d, 2H, J = 8.8), 7.14 (d, 2H, J = 9.3), 7.52 (d, 2H, J = 8.6), 7.62–7.59 (m, 6H), 8.17 (d, 2H, J = 9.1), 9.55 (s, 1H) ppm. 13C-NMR (75 MHz, d6-DMSO, 25 °C): δ = 67.38, 68.43, 68.80, 69.14, 70.03, 70.13, 115.09, 115.23, 115.95, 126.07, 126.53, 126.67, 127.69, 127.75, 130.59, 132.30, 137.85, 138.67, 140.98, 157.33, 158.24, 164.04 ppm. MALDI-TOF (dithranol): m/z = 559.02 [M]+, 582.03 [M + Na]+. Calculated for C32H33NO8: %C, 68.68; %H, 5.94; %N, 2.50. Found: %C, 68.31; %H, 6.32; %N, 2.48.
2.3.8. N-[11-(4’’-Hydroxy-1’’-phenyl-1’,4’-phenyl-1,4-phenoxy)-3,6,9-trioxaundecyl]naphthalene-1,8-dicarboxylic acid imide (2).
1H-NMR (300 MHz, d6-DMSO, 25 °C): δ = 3.56–3.47 (m, H), 3.68–3.64 (m, H), 4.09–4.06 (m, 4H), 4.25 (t, 2H, J = 6.5), 6.84 (d, 2H, J = 8.6), 6.99 (d, 2H, J = 8.8), 7.51 (d, 2H, J = 8.6), 7.61–7.57 (m, 6H), 7.86 (t, 2H, J = 4.0), 8.46 (d, 2H, J = 7.5), 8.5087 (d, 2H, J = 7.3), 9.55 (s, 1H) ppm. 13C-NMR (75 MHz, d6-DMSO, 25 °C): δ = 67.11, 67.32, 69.08, 69.85, 69.89, 70.00, 70.05, 115.08, 115.94, 122.21, 126.51, 126.66, 127.46, 127.60, 127.66, 127.74, 130.59, 131.01, 131.08, 131.52, 132.26, 134.60, 137.85, 138.65, 157.31, 158.21, 163.64 ppm. MALDI-TOF (dithranol): m/z = 617.57 [M]+, 640.58 [M + Na]+, 656.54 [M + K]+. HRMS (ESI) m/z calculated for C38H35NNaO7 [M + Na]+: 640.2311, found 640.2292.
2.3.9. 2-Methoxyethyl-(4-nitrophenyl)ether (10).
K2CO3 (0.552 g, 4.0 mmol) was added to a solution of a mixture of 4-nitrophenol (0.278 g, 2.0 mmol) and 2-methoxyethoxy-4-toluenesulfonate (0.921 g, 4.0 mmol) in dimethylformamide (DMF, 10 ml), and the resulting suspension was refluxed for 12 h. The reaction mixture was allowed to cool to room temperature and evaporated to dryness. CH2Cl2 solution of the residue was washed with water, dried over anhydrous Na2SO4, and evaporated to dryness. The residue and NaI (1.20 g, 8.0 mmol) were suspended in acetone (5 ml), and heated at reflux for 3 h. The reaction mixture was allowed to cool to room temperature and solvents removed under reduced pressure. The residue was dissolved in CH2Cl2, washed with water, dried over anhydrous Na2SO4, and then solvent was removed under reduced pressure. The product was obtained as colorless yellow needles (0.065 g) in a 17% yield. 1H-NMR (300 MHz, CDCl3, 25 °C): δ = 3.46 (s, 3H), 3.79 (t, 2H, J = 3.0), 4.22 (t, 2H, J = 6.0), 7.00 (d, 2H, J = 9.0), 8.20 (d, 2H, J = 6.0) ppm. 13C-NMR (75 MHz, CDCl3, 25 °C): δ = 59.28, 68.04, 70.58, 114.53, 125.85, 141.62, 163.78 ppm.
