
Abstract
Precious metal nanoparticles are commonly used as the main active components of various catalysts. Given their high cost, limited quantity, and easy loss of catalytic activity under severe conditions, precious metals should be used in catalysts at low volumes and be protected from damaging environments. Accordingly, reducing the amount of precious metals without compromising their catalytic performance is difficult, particularly under challenging conditions. As multifunctional materials, core–shell nanoparticles are highly important owing to their wide range of applications in chemistry, physics, biology, and environmental areas. Compared with their single-component counterparts and other composites, core–shell nanoparticles offer a new active interface and a potential synergistic effect between the core and shell, making these materials highly attractive in catalytic application. On one hand, when a precious metal is used as the shell material, the catalytic activity can be greatly improved because of the increased surface area and the closed interfacial interaction between the core and the shell. On the other hand, when a precious metal is applied as the core material, the catalytic stability can be remarkably improved because of the protection conferred by the shell material. Therefore, a reasonable design of the core–shell catalyst for target applications must be developed. We summarize the latest advances in the fabrications, properties, and applications of core–shell nanoparticles in this paper. The current research trends of these core–shell catalysts are also highlighted.
1. Introduction
A catalyst is a substance that can change the reaction rate by decreasing the activation energy of a reaction and of itself, but never take part in such reaction. These substances create a convenient surface for the reaction to occur. Precious metals are frequently used as active components of various catalysts. Increasing the catalytic activity and stability of precious metals has always been an important research topic because of the limited quantity of these materials. Previous studies show that the designing of nanoparticles with a core–shell architecture can improve the physical and chemical properties of the material by combining multiple functionalities on a nanoscopic length scale and by providing new active interfaces as well as synergistic effects among different components [Citation1–Citation5]. The development of core–shell particles has been reviewed previously [Citation6–Citation13] and the related materials have also been used broadly in the catalytic area [Citation14–Citation22].
Extensive efforts have been made over the recent years in the designing and the controlled preparation of core–shell structure catalysts with precious metals. These metals can be used both as the core and the shell material according to different purposes.
To preserve these materials, precious metals are mainly used as shell materials of the core–shell structure. The surface area of the precious metal can greatly increase in this structure, which subsequently improves the catalytic activity. A typical example that demonstrates such phenomena is the use of a thin Pt shell in engineered core–shell nanoparticle catalysts, which is among the best solutions to reduce the cost of the catalyst in next-generation fuel cell systems [Citation23].
When precious metals are used as core materials, the stability of the catalyst could be improved because of the protection from the shell material. Silica is considered the most popular shell material for many decades because of its simple and robust synthesis from tetraethoxysilane (TEOS) [Citation24]. However, the chemical inertness of silica has made this material an unsuitable catalyst component in some reaction systems. Metal oxides have also been used as shell materials to improve the activity of the core–shell catalyst, but the fabrication of this catalyst remains a challenging task [Citation25].
The core–shell structure can improve the catalytic activity or the stability of the precious metal catalysts as well as provide new functions to the catalyst. Based on their structure, precious metal core–shell catalysts can be mainly classified into core–precious metal shell and precious metal core–shell. In the core–precious metal shell, the precious metal shell plays an active part and the core material takes multiple functions. The core material can offer significant improvements in the activity and selectivity of the catalysts by increasing the surface area of the active part and enhancing the synergistic effects. Using magnetic metal as a core material also facilitates the separation of the catalysts from the reaction mixtures by using a magnet. The core–shell catalysts with a thin precious metal shell or monolayer-based precious metal nanoparticles are of great interest in most cases [Citation26–Citation33]. However, the precise fabrication of this kind of core–shell catalyst remains a great challenge. In the precious metal core–shell, the precious metal core plays an active part and the shell materials play a protective role. The catalytic stability of the precious metal in this catalyst can be improved by confining the transfer of the active particles and by protecting these particles from a damaging environment. Unfortunately, such activity is often degraded because of the coverage of the shell layer. Therefore, maintaining the high activity of this core–shell catalyst remains a problem.
Many approaches for the preparation of core–shell catalysts have been developed recently. These approaches are generally categorized into ‘top-down’ and ‘bottom-up’ approaches. The former includes laser-beam processing and mechanical techniques, whereas the latter includes layer-by-layer coating, chemical reduction, electrochemical deposition, and sol-gel processes. The sol-gel method is regarded as the most versatile of these approaches because such method does not require any complicated processes or instruments to synthesize the core–shell catalyst with complex structures. However, this approach still has its own limitations and can only be used for synthesizing certain materials.
We mainly focus on the two abovementioned dominant precious metal core–shell catalysts in this paper. The fabrication methods and the properties of these catalysts are summarized and described in detail. Afterwards, the design and fabrication of the novel core–shell catalysts according to the special requirement are briefly highlighted.
2. Core–shell catalysts with precious metal shell
Core–shell catalysts with precious metal shell and various cores have been fabricated for special purposes. As a typical noble metal, platinum (Pt) becomes very important in fundamental studies and industrial applications. Pt was used as the representative metal in this study to interpret the recent advances in the properties, applications, and synthesis of core–shell catalysts with precious metal shell.
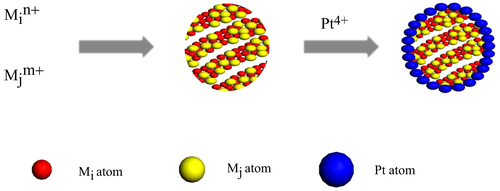
Pt is often used in the form of nanoparticles to create a high surface area for catalysis application. Therefore, Pt is identified as the best catalyst for both the anode and cathode of the proton exchange membrane fuel cell (PEMFC). However, the high cost and instability of Pt prevent PEMFC from having widespread applications. Moreover, the sluggish kinetics at the cathode has a profound effect on the degradation of fuel cell efficiency. Therefore, the improvement of the electrocatalytic activity and durability of a Pt-based catalyst with a low Pt content has been thoroughly investigated. Several strategies have been formulated to address this problem, such as the production of Pt nanostructures with high surface areas and a high catalytic performance as well as the combination of Pt with other materials to improve its performance by modifying the electronic and geometric structures of the surface Pt atoms. In this regard, the synthesis of the core–shell and the core–shell-like catalysts with Pt shell has also attracted significant research interest. Figure illustrates the synthetic procedure of the MiMj–Pt core–shell structure, where Mi and Mj represent metallic elements. The illustration shows that the specific surface area of Pt can be greatly improved by combining Pt with other materials. Zhang et al [Citation34] prepared the catalyst of Pd–Sn core and Pt shell, which demonstrated a high oxygen reduction reaction (ORR) performance. In principle, many materials can be used as the core. These materials can be mainly classified into three types, namely, precious metal, non-noble metal, and mixture or alloy.
Figure 1. Schematic of the synthetic procedure of the MiMj–Pt core–shell catalyst.
