
Abstract
In recent decades, the substitution of non-renewable fossil resources by renewable biomass as a sustainable feedstock has been extensively investigated for the manufacture of high value-added products such as biofuels, commodity chemicals, and new bio-based materials such as bioplastics. Numerous solid catalyst systems for the effective conversion of biomass feedstocks into value-added chemicals and fuels have been developed. Solid catalysts are classified into four main groups with respect to their structures and substrate activation properties: (a) micro- and mesoporous materials, (b) metal oxides, (c) supported metal catalysts, and (d) sulfonated polymers. This review article focuses on the activation of substrates and/or reagents on the basis of groups (a)–(d), and the corresponding reaction mechanisms. In addition, recent progress in chemocatalytic processes for the production of five industrially important products (5-hydroxymethylfurfural, lactic acid, glyceraldehyde, 1,3-dihydroxyacetone, and furan-2,5-dicarboxylic acid) as bio-based plastic monomers and their intermediates is comprehensively summarized.
1. Introduction
1.1. Role of solid catalysts in green and sustainable chemistry
Although the petrochemical industry has contributed to worldwide economic development over the past century, many serious environmental problems still sometimes arise. Therefore, to establish environment-friendly chemical processes requires the development of novel and cost-effective approaches to pollution prevention. Green chemistry is one of the most attractive concepts for pollution prevention, as developed by Anastas and Warner in their 12 principles [Citation1]. A brief definition is that green chemistry reduces and/or eliminates the use or generation of hazardous substances in the design, manufacture, and application of chemical products. Advances in green chemistry will contribute to the abatement of obvious hazards associated with global issues such as climate change, energy production, availability of safe and adequate water supplies, food production, and the presence of toxic substances in the environment. The challenges faced to achieve sustainability will lead to new technologies that provide society with products that we can depend on in an environmentally responsible manner [Citation1–Citation4].
For the manufacture of chemicals (especially high value-added chemicals), antiquated stoichiometric technologies are still widely used, such as acid/base-catalyzed reactions with mineral acids (H2SO4, H3PO4, etc), Lewis acids (AlCl3, ZnCl2, etc), and inorganic bases (NaOH, KOH, etc), reduction with metals (Na, Mg, Fe, Zn) and metal hydrides (LiAlH4, NaBH4), and oxidation with permanganate or chromium(VI) reagents. However, these stoichiometric reagents cannot be recovered and recycled. Therefore, the use and generation of toxic and hazardous substances can be reduced by replacement of these stoichiometric methodologies with cleaner catalytic alternatives [Citation1–Citation7]. Many soluble homogeneous catalysts have been developed to date for the green synthesis of various chemicals. These catalysts are usually dissolved in reaction media, which makes all catalytic sites accessible to substrates, and exhibit high catalytic activity and selectivity. Despite these advantages, homogeneous catalysts have a share of only ca. 20% in industrial processes because catalyst/product(s) separation (i.e., product contamination) and reuse of expensive catalysts are very difficult to achieve efficiently and cost-effectively [Citation8]. In this context, the development of easily recoverable and recyclable solid catalysts has received particular interest for environment-friendly syntheses of high value-added chemicals.
As an example, the AlCl3- and zeolite-catalyzed Friedel–Crafts acylation of anisole are compared (figure ) [Citation7]. Zeolite-catalyzed acylation uses acetic anhydride as the acylating agent and requires no solvent. In contrast, the AlCl3-catalyzed process uses acetyl chloride and requires more than one equivalent of AlCl3 (the use of anhydride would require >2 equivalents of AlCl3) and a chlorinated hydrocarbon solvent. Thus, the zeolite-catalyzed process avoids the generation of HCl and the use of acetyl chloride for synthesis. In addition, the zeolite-catalyzed process has the following advantages: (i) the amount of aqueous effluent is >100 times less than that for the classical AlCl3-catalyzed process, (ii) a higher chemical yield of p-methoxy acetophenone is achieved, (iii) the catalyst is recyclable, and (iv) the number of unit operations is reduced from 12 to 2. In this context, heterogeneous catalysis can be expected to remain a cornerstone in building a sustainable chemical community through green chemistry [Citation8–Citation13].
Figure 1. Comparison of the AlCl3- and zeolite-catalyzed Friedel–Crafts acylation processes [Citation7].