2.3.10. N-(2-Methoxyethyl)naphthalene-1,8-dicarboxylic acid imide (11).
1H-NMR (300 MHz, CDCl3, 25 °C): δ = 3.39 (s, 3H), 3.74 (t, 2H, J = 6.0), 4.46 (t, 2H, J = 6.0), 7.76 (t, 2H, J = 9.0), 8.22 (d, 2H, J = 9.0), 8.62 (d, 2H, J = 6.0) ppm. 13C-NMR (75 MHz, CDCl3, δ, ppm): 38.81, 58.38, 69.21, 122.15, 126.47, 127.89, 130.92, 131.14, 133.54, 163.88 ppm. MALDI-TOF (dithranol): m/z = 255.09 [M]+. HRMS (ESI) m/z calculated for C15H13NNaO3 [M + Na]+: 278.0793, found 278.0790.
2.3.11. 2-[2-(2-Methoxyethoxy)ethoxy]ethyl-4-methylbenzenesulfonate (12) [Citation37].
Sodium hydroxide (0.9 g, 20 mmol) dissolved in water (20 ml) and triethylene glycol monomethyl ether (2.46 g, 15 mmol) in THF (20 ml) were placed in a flask and the mixture was cooled on an ice bath with magnetic stirring. To the mixture was added dropwise p-toluenesulfonyl chloride (2.88 g, 14 mmol) in THF (20 ml) over 1 h with continuous stirring and cooling of the mixture below 5 °C. The solution was stirred at 0–5 °C for an additional 4 h, and then poured into ice water (70 ml). The mixture was extracted twice with dichloromethane. The combined organic extracts were washed with saturated aqueous sodium chloride solution, and then dried over anhydrous magnesium sulfate. After evaporation of the solvent, the resulting crude product was purified by chromatography on silica gel with ethyl acetate to give 12 as a colorless oil. 1H-NMR (300 MHz, CDCl3, 25 °C): δ = 2.44 (s, 3H), 3.36 (s, 3H), 3.36–3.54 (m, 2H), 3.58–3.62 (m, 6H), 3.66–3.70 (m, 2H), 4.10–4.17 (m, 2H), 7.34 (d, 2H, J = 8.6), 7.79 (d, 2H J = 8.3) ppm. 13C-NMR (75 MHz, CDCl3, 25 °C): δ = 21.60, 58.99, 68.61, 69.19, 70.48, 70.51, 71.84, 127.92, 129.77, 132.91, 144.74 ppm. MALDI-TOF (dithranol): m/z = 341.0 [M + Na]+, 357.0 [M + K]+.
2.3.12. 1-(2-(2-(2-Methoxyethoxy)-ethoxy)ethoxy)-4-bromobenzene (13).
To a THF (15 ml) solution of mixture of 4-bromophenol (0.692 g, 4 mmol) and 12 (1.12 g, 3.5 mmol) was added KOH (1.0 g, 16 mmol), and the resulting suspension was refluxed for 15 h under N2. The reaction mixture was allowed to cool to room temperature and evaporated to dryness. The AcOEt solution of the residue was washed with water, dried over anhydrous Na2SO4, and then the solvent was removed under reduced pressure. The residue was subjected to column chromatography (SiO2, AcOEt/hexane 1/1 v/v) to allow isolation of 13 as a colorless oil (0.7 g) in a 62% yield. 1H-NMR (300 MHz, CDCl3, 25 °C): δ = 3.37 (s, 3H), 3.54 (m, 2H), 3.72 (m, 6H), 3.84 (m, 2H), 4.09 (m, 2H), 6.80 (d, 2H, J = 9.0), 7.35 (d, 2H, J = 9.2) ppm. MALDI-TOF (dithranol): m/z = 356.94 [M + K]+.
2.3.13. 4-{2-[2-(2-Methoxy-ethoxy)-ethoxy]-ethoxy}-[1,1';4',1'']terphenyl-4''-ol (3TEG) [Citation38].