2.1. Core–shell catalyst with platinum shell and precious metal core
Metal–metal, such as precious metal–platinum core–shell nanoparticles, are considered important catalysts because a metal nanoparticles shell, especially mesoporous shell, provides many catalytically active sites and a high accessibility of guest to the core surface for catalytic reaction [Citation35, Citation36]. The core–shell structure can also generate new catalytic properties that are driven by the electronic interactions between the Pt shell and the second core metal [Citation36, Citation37]. Therefore, the synthesis of the Pt-based core–shell catalyst is of great significance. As shown in figure [Citation38], the Pd–Pt core–shell nanoparticles with dendritic shell can be synthesized via a sophisticated chemical etching approach. These nanoparticles possess a superior catalytic activity for methanol oxidation reaction as compared to other Pt catalysts. Other metals, such as Au and Ag, can also be used as sacrificial templates for the preparation of hollow Pt nanoparticles or core–shell particles [Citation36, Citation39].
Figure 2. (a) Schematic of the formation of a bimetallic dendritic nanocage with a hollow interior and a porous dendritic wall. (b)–(e) Transmission electron microscopy (TEM) and high-angle annular dark-field scanning TEM (HAADF-STEM) images of dendritic Pt-on-Pd nanoparticles. (b)–(c) Before chemical etching; (d) and (e) after chemical etching [Citation38].

Table lists the applications, performance advantages, defects, and catalytic activities of typical core–shell catalysts with platinum shell and precious metal core. The prices of some precious metals are also listed on this table. Among these metals, Pt is the most expensive, Au and Rh are cheaper, and Ru is the cheapest. Given that Pd, Ru, and Rh are all VIIIB group elements, which properties are analogous with those of Pt, they may be considered as highly suitable replacements for Pt. However, given the expensiveness of Rh, Rh–Pt fails to attract much interest as a research topic. Recent reports suggest the Pd–Pt bimetallic catalyst, especially the Pd–Pt core–shell catalyst, as a promising replacement for the Pt catalyst because of its unique characteristics, which are not merely combined from the properties of the two constituent metals [Citation40–Citation49]. Table shows that the methanol electron–oxidation reaction (MOR) activity on Pd–Pt can be greatly improved. The peak specific area and mass current density for MOR on the Pd–Pt catalyst are approximately 50 mA cm–2 and 376 mA mg–1, respectively, which are ten times greater than those on the pure Pt catalyst. At the same time, Ru–Pt [Citation22, Citation50–Citation54], Rh–Pt [Citation55], Au–Pt [Citation56–Citation77], and Ag–Pt [Citation36] also possess a superior catalytic performance in terms of CO tolerance.
Table 1. The applications, advantages, defects and catalytic activities for the fuel cell of typical precious metal–Pt core–shell catalysts.
In summary, the core–shell catalysts prepared by depositing Pt on other precious metals have attracted significant research interest because the Pt shells supported on such cores demonstrate a catalytic activity and durability that are remarkably greater as compared to those of the pure Pt catalyst; such enhancement is a result of the modification of the electronic and geometric structures of the surface Pt atoms [Citation19, Citation20, Citation23, Citation78, Citation79, Citation80–Citation82].
2.2. Core–shell catalyst with platinum shell and non-precious metal core
Although precious metal–Pt core–shell catalyst is a fine replacement for the Pt catalyst, this material is extremely expensive. Recent studies show that the Pt electrocatalyst can achieve a high catalytic activity and utilization efficiency by replacing the core of the nanoparticles of the precious metal electrocatalysts with low-cost or non-precious metals or by reproducing a nanosphere with high surface-to-volume ratios. Theoretical and experimental studies show that arranging Pt metals as thin shells on the proper non-noble metal (e.g., Fe, Co, Ni, Cu, Cr, and V) cores not only reduces their usage but also significantly enhances their catalytic activity [Citation83–Citation85]. The atomic neighborhood of two dissimilar metals can modify the monometallic behavior and generate the desired new properties because of the short-range electronic charge transfer (ligand effects [Citation86–Citation90]) and the altered lattice constants (geometric effects [Citation89, Citation91]).
Table shows the applications, performance advantages, defects, and catalytic activities of typical core–shell catalysts with platinum shell and non-noble metal core. Given that Fe, Co, and Ni are VIIIB group elements that possess properties that are analogous with those of Pt, they are deemed suitable candidates to replace Pt as the core material. The mass activity of these core–shell catalysts toward ORR has been improved 4–22 times, which has subsequently decreased the cost of the catalyst [Citation92–Citation95]. Furthermore, the magnetism of these elements can also facilitate the recovery of the catalysts. Aside from the abovementioned non-noble metals, Cu is also a favorable candidate for improving the catalytic activity and decreasing the cost of the Pt catalyst because Cu is a member of IB group elements that possess properties that are similar with those of Au and Ag.
Table 2. The applications, advantages, defects and catalytic activities for the fuel cell of typical non-noble metal–Pt core–shell catalysts.
Other metals, such as Cr, V, and Sn, have also been used as core materials to fabricate non-noble metal–Pt core–shell catalysts. Chen et al [Citation96, Citation97] synthesized the Sn–Pt core–shell nanospheres that were used for electrochemical catalysts. Given their low electrical resistivity, low melting point, and high surface tension, the structure of these catalysts can be easily tuned.
2.3. Core–shell catalyst with platinum shell and bi-metal core
The properties of the core–shell catalyst with platinum shell can be tuned by changing the composition of the core materials. Given that metals demonstrate different advantages, the catalytic performance of the core–shell catalyst may be improved by combining one metal with another to serve as the core. Therefore, a core–shell catalyst with platinum shell and bi-metal core has been fabricated. Pd–M (precious metal or non-precious metal)–Pt has attracted much research interest as the Pd–Pt core–shell catalyst is a promising replacement for the Pt catalyst. Wang et al [Citation98] investigated the ORR activity of Pt, Pd–Pt, and Pd–Co–Pt core–shell nanoparticles and found that the Pt-monolayer catalyst on the 4 nm Pd and 4.6 nm Pd3Co cores exhibited 1.0 mA mg–1 and 1.6 mA mg–1 Pt mass activities, respectively, which were about five and nine times greater than those of 3 nm Pt nanoparticles. The Pd–Sn–Pt catalyst has also been synthesized by Zhang et al [Citation34] to reproduce a nanosphere shell with high surface-to-volume ratios. Pd–Sn is considered a favorable core for the following reasons: (1) Pd is a fine type of electrocatalyst that can be used under acid conditions and has a surface that favors the reductive deposition of Pt [Citation34, Citation99]; and (2) Sn is an active metal that can contribute to decreasing the cost of catalysts and can be easily replaced by Pt [Citation34]. For the same purpose, Hui et al [Citation100] proposed a general strategy to prepare carbon-supported Pd–Cu–Pt nanoparticles with an intimate contact between Pt and Pd–Cu. The MOR and ORR catalytic activities of the catalyst is higher than those of the Pt/C. Yang et al [Citation101] prepared Pd–Ag–Pt core–shell catalyst that exhibited a superior catalytic activity toward the ORR in fuel cells. The Ag component is crucial for constructing the multiple-twinned structure of the core–shell nanoparticles, whereas the Pd component is used to reduce the tensile strain effect of the Ag on the deposited Pt layer, which renders the Pd binding energy in core–shell Pd–Ag–Pt nanoparticles. Aside from the above examples, the Pd–Pt–Pt, Mo–Pt–Pt, and Pb–Pt–Pt core–shell catalysts have also been prepared to improve the performance of the Pt-based catalyst [Citation102–Citation105]. It is obvious that, the core material composition is a key factor that affects catalytic activity. Choosing and tuning the composition of the core material may be another interesting research topic.