![Figure 1. Comparison of the AlCl3- and zeolite-catalyzed Friedel–Crafts acylation processes [Citation7].](/cms/asset/05d33dfe-38ef-4f0e-ad21-a935a400f5dd/tsta_a_11661295_f0001_ob.jpg)
1.2. Biomass conversion into value-added chemicals and fuels
There has been continued focus on waste minimization and the avoidance of toxic and/or hazardous reagents and solvents, particularly in the fine chemicals and related industries, as discussed in section 1.1. In recent decades, the substitution of non-renewable fossil resources such as crude oil, coal, and natural gas by renewable biomass including lignocellulose and triglycerides as a sustainable feedstock has been extensively investigated for the manufacture of high value-added products such as biofuels, commodity chemicals, and new bio-based materials such as bioplastics [Citation14–Citation53]. Representative chemocatalytic processes for the manufacture of such high value-added products are summarized in figure .
Figure 2. Representative processes for biomass conversion into chemicals and fuels.
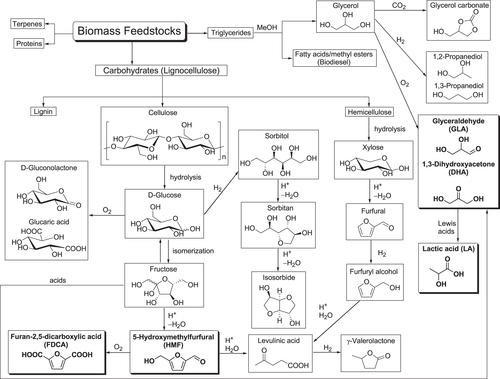
In contrast with edible biomass such as starches, sugars, and vegetable oils, the most abundant lignocellulose, the fibrous material that constitutes the cell walls of plants, is an inexpensive nonedible biomass that could be an excellent source of fuels and chemicals without affecting food supplies. Lignocellulose is composed of ca. 20% lignin, ca. 25% hemicellulose, and ca. 40% cellulose [Citation14–Citation32]. Lignin is a three-dimensional polyphenolic biopolymer with a non-uniform structure that imparts rigidity and recalcitrance to plant cell walls (figure ), but its selective conversion into chemically useful aromatic components remains a challenge [Citation40–Citation42]. The hemicellulose polymer is formed by pentose and hexoses (e.g., D-xylose, D-galactose, D-arabinose, D-glucose, and D-mannose), of which xylose is the most abundant [Citation38, Citation39]. Cellulose is also a polymer of glucose units linked by β-glycosidic bonds [Citation37]. While lignocellulose is abundant as a resource from plant material, its exploitation has been limited due to its composite nature and rigid structure. Therefore, pretreatment to liberate cellulose and hemicellulose from the lignin composite is required.
Figure 3. Schematic representation of a hardwood lignin structure and common linkages found in the lignin polymer [Citation40, Citation41].
![Figure 3. Schematic representation of a hardwood lignin structure and common linkages found in the lignin polymer [Citation40, Citation41].](/cms/asset/81a7a272-294f-48ce-95bb-cef18f85d646/tsta_a_11661295_f0003_ob.jpg)
The three main catalytic routes used to transform biomass into fuels and chemicals are gasification, pyrolysis and hydrolysis [Citation33–Citation36]. In this review article, we mainly focus on hydrolysis processes used to break the lignocellulose into its constituent parts, and hydrolysis-related processes. The cellulose and hemicellulose can be hydrolytically converted into their constituent building blocks, such as C5 and C6 monosaccharides, and these pentoses and hexoses can be subsequently used as raw materials for chemocatalytic conversion processes to biofuels or platform commodity chemicals such as polyols, furans, and acids (figure ) [Citation37]. Cellulose is hydrolyzed into glucose monomers under severe reaction conditions (i.e., at high reaction temperature with sulfuric acid as a catalyst). Furthermore, glucose can be converted into various useful chemicals by oxidation, hydrogenation, and dehydration. The oxidation of glucose affords gluconic acid, which is successfully oxidized to glucaric acid as a monomer for the preparation of biodegradable polyamides with unique properties. Glucose is hydrogenated with H2 to form sorbitol, followed by acid-catalyzed dehydration to sorbitan and subsequently isosorbide as an industrial monomer. Acid-catalyzed dehydration of fructose, which is formed by the isomerization of glucose, gives 5-hydroxymethylfurfural (HMF) as a raw material for the production of chemicals, polymers and biofuels [Citation52, Citation53]. However, to achieve commercially viable selectivity toward HMF is a challenge because of the propensity of HMF towards further reaction under the acidic reaction conditions. HMF is oxidized to furan-2,5-dicarboxylic acid (FDCA), which is a structural analogue of the terephthalic acid monomer. Polyethylene furandicarboxylate can be an alternative bio-based plastic to replace polyethylene terephthalate. Further reaction of HMF with water under acidic conditions yields levulinic acid with the concomitant formation of formic acid. Levulinic acid is an attractive platform chemical itself and subsequent hydrogenation affords γ-valerolactone, which is a potential building block for polyesters. A polymeric xylan, which is obtained from the hemicellulose fraction, can be depolymerized to form a xylose monomer through diluted acid hydrolysis (e.g., with sulfuric acid) [Citation38, Citation39]. Pentoses including xylose can also be converted to levulinic acid. This process involves the dehydration of xylose to furfural and subsequent hydrogenation to furfuryl alcohol, which is finally hydrolyzed to levulinic acid.