A mixture of Na2CO3 (0.158 g, 1.5 mmol), Pd(OAc)2 (0.003 g, 0.5 mol.%), 4 (0.236 g, 0.8 mmol), 13 (0.226 g, 0.7 mmol), distilled water (7 ml) and acetone (6 ml) was stirred for 3 h at room temperature. Afterward, the reaction mixture was evaporated to dryness, and the solution was quenched with dilute aqueous hydrochloric acid. The precipitate was washed with water. The residue was subjected to column chromatography (SiO2, AcOEt/hexane 1/1 v/v) to allow isolation of 3TEG as a white powder (0.414 g) in a 72% yield.1H-NMR (300 MHz, d6-DMSO, 25 °C): δ = 3.30 (s, 3H), 3.43 (m, 2H), 3.51 (m, 4H), 3.58 (m, 2H), 3.74 (m, 2H), 4.13 (m, 2H), 6.87 (d, 2H, J = 8.6), 7.03 (d, 2H, J = 8.8), 7.52 (d, 2H, J = 8.4), 7.62 (d, 6H, J = 8.1), 9.52 (s, 1H) ppm. 13C-NMR (75 MHz, d6-DMSO, 25 °C): δ = 58.27, 67.41, 69.18, 69.83, 70.03, 70.17, 71.49, 115.14, 115.98, 126.56, 126.71, 127.73, 127.77, 130.61, 132.33, 137.90, 138.69, 157.35, 158.28 ppm. MALDI-TOF (dithranol): m/z = 408.0 [M]+, 431.0 [M + Na]+. HRMS (ESI) m/z calculated for C25H28NaO5 [M + Na]+: 431.1834, found 431.1831.
3. Results and discussion
Figure illustrates the selective sensing activity of 1 including its solid-state fluorescence response to the presence of Cs+ (figure (a)). Figure (b) shows the difference in fluorescence emission color from 1 in the presence of Li+, Na+, K+ or Cs+ (as their carbonate salts), following exposure to methanol by spraying a small amount of the solvent on the powdered solidFootnote6. The absence of fluorescence in the presence of Mg2+ and Ca2+ is also shown (figure (b)). Green fluorescence due to the presence of Cs+ on dry soil is shown after spraying of a solution of 1 in methanol so that the contaminated site can be easily identified and removed from the surrounding material (figure (c)). In addition, Cs+ particles can be visualized after spraying with 1 in methanol (see figure (d) and the supplementary information available at stacks.iop.org/STAM/14/015002/mmedia).
The limit of detection for Cs+ should depend on several parameters including counteranion, local pH and intercalation of Cs+ into clay contained in ground soils. We estimate a detection limit of 1 ppm under ideal conditions (see figure ). This limit is significant since caesium is present in the Earth's crust at 1 ppm: detection of caesium concentrations above this level would indicate possible local contamination. From figure it is evident that the wavelength of the fluorescence emission maximum is dependent on the concentration of Cs+ present making construction of a calibration curve possible. The form of the calibration curve suggests that concentrations of Cs+ far lower than 1 ppm could be detected, although this is not necessarily useful from a practical perspective. The importance of the connectivity of 4-NPE in 1 was investigated by comparing its response to that of a physical mixture of a triethylene glycol-substituted terphenol (3TEG), a simple ether of 4-nitrophenol (10) and the respective alkali metal carbonates in the presence of methanol (see figure ). Notably, the green fluorescence characteristic of the presence of Cs+ in 1 was not observed, and all metal carbonates (Na+, K+ or Cs+) gave the standard blue fluorescence in the solid state moistened with methanol. This further demonstrates that Cs+ binding to 1 differs from that of the other alkali metal cations leading to the specificity in its fluorometric response.