2.4. The fabrication of the core–shell catalyst with platinum shell
In summary, the activity and stability of the Pt-based electrocatalyst can both be improved by the fabrication of the core–shell structure. Numerous methods have been developed for the preparation of core–shell catalysts with Pt shell.
Table shows the basic operations and features of different fabrication methods. The fabrication of the core–shell catalyst with Pt shell follows three methods mainly. The general method is the wet chemical method, which includes the one-step and two-step (sequential reduction) methods [Citation48, Citation66, Citation69, Citation73, Citation75]. These methods adopt many techniques (such as microwave heating [Citation48] and ultraviolet irradiation [Citation75]) and sometimes use surfactant as a template for modifying the surface morphology of the material. The advantages of this method, especially the one-step method, lie in their simple operation and gentle operation condition.
Table 3. Fabrication methods for the Pt-based core–shell catalysts.
Electrochemical synthesis is another important method for fabricating the Pt shell catalyst. This method includes the general electrochemical method [Citation106] and the Galvanic displacement method [Citation23, Citation49, Citation107, Citation108]. Electrochemical synthesis requires conductive substrates and an electrochemical cell, and its process is more complicated than that of the wet chemical method.
Atomic layer deposition can also be used to synthesize the Pt shell catalyst [Citation109]. This technique has promising uses for the production of uniform precious metal nanoparticles on high surface area supports because of its sequential and self-limiting surface reactions. However, this method has a high instrument requirement and a severe operation condition.
3. Core–shell catalyst with precious metal core
Supported precious metal catalysts have been extensively studied because of their unique properties that greatly enhance their catalytic activity for various reactions. The main function of the catalyst support is to offer an effective surface and a suitable porous structure. The interaction between the active component and the support also plays an important role in the catalytic reaction. However, the aggregation and poisoning of metal nanoparticles will minimize their surface area and surface energy, which results in a loss of nanostructure-specific properties. The encapsulation of precious metal nanoparticles as a core inside of a supported material as a shell has recently attracted much research attention. This core–shell structure can maintain the size and shape of the precious metal nanoparticles as well as protect them from damage. The stability and compatibility of the particles can be enhanced by encapsulating nano precious metal particles in a stable shell. At the same time, the electron charge, reactivity, and functionality of the enwrapped materials can also be changed in the process [Citation110–Citation112]. Several materials can be used as shell materials, such as mesoporous silica, metal oxides, and other porous materials. Mesoporous silica and metal oxides, which are considered the general catalyst supports, are also identified as the most important among these materials.
3.1. Core–shell catalyst with precious metal core and mesoporous silica shell
Ordered mesoporous silica materials were fist synthesized in 1992 [Citation113] and the interest has been extended from the mesoporous silica nanoparticles [Citation114] to the thin films [Citation115–Citation118]. The progress in this area has been reviewed [Citation119–Citation121]. Since its discovery, mesoporous silica has been extensively used as a catalyst support because of its high thermal stability, controllable morphology, and desirable surface area. Silica particles also play an important role in the preparation of core–shell nanoparticle systems because of their easy functionalization and simple synthesis from the TEOS monomer. Liz-Marzán et al [Citation122] were the first to report the synthesis of gold–silica core–shell particles using the Stöber method. However, given that the silica synthesized by the classical Stöber method is usually non-porous, this property restricts the application of the core–silica shell in the catalytic area. This problem can be effectively resolved by preparing mesoporous silica (mSiO2) [Citation123–Citation125]. The first core–mSiO2 structure that was formed by the Stöber process was reported by Büchel et al [Citation126], which was followed by an increasing attention from many researchers, such as Somorjai et al [Citation24] and Zhao et al [Citation127–Citation131]. The core material has been extended to all kinds of materials, such as metals and oxides. Numerous studies have been conducted to prepare various precious metal core–silica shell nanostructures for a wide range of applications. The categories, features, applications, and fabrications of the core–shell catalysts with precious metal core and silica shell are summarized in table [Citation16, Citation24, Citation112, Citation132–Citation140, Citation141–Citation144, Citation145–Citation150, Citation151].
Table 4. Features of the core–shell catalysts with precious metal core and silica shell.
As shown in table , the core–shell material with precious metal core and silica shell can be used as the catalyst of many reduction or oxidation reactions. This material has an easy fabrication process, high activity, high stability, and is generally fabricated using colloid methods, which involve three operation steps (table ). The formation mechanism of the precious metal–mSiO2 core–shell structured nanoparticles has been depicted in detail by Somorjai et al [Citation24] (figure ), who also reported the synthesis of the thermally stable Pt–mSiO2 core–shell nanocatalyst for a high-temperature reaction. The TEM image of the material that is prepared by the above method is shown in figure [Citation24]. This material can maintain its core–shell configuration up to 750 °C, and the ignition temperature of CO on the catalyst is about 300 °C. Although the catalytic activity of the precious metal–silica core–shell catalyst is higher than that of the general silica-supported precious metal catalyst, such activity remains very low because of the inertness of silica. Therefore, replacing silica with other active materials, such as metal oxide, may present a favorable alternative.
Figure 3. Schematic of the synthesis of Pt–mSiO2 nanoparticles. Pt nanoparticles were synthesized using the tetradecyltrimethyl ammonium bromide (TTAB) surfactant as the capping agent, and used as the core particles. Second, as-synthesized Pt–SiO2 particles were prepared by polymerizing TEOS around the TTAB [Citation24]. RT stands for room temperature.
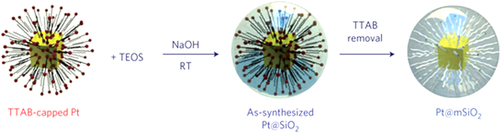
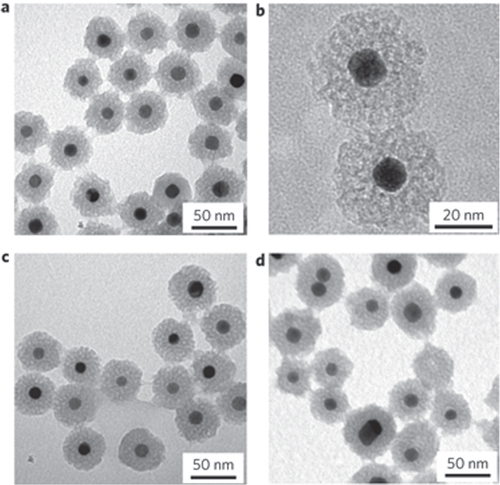
Figure 4. Thermal stability of Pt–mSiO2 nanoparticles. (a)–(d) TEM images of Pt–mSiO2 nanoparticles after calcination at 350 °C (a) and (b), 550 °C (c) and 750 °C (d) [Citation24].
3.2. Core–shell catalyst with precious metal core and metal oxide shell
The precious metal nanoparticles that are encapsulated in metal oxide are extremely active and stable in many reactions because a group of ‘active’ support materials, such as Fe2O3, TiO2, SnO2, NiOx, CoOx, MnOx, CeOx, and CuOx, show a higher catalytic activity than that of ‘inert’ materials, such as SiO2, when combined with precious metal nanoparticles [Citation152]. These ‘active’ reducible transition metal oxides serve as an oxygen supply during reactions. A synergistic mechanism may occur at the precious metal–metal oxide interface, and with the metal oxide being part of the catalytic process, the electrons are transferred from the oxide support to the precious metal nanoparticles [Citation153]. Table shows the features, applications, and fabrications of typical core–shell catalysts with precious metal core and metal oxide shell.
Table 5. Features of typical core–shell catalysts with precious metal core and metal oxide shell.
TiO2 semiconductor has been regarded as one of the most popular photocatalysts, especially in the cleaning of water and the removal of volatile organic compounds (VOC) in air [Citation154–Citation159]. This material has also been used extensively as the support of CO oxidation or water–gas shift reaction catalysts [Citation160–Citation165]. Several studies prove that, in metal–TiO2 core–shell particles, the high Fermi energy level of metal cores can efficiently separate the photoexcited electron-hole pairs of the TiO2 conductor, which heightens the redox capability of the metal core [Citation166–Citation168]. Table shows that Pt, Au, and Pd–TiO2 core–shell catalysts have all been prepared and used for photocatalytic reactions, CO oxidation, and water–gas shift reactions [Citation169–Citation175]. Although these catalysts are very active and stable, the fabrication of metal–TiO2 structures remains a great challenge. The hydrothermal reaction method remains as the general method and only few successful examples have been reported to date [Citation174, Citation176]. Figure shows the flower-like Au–TiO2 nanoparticles with shell that are built by packed TiO2 nanoparticles [Citation174].
Figure 5. TEM images of the as-prepared core–shell Au–TiO2 nanoparticles (a) and individual particle image (b). The inset in (a) is the electronic diffraction (ED) pattern of the individual particle. Local HRTEM images of individual TiO2 antenna: root region (c) and external edge of individual antenna (d); the insets in(c) and (d) are their corresponding FFT patterns [Citation174].

As shown in figures and , the main difference between the silica shell and metal oxide shell catalysts lies in their particle size and morphology, which can be ascribed to the fact that the two systems follow different particle growth theories. The large precious metal particles and thick shells of metal–TiO2 may lead to a low catalytic activity. However, the flower-like morphology is more suitable than the spherical morphology as a catalyst. Therefore, combining the advantages of the two systems may present another topic that is worth investigating. Lekeufack et al [Citation177] obtained different morphologies of the particles by adjusting the proportion of the TiO2 precursor in the silica precursor and by displacing the gold particles from the center to eccentric positions, which would generate acron- and raspberry-like structures.
Fe2O3 serves as a favorable catalyst support because of its fair oxidation and reduction properties [Citation178, Citation179]. Yin et al [Citation179] prepared Au–Fe2O3 core–shell structures on silica supports and investigated their catalytic activity. The Fe2O3 shell has a thickness of about 2 nm, and the Au–Fe2O3 interface is active for CO oxidation. The catalytic activity of silica-supported Au–Fe2O3 core–shell structure catalyst is greater than that of the Fe2O3-supported Au catalyst with a comparable gold loading. The silica-supported Au–Fe2O3 catalysts are stable in CO oxidation at room temperature after 20 h on stream.
CeO2 has been identified as an important component of catalysts for many reactions, such as water–gas shift reaction and automotive off-gas purification. This material can store and release both oxygen and hydrogen by forming a surface, bulk vacancies, or an inter-metallic M–Ce compound. Ceria can also serve as a stabilizer for metal and alumina supports by maintaining a high dispersion of the catalytic metals. Table shows that Pt, Au, Pd, and Ag can all be encapsulated into CeO2 to form the core–shell structure [Citation25, Citation180–Citation184]. Mitsudome et al [Citation181] successfully synthesized a core–shell Ag–CeO2 nanocomposite that comprised Ag nanoparticles with a 10 nm diameter in the core as well as assembled a spherical CeO2 with a 3 nm diameter in the shell. This catalyst is easily separable and reusable, with retention of its high catalytic performance.
Sn belongs to the same IVA group as Si, and support an easy fabrication of M–SnO2 core–shell structures. In addition, the catalytic activity of SnO2 is much higher than that of SiO2. Therefore, the M–SnO2 core–shell catalyst presents a very interesting phenomenon [Citation153, Citation185, Citation186, Citation187]. Yu et al [Citation153] prepared Au–SnO2 catalyst for CO oxidation and found that its catalytic activity and stability was much higher than that of the non-encapsulated Au–SnO2.
Cu2O can serve as a photocatalyst or redox catalyst owing to its optical and chemical properties. Therefore, Au–Cu2O becomes a favorable catalyst [Citation188–Citation191]. To date, the Au–Cu2O core–shell particle remains the only particle with a perfect core–shell structure that is comparable with that of M–SiO2 (figure ) [Citation191].
Figure 6. Formation of rattle-like Au–Cu2O yolk–shell nanoparticles. (A) Schematic illustration of the structural evolution of Au–Cu2O core–shell nanoparticles during the hollowing of Cu2O shell. (B)–(M) Bright-field TEM images showing the structural evolution of Au–Cu2O core–shell particles with three different outer radii: particles with an average outer radius of 183 nm obtained at (B) 5, (C) 30, (D) 60, and (E) 90 min; particles with an average outer radius of 130 nm obtained at (F) 5, (G) 20, (H) 40, and (I) 60 min; particles with an average outer radius of 98 nm obtained at (J) 5, (K) 20, (L) 40, and (M) 60 min. The scale of all TEM images is the same as in B [Citation191].

Overall, the morphology of the core–shell catalyst with precious metal core and metal oxide shell is often irregular. The activity and stability of this catalyst are much higher. Nevertheless, this catalyst has a complicated preparation process and a lower yield. Therefore, a convenient method for increasing the yield of the core–shell catalyst must be developed.
4. Problems and challenges
Although the study of precious metal core–shell catalysts has only started a few years ago, they have exhibited a great potential for application in heterogeneous catalysis area owing to their high activity, high stability, high selectivity and other functions. Despite the great advancements in this field, several problems remain unsolvable, which are listed as follows: (1)The efficient fabrication methods of the core–shell structural catalyst are still limited, and each method can only be used to prepare certain materials. (2)The core size and the shell thickness remains difficult to control precisely. (3)The yield of the core–shell catalyst remains low. (4)When a precious metal core is used as the active part of the catalyst, its catalytic activity is always degraded by the shell-block effect. These facts have restricted the application of the core–shell structure material and meanwhile it offers a lot of room for the extension of the fabrication of core–shell catalyst.