Figure 2. Detection limit of Cs2CO3 by using 1. (a) Photographs and plot of fluorescence emission maxima wavelengths for mixtures between 1 and Cs2CO3/K2CO3 under UV irradiation (365 nm) after addition of a drop of methanol. (b) Fluorescence spectra of mixtures used to construct the plot in (a). (c) Photographs and emission maxima for mixtures between 1 and Cs2CO3/K2CO3 under UV irradiation (365 nm) after addition of a drop of methanol.
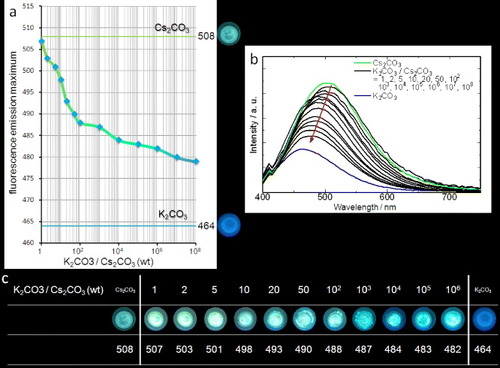
Figure 3. Interaction of 1 (a) or its component chromophore moieties (3TEG) (b) with alkali metal cations. (c) Fluorescence spectra of the mixtures revealing the different spectrum in the presence of Cs+. (d) In the simple mixture of component chromophores the shift in fluorescence maximum is rather small and is not sufficient for sensing activity. Numbers above the spectra in panels (c) and (d) indicate the wavelength of the fluorescence maximum.
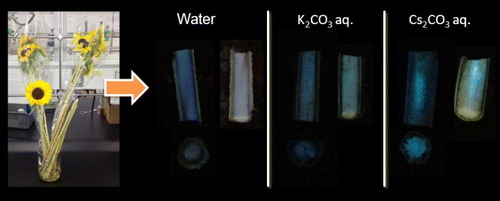
Recent research suggests that sulfate aerosols were the transport medium of radiocaesium from the Fukushima nuclear accident [Citation8]. Accordingly, a mixture of 1 and caesium sulfate exhibited green fluorescence under basic conditions after spraying with methanol (figure ). Diffusion behavior and the bioaccumulation processes of caesium can be easily and safely studied by using 1 and non-radioactive Cs-133. For instance, cut sunflowers were immersed in 1 wt% aqueous solution of caesium carbonate for several days. The sunflowers were subsequently freeze-dried at −40 °C. A sunflower stem cross section exhibited green fluorescence after spraying with 1 in methanol (figure ; see also the supplementary information available at stacks.iop.org/STAM/14/015002/mmedia).
Figure 4. Photographs (a)–(c) and spectra of fluorescence change (d)–(f) of a mixture of 1 and alkali metal salt under UV irradiation (365 nm) after addition of a drop of methanol. Fluorescence change of a powder mixture of 1 and Cs+ under basic conditions after spraying with methanol. Numbers in panels (a)–(c) indicate the wavelength of the fluorescence maximum.
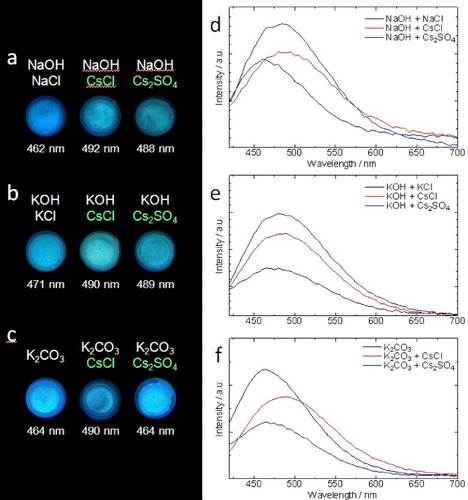
Figure 5. Photographs of sunflowers and cross sections of their freeze-dried stems after immersion in water or an aqueous solution of potassium or caesium carbonate. The photographs were taken with the stems under UV irradiation (365 nm) after spraying only with methanol and with 1 in methanol.