5. Conclusions
Studies on the core–shell structure catalyst are often conducted to reduce the cost of such catalyst by improving the performance of precious metals. The efficiency of precious metals may be improved by fabricating the precious metal shell as thin as possible, decreasing the size of the precious metal core nanoparticles, and dispersing the core–shell structure catalysts on the support. Therefore, future studies on core–shell structure fabrication must consider controlling the core size and tuning the shell thickness. An increase in the shell pores can also improve the catalytic activity of the precious metal core. These goals can only be achieved by developing new methods or optimizing the technology of the existing methods. Moreover, using the mixture of the easy shaping and chemically active materials (such as silica and metal oxide) as a shell material may also contribute to the fabrication of a high-performance core–shell catalyst that has the combined advantages of different materials.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (21103104).
Reference
- FeyenMWeidenthalerCGuettelRSchlichteKHolleULuA-HSchuethF 2011 Chem.-a Eur. J. 17 598 10.1002/chem.201001827
- HuangXTangSLiuBRenBZhengN 2011 Adv. Mater. 23 3420 10.1002/adma.201100905
- SindoroMFengYXingSLiHXuJHuHLiuCWangYZhangHShenZChenH 2011 Angew. Chem., Int. Ed. 50 9898 10.1002/anie.v50.42
- ZhangFCheRLiXYaoCYangJShenDHuPLiWZhaoD 2012 Nano Lett. 12 2852 10.1021/nl300421n
- ZhangZWangLWangJJiangXLiXHuZJiYWuXChenC 2012 Adv. Mater. 24 1418 10.1002/adma.201104714
- CaoSChenJHuJ 2009 Aust. J. Chem. 62 1561 10.1071/CH08420
- JankiewiczB JJamiolaDChomaJJaroniecM 2012 Adv. Colloid Interface Sci. 170 28 10.1016/j.cis.2011.11.002
- LeiBHanSLiCZhangDLiuZZhouC 2007 Nanotechnology 18 040019 10.1088/0957-4484/18/17/175605
- LiWZhaoD 2013 Adv. Mater. 25 142 10.1002/adma.201203547
- ReissPProtiereMLiL 2009 Small 5 154 10.1002/smll.200800841
- SchaertlW 2010 Nanoscale 2 829 10.1039/c0nr00028k
- van EmbdenJJasieniakJGomezD EMulvaneyPGiersigM 2007 Aust. J. Chem. 60 457 10.1071/CH07046
- WeiSWangQZhuJSunLLinHGuoZ 2011 Nanoscale 3 4474 10.1039/c1nr11000d
- AlayogluSNilekarA UMavrikakisMEichhornB 2008 Nat. Mater. 7 333 10.1038/nmat2156
- DengYCaiYSunZLiuJLiuCWeiJLiWLiuCWangYZhaoD 2010 J. Am. Chem. Soc. 132 8466 10.1021/ja1025744
- MitsudomeTTakahashiYIchikawaSMizugakiTJitsukawaKKanedaK 2013 Angew. Chem., Int. Ed. 52 1481 10.1002/anie.201207845
- MoriKSugiharaKKondoYTakeuchiTMorimotoSYamashitaH 2008 J. Phys. Chem. C 112 16478 10.1021/jp709571v
- Nguyen VietLCao MinhTNogamiMOhtakiM 2012 Recent Pat. Mater. Sci. 5 175 10.2174/1874464811205030175
- ShaoMShoemakerKPelesAKanekoKProtsailoL 2010 J. Am. Chem. Soc. 132 9253 10.1021/ja101966a
- StrasserP 2010 Nat. Chem. 2 454 10.1038/nchem.623
- ZhaiQXieSFanWZhangQWangYDengWWangY 2013 Angew. Chem., Int. Ed. Eng. 52 5776 10.1002/anie.v52.22
- ZhangLKimJZhangJNanFGauquelinNBottonG AHePBashyamRKnightsS 2013 Appl. Energy 103 507 10.1016/j.apenergy.2012.10.017
- SasakiKNaoharaHCaiYChoiY MLiuPVukmirovicM BWangJ XAdzicR R 2010 Angew. Chem., Int. Ed. 49 8602 10.1002/anie.201004287
- Sang HoonJJeong YoungPChia-KuangTYamadaYPeidongYSomorjaiG A 2009 Nat. Mater. 8 126 10.1038/nmat2329
- AdijantoLBennettD AChenCYuA SCargnelloMFornasieroPGorteR JVohsJ M 2013 Nano Lett. 13 2252 10.1021/nl4008216
- GuptaGSlanacD AKumarPWiggins-CamachoJ DWangXSwinneaSMoreK LDaiSStevensonK JJohnstonK P 2009 Chem. Mater. 21 4515 10.1021/cm901203n
- KohSStrasserP 2007 J. Am. Chem. Soc. 129 12624 10.1021/ja0742784
- ManiPSrivastavaRStrasserP 2008 J. Phys. Chem. C 112 2770 10.1021/jp0776412
- MuravievD NMacanasJRuizPMunozM 2008 Physica Status Solidi a 205 1460 10.1002/pssa.v205:6
- NilekarA UMavrikakisM 2008 Surf. Sci. 602 L89 10.1016/j.susc.2008.05.036
- ShaoMSasakiKMarinkovicN SZhangLAdzicR R 2007 Electrochem. Commun. 9 2848 10.1016/j.elecom.2007.10.009
- WangGWuHWexlerDLiuHSavadogoO 2010 J. Alloys Compd. 503 L1 10.1016/j.jallcom.2010.04.236
- ZhouSVarugheseBEichhornBJacksonGMcIlwrathK 2005 Angew. Chem., Int. Ed. 44 4539 10.1002/(ISSN)1521-3773
- ZhangWWangRWangHLeiZ 2010 Fuel Cells 10 734 10.1002/fuce.v10:4
- Ataee-EsfahaniHImuraMYamauchiY 2013 Angew. Chem., Int. Ed. 52 13611 10.1002/anie.201307126
- LiCYamauchiY 2013 Phys. Chem. Chem. Phys. 15 3490 10.1039/c3cp44313b
- WangLYamauchiY 2010 J. Am. Chem. Soc. 132 13636 10.1021/ja100329d
- WangLYamauchiY 2013 J. Am. Chem. Soc. 135 16762 10.1021/ja3096762
- ShaojunGLiangWShaojunDErkangW 2008 J. Phys. Chem. C 112 13510
- GolikandA NLohrasbiEMaraghehM GAsgariM 2009 J. Appl. Electrochem. 39 2421 10.1007/s10800-009-9930-2
- KadirganFBeyhanSAtilanT 2009 Int. J. Hydrog. Energy 34 4312 10.1016/j.ijhydene.2009.03.024
- LimBJiangMCamargoP H CChoE CTaoJLuXZhuYXiaY 2009 Science 324 1302 10.1126/science.1170377
- PengZYangH 2009 J. Am. Chem. Soc. 131 7542 10.1021/ja902256a
- WangHXuCChengFZhangMWangSJiangS P 2008 Electrochem. Commun. 10 1575 10.1016/j.elecom.2008.08.011
- WeiHJuanyingLYongjinQZhiqingZXiaogangZAkinsD LHuiY 2010 J. Power Sources 195 1046 10.1016/j.jpowsour.2009.08.027
- XuYLinX 2007 J. Power Sources 170 13 10.1016/j.jpowsour.2007.03.064
- YuanQZhouZZhuangJWangX 2010 Chem. Commun. 46 1491 10.1039/b922792j
- ZhangHYinYHuYLiCWuPWeiSCaiC 2010 J. Phys. Chem. C 114 11861 10.1021/jp100354z
- ZhangJMoYVukmirovicM BKlieRSasakiKAdzicR R 2004 J. Phys. Chem. B 108 10955 10.1021/jp049933y
- AlayogluSZavalijPEichhornBWangQFrenkelA IChupasP 2009 Acs Nano 3 3127 10.1021/nn900242v
- ChenD-JHofstead-DuffyA MParkI-SAtienzaD OSusutCSunS-GTongY J 2011 J. Phys. Chem. C 115 8735 10.1021/jp200557m
- LeiZKimJHao MingCFeihongNDudeckKRu-ShiLBottonG AJiujunZ 2011 J. Power Sources 196 9117 10.1016/j.jpowsour.2011.05.020
- OchalP 2011 J. Electroanal. Chem. 655 140 10.1016/j.jelechem.2011.02.027
- YangLVukmirovicM BSuDSasakiKHerronJ AMavrikakisMLiaoSAdzicR R 2013 J. Phys. Chem. C 117 1748 10.1021/jp309990e
- AlayogluSEichhornB 2008 J. Am. Chem. Soc. 130 17479 10.1021/ja8061425
- AtienzaD OAllisonT CTongY J 2012 J. Phys. Chem. C 116 26480 10.1021/jp310313k
- ChenLKuaiLGengB 2013 Crystengcomm 15 2133 10.1039/c3ce27058k
- DuanM-YLiangRTianNLiY-JYeungE S 2013 Electrochim. Acta 87 432 10.1016/j.electacta.2012.09.076
- FengRLiMLiuJ 2012 Rare Metals 31 451 10.1007/s12598-012-0538-z
- HartlKMayrhoferK J JLopezMGoiaDArenzM 2010 Electrochem. Commun. 12 1487 10.1016/j.elecom.2010.08.013
- JianhuangZJunYLeeJ YWeijiangZ 2006 J. Phys. Chem. B 110 24606
- KhalidMWasioNChaseTBandyopadhyayK 2010 Nanoscale Res. Lett. 5 61 10.1007/s11671-009-9443-2
- KuaiLGengBWangSSangY 2012 Chem.-a Eur. J. 18 9423 10.1002/chem.v18.30
- LeeYKimJYunD SNamY SShao-HornYBelcherA M 2012 Energy Environ. Sci. 5 8328 10.1039/c2ee21156d
- LiSDongYBiXTangM 2013 Int. J. Electrochem. Sci. 8 8662
- LiXLiuJHeWHuangQYangH 2010 J. Colloid Interface Sci. 344 132 10.1016/j.jcis.2009.12.019
- LuYYuanJPolzerFDrechslerMPreussnersJ 2010 Acs Nano 4 7078 10.1021/nn102622d
- MonteroM AGennero de ChialvoM RChialvoA C 2011 Int. J. Hydrog. Energy 36 3811 10.1016/j.ijhydene.2010.12.115
- RongjuanFMinLJiaxiangL 2012 Colloids Surf. 406 6 10.1016/j.colsurfa.2012.04.030
- ShaojunGYouxingFShaojunDErkangW 2007 J. Phys. Chem. C 111 17104
- ShuangyinWKristianNSanpingJXinW 2009 Nanotechnology 20 025605
- SongH MAnjumD HSougratRHedhiliM NKhashabN M 2012 J. Mater. Chem. 22 25003 10.1039/c2jm31582c
- ToshimaNNaoharaHYoshimotoTOkamotoY 2011 Chem. Lett. 40 1095 10.1246/cl.2011.1095
- XiuCShengnanWJungwirthSZhibingCZhenghuaWLunWYongxinL 2013 Nanotechnology 24 295402
- XuYDongYShiJXuMZhangZYangX 2011 Catal. Commun. 13 54 10.1016/j.catcom.2011.06.018
- YamaguchiWTaiY 2013 J. Fuel Cell Sci. Technol. 10 041006 10.1115/1.4024574
- ZhaiJHuangMDongS 2007 Electroanalysis 19 506 10.1002/(ISSN)1521-4109
- ChenYLiangZYangFLiuYChenS 2011 J. Phys. Chem. C 115 24073 10.1021/jp1112944
- MalheiroA RPerezJSantiagoE IMercedes VillullasH 2010 J. Phys. Chem. C 114 20267 10.1021/jp107054u
- TaufanyF 2011 Acs Nano 5 9370 10.1021/nn202545a
- WangC 2011 J. Am. Chem. Soc. 133 14396 10.1021/ja202055v
- ZhangJ LVukmirovicM BXuYMavrikakisMAdzicR R 2005 Angew. Chem., Int. Ed. 44 2132 10.1002/(ISSN)1521-3773
- MazumderVChiMMoreK LSunS 2010 Angew. Chem., Int. Ed. 49 9368 10.1002/anie.201003903
- MazumderVChiMMoreK LSunS 2010 J. Am. Chem. Soc. 132 7848 10.1021/ja1024436
- ZhangX-BYanJ-MHanSShioyamaHXuQ 2009 J. Am. Chem. Soc. 131 2778 10.1021/ja808830a
- GreeleyJNorskovJ KMavrikakisM 2002 Annu. Rev. Phys. Chem. 53 319 10.1146/annurev.physchem.53.100301.131630
- HeggenMOezaslanMHoubenLStrasserP 2012 J. Phys. Chem. C 116 19073 10.1021/jp306426a
- KitchinJ RNorskovJ KBarteauM AChenJ G 2004 J. Chem. Phys. 120 10240 10.1063/1.1737365
- KitchinJ RNrskovJ KBarteauM AChenJ G 2004 Phys. Rev. Lett. 93 156801/1 10.1103/PhysRevLett.93.156801