In ascertaining whether the fluorimetric response or selectivity of 1 could be varied, we prepared compound 2 where the 4-NPE group of 1 had been replaced by a fluorescent naphthalene-1,8-dicarboxylic imide unit. Compound 2 unexpectedly behaves as a selective sensor for Rb+ yielding a similar green fluorescence to that obtained for 1 and Cs+ under basic conditions (figure ). Fluorescence lifetimes for 1 and 1 with K+ or Cs+ in methanol are summarized in table and figure S2 (see the supplementary information at stacks.iop.org/STAM/14/015002/mmedia). Lifetimes of intermolecular fluorescence (i.e. that involving two chromophores linked in the same molecule), such as that occurring due to FRET or exciplex mechanisms, are an order of magnitude longer than those of intramolecular fluorescence (i.e. that involving an isolated chromophore). For instance, the fluorescence lifetime of the naphthalimide monomer is about 1 ns. If a naphthalimide dimer forms an excimer then the fluorescence lifetime is longer (about 20 ns). The difference in fluorescence lifetime between [1 + K2CO3] and [1 + Cs2CO3] is small. Fluorescence lifetimes for 1 in the presence of alkali metal cations are an order of magnitude shorter than those expected for FRET and exciplex fluorescence emission mechanisms and suggest an intra-moiety mechanism involving the deprotonated terphenol group.
Figure 6. Photographs and spectra illustrating the differences in fluorescence emission from (a) 1 and (b) 2 in the presence of carbonate salts of alkali metal cations after spraying with methanol.
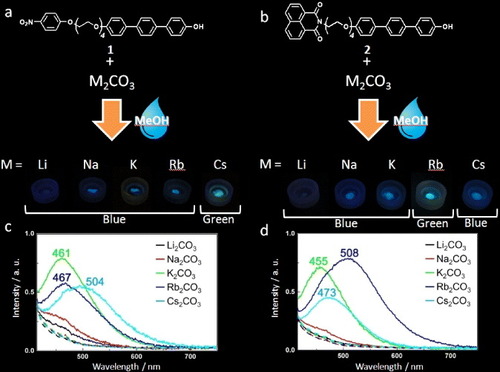
Table 1. Fluorescence lifetime of the mixtures of phenols and alkali metal carbonate after addition of methanol.
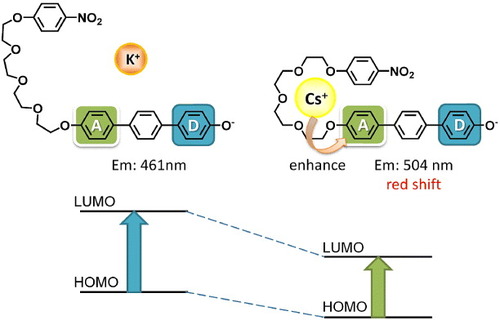
To explain this disparity in the responses of the two sensor molecules, we considered the respective geometries of 1 and 2 during interaction with the analyte cations by applying density functional theory (DFT) [Citation27]Footnote7 geometry optimization to the resulting complexes. The results (figure ) show that the forms of the complexes of the cations with each ligand are very similar, with the electropositive metal cation being coordinated by oxygen atoms of the tetraethylene glycol chain, similar to the situation in the crown ether complexes. It is important to note that a weak hydrogen bond between the 4-nitro group of the NPE moiety and one methine of the terphenol is predicted and in fact could be detected in infrared spectra of 1 in the presence of alkali metal cations (see figure S3 in the supplementary information available at stacks.iop.org/STAM/14/015002/mmedia). This hydrogen bond apparently stabilizes a quasi-macrocyclic conformation of 1 while no similar interaction occurs in 2. This also explains why for the previously mentioned physical mixture involving disconnected components of 1 there is no differentiation between cations, as macrocyclization of the host's component moieties is not possible. On the other hand, in 2 there are apparent CH…π interactions between the naphthalenedicarboxylic imide unit and the π-electronic system of its terphenyl. Now, considering the departure from ideal geometry in each case, measured by summing the angular departures from ideal tetrahedral geometry