- NilssonAPetterssonL G MHammerBBligaardTChristensenC HNorskovJ K 2005 Catal. Lett. 100 111 10.1007/s10562-004-3434-9
- MavrikakisMHammerBNorskovJ K 1998 Phys. Rev. Lett. 81 2819 10.1103/PhysRevLett.81.2819
- Godinez-SalomonFHallen-LopezMSolorza-FeriaO 2012 Int. J. Hydrog. Energy 37 14902 10.1016/j.ijhydene.2012.01.157
- SarkarAManthiramA 2010 J. Phys. Chem. C 114 4725 10.1021/jp908933r
- ShimizuKChengI FWaiC M 2009 Electrochem. Commun. 11 691 10.1016/j.elecom.2009.01.016
- ZeshengLChunyongHMeiCShuaiKPei KangS 2012 Int. J. Hydrog. Energy 37 14152
- ChenC CChenC LLaiY-S 2011 Mater. Chem. Phys. 131 250 10.1016/j.matchemphys.2011.09.030
- ChenC CChenC LLaiY S 2013 Ceram. Int. 39 4369 10.1016/j.ceramint.2012.11.023
- WangJ XInadaHWuLZhuYChoiYLiuPZhouW-PAdzicR R 2009 J. Am. Chem. Soc. 131 17298 10.1021/ja807202m
- XiaoLZhuangLLiuYLuJAbrunaH D 2009 J. Am. Chem. Soc. 131 602 10.1021/ja8063765
- HuiWRongfangWHaoLQunfangWJianKZiqiangL 2011 Int. J. Hydrog. Energy 36 839 10.1016/j.energy.2010.12.031
- YangJYangJYingJ Y 2012 Acs Nano 6 9373 10.1021/nn303298s
- KangYQiLLiMDiazR ESuDAdzicR RStachELiJMurrayC B 2012 Acs Nano 6 2818 10.1021/nn3003373
- LiuZHuJ EWangQGaskellKFrenkelA IJacksonG SEichhornB 2009 J. Am. Chem. Soc. 131 6924 10.1021/ja901303d
- WuY-NLiaoS-JGuoH-FHaoX-Y 2013 J. Power Sources 235 135 10.1016/j.jpowsour.2013.02.011
- WuY-NLiaoS-JLiangZ-XYangL-JWangR-F 2009 J. Power Sources 194 805 10.1016/j.jpowsour.2009.06.069
- LublowMBouabadiBKubalaS 2012 Sol. Energy Mater. Sol. Cells 107 56 10.1016/j.solmat.2012.07.018
- SasakiKWangJ XNaoharaHMarinkovicNMoreKInadaHAdzicR R 2010 Electrochim. Acta 55 2645 10.1016/j.electacta.2009.11.106
- YangH 2011 Angew. Chem., Int. Ed. 50 2674 10.1002/anie.201005868
- LeiY 2012 Chem. Mater. 24 3525 10.1021/cm300080w
- GrassetF 2002 Langmuir 18 8209 10.1021/la020322b
- VelikovK PMorozAvan BlaaderenA 2002 Appl. Phys. Lett. 80 49 10.1063/1.1431698
- YaoLShiTLiYZhaoJJiWAuC-T 2011 Catal. Today 164 112 10.1016/j.cattod.2010.10.056
- Kresge M E LC TRothW JVartuliJ CBeckJ S 1992 Nature 359 710 10.1038/359710a0
- WangS-GWuC-WChenKLinV S Y 2009 Chem.- Asian J. 4 658 10.1002/asia.v4:5
- Chia-WenWAokiTKuwabaraM 2004 Nanotechnology 15 1886 10.1088/0957-4484/15/12/035
- Chia-WenWKuwabaraM 2003 Sci. Technol. Adv. Mater. 4 593 10.1016/j.stam.2003.12.004
- Chia-WenWOhsunaTEduraTKurodaK 2007 Angew. Chem., Int. Ed. 46 5364 10.1002/(ISSN)1521-3773
- Chia-WenWYamauchiYOhsunaTKurodaK 2006 J. Mater. Chem. 16 3091 10.1039/b604062d
- SlowingI IVivero-EscotoJ LWuC-WLinV S Y 2008 Adv. Drug Deliv. Rev. 60 1278 10.1016/j.addr.2008.03.012
- Vivero-EscotoJ LChiangY-DWuC-W KYamauchiY 2012 Sci. Technol. Adv. Mater. 13 013003 10.1088/1468-6996/13/1/013003
- YamauchiY 2013 J. Ceram. Soc. Japan 121 831 10.2109/jcersj2.121.831
- Liz-MarzanL MGiersigMMulvaneyP 1996 Langmuir 12 4329 10.1021/la9601871
- LiuJQiaoS ZHuQ HLuG Q 2011 Small 7 425 10.1002/smll.201001402
- ShokouhimehrMPiaoYKimJJangYHyeonT 2007 Angew. Chem., Int. Ed. 46 7039 10.1002/(ISSN)1521-3773
- ZhangQZhangTGeJYinY 2008 Nano Lett. 8 2867 10.1021/nl8016187
- BuchelGUngerK KMatsumotoATsutsumiK 1998 Adv. Mater. 10 1036 10.1002/(ISSN)1521-4095
- GuoXDengYGuDCheRZhaoD 2009 J. Mater. Chem. 19 6706 10.1039/b910606e
- QianX FLiBHuY YNiuG XZhangD Y HCheR CTangYSuD SAsiriA MZhaoD Y 2012 Chem.- Eur. J. 18 931 10.1002/chem.v18.3
- XufangQYingyingLWeiLYongyaoXDongyuanZ 2011 J. Mater. Chem. 21 13025
- YangJDengYWuQZhouJBaoHLiQZhangFLiFTuBZhaoD 2010 Langmuir 26 8850 10.1021/la904596x
- YangJ 2011 Chem. Commun. 47 11618 10.1039/c1cc10584a
- BellC SYuS SGiorgioT D 2011 Small 7 1157 10.1002/smll.v7.9
- Cavaliere-JaricotSDarbandiMNannT 2007 Chem. Commun. 20 2031 10.1039/b703811a
- ChenFChenQFangSSunY AChenZXieGDuY 2011 Dalton Trans. 40 10857 10.1039/c0dt01616k
- Ching-ChichLShih-TangCFu-KenLCheng-XuanW 2012 J. Mater. Chem. 22 2089 10.1039/c1jm13984c
- DasSAsefaT 2012 Top. Catal. 55 587 10.1007/s11244-012-9835-x
- JoonsungPHyojongY 2013 J. Mater. Chem. A 1 5408 10.1039/c3ta10613f
- KobayashiYInoseHNakagawaTGondaKTakedaMOhuchiNKasuyaA 2012 Surf. Eng. 28 129 10.1179/1743294411Y.0000000069
- LinK JChenL JPrasadM RChengC Y 2004 Adv. Mater. 16 1845 10.1002/(ISSN)1521-4095
- MinY-LWanYLiuRYuS-H 2008 Mater. Chem. Phys. 111 364 10.1016/j.matchemphys.2008.04.030
- OhJ-GKimH 2013 Curr. Appl. Phys. 13 130 10.1016/j.cap.2012.06.025
- OkadaSIkurumiSKamegawaTMoriKYamashitaH 2012 J. Phys. Chem. C 116 14360 10.1021/jp3025073
- QiYChenMLiangSYangWZhaoJ 2008 Appl. Surf. Sci. 254 1684 10.1016/j.apsusc.2007.07.136
- RuffinoFPugliaraACarriaERomanoLBongiornoCSpinellaCGrimaldiM G 2012 Nanotechnology 23 045601 10.1088/0957-4484/23/4/045601
- SangermanoMPerruchasSGacoinTRizzaG 2008 Macromol. Chem. Phys. 209 2343 10.1002/macp.v209:22
- SchulzendorfMCaveliusCBornPMurrayEKrausT 2011 Langmuir 27 727 10.1021/la102630y
- SouleSAlloucheJDupinJ-CMartinezH 2013 Microporous Mesoporous Mater. 171 72 10.1016/j.micromeso.2012.12.034
- TanLWuXChenDLiuHMengXTangF 2013 J. Mater. Chem. A 1 10382 10.1039/c2ta00325b
- WangYZhaiYPierreDFlytzani-StephanopoulosM 2012 Appl. Catal. B-Environ. 127 342 10.1016/j.apcatb.2012.08.038
- WuCLimZ-YZhouCWangW GZhouSYinHZhuY 2013 Chem. Commun. 49 3215 10.1039/c3cc39202c
- ZhangCLeeJ Y 2013 J. Phys. Chem. C 117 15253 10.1021/jp308442t
- SchubertM MHackenbergSvan VeenA CMuhlerMPlzakVBehmR J 2001 J. Catal. 197 113 10.1006/jcat.2000.3069
- YuKWuZZhaoQLiBXieY 2008 J. Phys. Chem. C 112 2244 10.1021/jp711880e
- AuvinenJWirtanenL 2008 Atmos. Environ. 42 4101 10.1016/j.atmosenv.2008.01.031
- KimHChoiW 2007 Appl. Catal. B-Environ. 69 127 10.1016/j.apcatb.2006.06.011
- LiuZZhangXNishimotoSMurakamiTFujishimaA 2008 Environ. Sci. Technol. 42 8547 10.1021/es8016842
- OhnoTMurakamiNTsubotaTNishimuraH 2008 Appl. Catal. A-General 349 70 10.1016/j.apcata.2008.07.016
- SchmidtC MBuchbinderA MWeitzEGeigerF M 2007 J. Phys. Chem. A 111 13023 10.1021/jp076745+
- YangXCaoCHohnKEricksonLMaghirangRHamalDKlabundeK 2007 J. Catal. 252 296 10.1016/j.jcat.2007.09.014
- ArunajatesanVViandtMMcIntoshSCruzMRennekeRChenB S 2005 Gold Bull. 38 133
- DenkwitzYGeserickJHoermannUPlzakVKaiserUHusingNBehmR J 2007 Catal. Lett. 119 199 10.1007/s10562-007-9229-z
- HammerNZarubovaSKvandeIChenDRonningM 2007 Gold Bull. 40 234 10.1007/BF03215586
- SchumacherBPlzakVKinneMBehmR J 2003 Catal. Lett. 89 109 10.1023/A:1024731812974
- SoaresJ M CHallMCristofoliniMBowkerM 2006 Catal. Lett. 109 103 10.1007/s10562-006-0064-4
- YuW-YLeeW-SYangC-PWanB-Z 2007 J. Chin. Inst. Chem. Eng. 38 151 10.1016/j.jcice.2007.01.002
- HirakawaTKamatP V 2005 J. Am. Chem. Soc. 127 3928 10.1021/ja042925a
- SubramanianVWolfE EKamatP V 2004 J. Am. Chem. Soc. 126 4943 10.1021/ja0315199
- WangWZhangJChenFHeDAnpoM 2008 J. Colloid Interface Sci. 323 182 10.1016/j.jcis.2008.03.043
- BianZZhuJCaoFLuYLiH 2009 Chem. Commun. 3789 10.1039/b906421d
- CargnelloMGrzelczakMRodriguez-GonzalezBSyrgiannisZBakhmutskyKLa ParolaVLiz-MarzanL MGorteR JPratoMFornasieroP 2012 J. Am. Chem. Soc. 134 11760 10.1021/ja304398b
- DillonR JJooJ-BZaeraFYinYBardeenC J 2013 Phys. Chem. Chem. Phys. 15 1488 10.1039/c2cp43666c
- DuJQiJWangDTangZ 2012 Energy Environ. Sci. 5 6914 10.1039/c2ee21264a
- SahuGGordonS WTarrM A 2012 Rsc Adv. 2 573 10.1039/c1ra00762a
- WuX-FSongH-YYoonJ-MYuY-TChenY-F 2009 Langmuir 25 6438 10.1021/la900035a
- ZhangNLiuSFuXXuY-J 2011 J. Phys. Chem. C 115 9136 10.1021/jp2009989
- LiJZengH C 2005 Angew. Chem., Int. Ed. 44 4342 10.1002/(ISSN)1521-3773
- LekeufackD DBrioudeAMoutiAAlauzunJ GStadelmannPColemanA WMieleP 2010 Chem. Commun. 46 4544 10.1039/c0cc00935k
- ChouK-SLinM-YWuH-H 2013 J. Taiwan Inst. Chem. Eng. 44 228 10.1016/j.jtice.2012.10.007
- YinHMaZChiMDaiS 2011 Catal. Today 160 87 10.1016/j.cattod.2010.05.013
- LiuBXuHZhangZ 2012 Catal. Commun. 26 159 10.1016/j.catcom.2012.05.013
- MitsudomeTMatobaMMizugakiTJitsukawaKKanedaK 2013 Chem.- Eur. J. 19 5255 10.1002/chem.201204160
- WiederN LCargnelloMBakhmutskyKMontiniTFornasieroPGorteR J 2011 J. Phys. Chem. C 115 915 10.1021/jp102965e
- YeungC M YTsangS C 2010 J. Mol. Catal. A-Chem. 322 17 10.1016/j.molcata.2010.02.001
- ZhuFChenGSunSSunX 2013 J. Mater. Chem. A 1 288 10.1039/c2ta00293k
- TripathyS KMishraAJhaS KWahabRAl-KhedhairyA A 2013 Anal. Methods 5 1456 10.1039/c3ay26549h
- YuY-TDuttaP 2011 J. Solid State Chem. 184 312 10.1016/j.jssc.2010.11.009
- YuY-TDuttaP 2011 Sensors and Actuators B 157 444 10.1016/j.snb.2011.04.088
- KuoC-HHuaT-EHuangM H 2009 J. Am. Chem. Soc. 131 17871 10.1021/ja9065333
- MeirNPlanteI J-LFlominKChocklerEMoshofskyBDiabMVolokhMMokariT 2013 J. Mater. Chem. A 1 1763 10.1039/c2ta00721e
- WangW-CLyuL-MHuangM H 2011 Chem. Mater. 23 2677 10.1021/cm200708q
- ZhangLBlomD AWangH 2011 Chem. Mater. 23 4587 10.1021/cm202078t