of the methylene groups of the tetraethylene glycol chain, we find that for 2, Rb+ is most easily accommodated, while for 1 Cs+ is favored. Perhaps serendipitously, the optimal binding geometry for 1.Cs+ and 2.Rb+ also leads to the respective cations being brought in closer proximity to the alkoxyphenyl end of the HTP group. To explain the selectivity of the response to Cs+ for 1, we consider that the deprotonated terphenol unit is formally a donor–acceptor (D–A) type moiety (figure ). Deprotonation of the HTP unit leads to an electronic donor state for the phenol part of the molecule while the opposing alkyloxyphenyl unit is relatively electron deficient. The fluorescence of the mixtures between 1 and alkali metal carbonates are bathochromically shifted by intramolecular charge transfer following addition of methanol by the mechanism proposed by Valeur [Citation28, Citation29]. Fluorescence lifetime is slightly longer for 1 + Cs2CO3 than 1 + K2CO3 suggesting that excitons of 1 + Cs2CO3 are more stable. When Cs+ interacts with the acceptor group of deprotonated 1 the excited state is stabilized more by Cs+ than the ground state, and this leads to a bathochromic shift of the fluorescence maximum (figure ). For 2, Rb+ also approaches more closely than other alkali metal cations the electron-deficient part of the deprotonated phenol.
Figure 7. Selected angles of the computed structures of the complexes of 1 with K+, Rb+ or Cs+ and 2 with K+, Rb+ or Cs+. Dihedral angles subtended at the indicated bonds are listed in the table on the right.
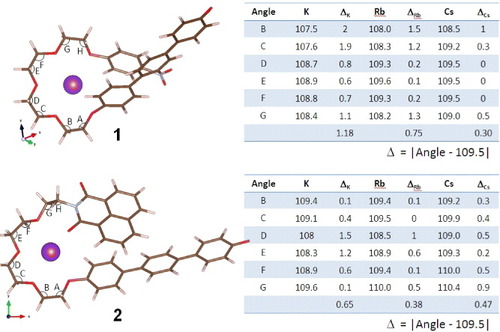
Figure 8. Origin of the donor–acceptor (D–A) character of terphenol. As illustrated in the lower part, in the phenol both the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are situated over the extent of the molecule. Upon deprotonation, HOMO is situated at the electron-rich phenoxide group while the LUMO is situated at the electron-deficient phenyl group remote from the phenol. Experimental and simulated electronic absorption spectra (top-right panel) reveal the accuracy of the model.
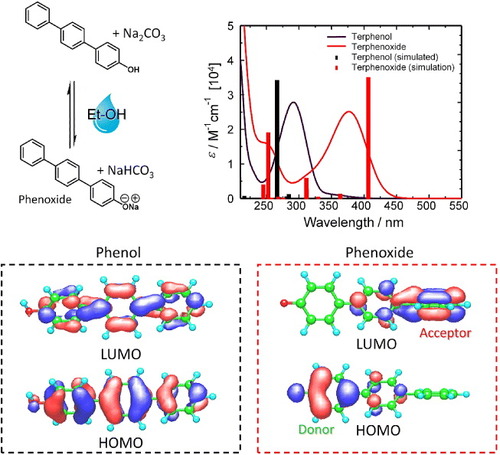
Figure 9. Schematic of the computed structures of the complexes of 1 with K+ or Cs+. Em stands for emission wavelength.
Although our initial molecular design was based on an expectation that FRET or exciplex phenomena might be observed in 1 due to intermolecular interaction, introduction of the 4-nitrophenyl ether group introduced a serendipitous point for intramolecular macrocyclization through hydrogen bonding (between the 4-nitro group and one HTP methane [Citation30]) with no optical effects due to this group, except for changes in the fluorescence upon binding different cations due to intramolecular effects. Moreover, we believe that it would anyway be extremely difficult to detect any difference between complexes of 1 with the alkali metal cations if FRET or exciplex mechanisms operated in this case, due to the substantial similarities in structure between the complexes, especially with regard to the relative positioning of the donor and acceptor chromophores. In fact, FRET or exciplex emission would likely obstruct cation differentiation in this case due to the subtlety of the effect of the proximity of the Cs+ to the HTP group.
4. Conclusions
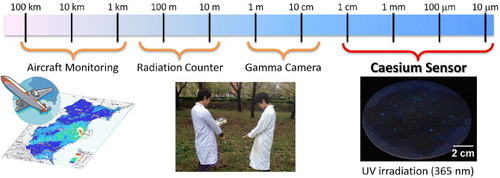
This simple optical probe 1 has a higher spatial resolution for Cs-137 than existing radioscopes and gamma ray cameras (figure ). Since only contaminated particles are detected by eye and can be easily removed, the volume of radioactively contaminated material for extraction is greatly reduced. The location of caesium in solid samples such as soil, foodstuffs and plants can be visualized by using this probe, allowing one to study its diffusion processes and accumulation behavior in those media. The availability of this and other similarly easily implemented tests for environmental contaminants is likely to increase the volume of data regarding rates of contamination around chemical and radiological hazards. This information will then be useful for constructing maps of contamination, which can be used to determine accommodation and agricultural policies involving local populations. Combination of the molecular optical probe described here with appropriate porous adsorptive materials [Citation31–Citation36] should also permit creation of materials for convenient detection–removal systems.
Figure 10. Schematic of spatial resolution of radioscopes and caesium sensor 1.
Acknowledgments
This research was supported by the World Premier International Research Center Initiative (WPI Initiative) on Materials Nanoarchitectonics, from MEXT Japan and the CREST program of the Japan Science and Technology Agency (JST), Japan.
Notes
4 For comparison, after the Three Mile Island reactor meltdown (Pennsylvania, USA, 1979) only noble gases and radioiodine were reportedly released into the surrounding environment [Citation7].
5 Caesium is a reactive metal that is never present in the environment in its elemental form.
6 Ethanol may also be used to circumvent use of toxic methanol.
7 All density functional theory (DFT) calculations were carried out using the B3LYP exchange correlation functional implemented in Gaussian 0921 with LanL2DZ basis set.
References
- EssenLParryJ V L 1995 Nature 176 280 10.1038/176280a0
- GlisinVCrkvenjakovRByusC 1974 Biochemistry 13 2633 10.1021/bi00709a025
- ButtermanW CBrooksW EReeseR GJr 2004 Mineral commodity profile: caesium Open-File Report 2004–1432 Virginia US Geological Survey p 5
- JohnsonG TLewisT RWagnerW D 1975 Toxicol. Appl. Pharmacol. 32 239 10.1016/0041-008X(75)90216-1
- International Atomic Energy Agency 2006 Environmental consequences of the Chernobyl accident and their remediation: twenty years of experience Report of the UN Chernobyl Forum Expert Group ‘Environment’ (Vienna: IAEA) STI/PUB/1239
- BrumfielG 2011 Nature 478 435 10.1038/478435a
- ManganoJ 2004 Bull. Atom. Sci. 60 30 10.2968/060005010
- KaneyasuNOhashiHSuzukiFOkudaTIkemoriF 2012 Environ. Sci. Technol. 46 5720 10.1021/es204667h
- LanghamW HAndersenE C 1959 Health Phys. 2 30 10.1097/00004032-195901000-00003
- RowanD JRasmussenJ B 1994 Can. J. Fish. Aquat. Sci. 51 2388 10.1139/f94-240
- RahmanM MVoigtG 2004 J. Environ. Radioact. 71 127 10.1016/S0265-931X(03)00163-2
- TurekianK KWedepohlK H 1961 Geol. Soc. Am. Bull. 72 175 10.1130/0016-7606(1961)72[175:DOTEIS]2.0.CO;2
- Visualization of Radioactive Substances with JAXA's ‘Ultra-Wide-Angle Compton Camera’ www.jaxa.jp/press/2012/03/20120329_compton_e.html 29 March 2012
- Toshiba Develops Camera to Detect Radiation Hotspots http://techon.nikkeibp.co.jp/english/NEWS_EN/20111214/202583/ 14 December 2011
- MatsunamiMTakakiAMaekawaHNishiguchiI 2005 Sci. Technol. Adv. Mater. 6 172 10.1016/j.stam.2004.11.012
- BureekaewSShimomuraSKitagawaS 2008 Sci. Technol. Adv. Mater. 9 014108 10.1088/1468-6996/9/1/014108
- ArigaKHillJ PLeeM VVinuACharvetRAcharyaS 2008 Sci. Technol. Adv. Mater. 9 014109 10.1088/1468-6996/9/1/014109
- LiMIshiharaSJiQAkadaMHillJ PArigaK 2012 Sci. Technol. Adv. Mater. 13 053001 10.1088/1468-6996/13/5/053001
- OhshimaAMomotakeAAraiT 2005 Sci. Technol. Adv. Mater. 6 633 10.1016/j.stam.2005.05.002
- KuroiwaKOdaNKimizukaN 2006 Sci. Technol. Adv. Mater. 7 629 10.1016/j.stam.2006.09.011
- XingZ CChangYKangI K 2010 Sci. Technol. Adv. Mater. 11 014101 10.1088/1468-6996/11/1/014101
- BrownW HFooteC SIversonB LAnslynE VNovakB M 2011 Organic Chemistry 6th edn Belmont, CA Brooks/Cole chapter 21, p 853
- YamaguchiIGotoKSatoM 2009 Tetrahedron 65 3645 10.1016/j.tet.2009.02.073
- YamaguchiIGotoKSatoM 2009 Macromolecules 42 7836 10.1021/ma9008884
- FörsterT 1965Delocalized excitation and excitation transfer Modern Quantum Chemistry. Istanbul Lectures. Part III: Action of Light and Organic Crystals SinanogluO New York Academic p 93
- BirksJ B 1975 Rep. Prog. Phys. 38 903 10.1088/0034-4885/38/8/001
- FrischM J et al 2009 Gaussian 09 revision A.01 Wallingford, CT Gaussian
- ValeurBLerayI 2000 Coord. Chem. Rev. 205 3 10.1016/S0010-8545(00)00246-0
- BoursonJValeurB 1989 J. Phys. Chem. 93 3871 10.1021/j100346a099
- SamshuddinSButcherR JAkkurtMNarayanaBSarojinidB KYathirajaneH S 2012 Acta Crystallogr. E68 o74
- AbeHSatohANishidaKAbeENakaTImaiMKitazawaH 2006 J. Solid State Chem. 179 1521 10.1016/j.jssc.2006.02.005
- ArigaKVinuAYamauchiYJiQHillJ P 2012 Bull. Chem. Soc. Japan 85 1 10.1246/bcsj.20110162
- ArigaKIshiharaSAbeHLiMHillJ P 2012 J. Mater. Chem. 22 2369 10.1039/c1jm14101e
- HuMSukegawaHNemotoY.ReboulJKitagawaSYamauchiY 2012 Angew. Chem., Int. Ed. Engl. 51 984 10.1002/anie.201105190
- Database of Promising Adsorbents for Decontamination of Radioactive Substances READS http://reads.nims.go.jp/index_en.html 13 December 2011
- IshiyamaTMurataMMiyauraN 1995 J. Org. Chem. 60 7508 10.1021/jo00128a024
- OuchiMInoueYLiuYNagamuneSNakamuraSWadaKHakushiT 1990 Bull. Chem. Soc. Japan 63 1260 10.1246/bcsj.63.1260
- LiuLZhangYXinB 2006 J. Org. Chem. 71 3994 10.1021/jo060122v