In addition to lignocellulose, conversion of triglyceride feedstocks (e.g., inedible plant oils, algal oil, or waste cooking oils and fats) into biodiesel and platform chemicals has attracted much attention as integrated triglyceride biorefinery technology [Citation43–Citation51]. Transesterification of triglycerides with methanol produces a mixture of glycerol and fatty acid methyl esters that can be commercially exploited as biodiesel, biolubricants, and biosurfactants. Glycerol can be converted to platform chemicals such as glycerol carbonate, propylene glycol, acrylic acid, and trioses (glyceraldehyde (GLA) and 1,3-dihydroxyacetone (DHA)). Trioses are also obtained by the retro-aldol fragmentation of fructose. In the presence of Lewis acids or bases, trioses are converted into lactic acid (LA) which is a bio-based commodity chemical with many applications, including the rapidly growing use of polylactate as a bioplastic.
Various types of chemicals can be obtained from renewable biomass as a sustainable feedstock. Recently, numerous solid catalyst systems for the effective conversion of biomass feedstocks into high value-added chemicals and fuels have been developed. Solid catalysts are classified into four main groups with respect to their structures and substrate activation properties: (a) micro- and mesoporous materials, (b) metal oxides, (c) supported metal catalysts, and (d) sulfonated polymers (figure ). This review article focuses on the activation of substrates and/or reagents on the basis of groups (a)–(d), including reaction mechanisms. In addition, recent progress in chemocatalytic processes for the production of five industrially important products (HMF, LA, GLA, DHA, and FDCA) as bio-based plastic monomers and their intermediates is comprehensively summarized. Details on the valorization of waste biomass by conversion to platform chemicals and fuels have been summarized in many excellent books and review articles [Citation14–Citation53]. Abbreviations of chemical compounds are summarized in table .
Figure 4. Examples of relevance to materials used in biomass conversion.
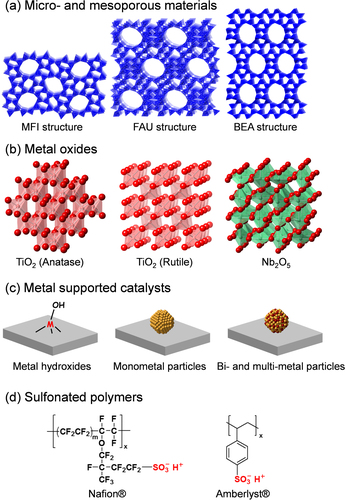
Table 1. Abbreviations of chemical compounds.
2. Solid catalysts for biomass conversion into value-added chemicals
In this section, we focus on the structures and chemical properties of the solid catalysts (a)–(d), and present catalytic processes for biomass conversion. Some reaction mechanisms, including the interaction between substrates and active sites, are also described. There are two main advantages of solid catalysts for liquid-phase reactions over homogeneous catalysts: (i) facile recovery from the reaction solution and (ii) reusability without significant loss of activity. The definitive test for heterogeneity is to filter the catalyst during the course of the reaction at the reaction temperature and allow the filtrate to react further. If the reaction mixture is first allowed to cool to ambient temperature before filtration, leached metal ion(s) can re-adsorb onto the catalyst [Citation54, Citation55]. Thus, the results for reuse experiments and leaching tests are also briefly summarized in tables – to discuss the heterogeneity and stability of each solid catalyst.
Table 2. Representative examples of the heterogeneously catalyzed formation of HMF from hexoses.
Table 3. Representative examples of the heterogeneously catalyzed formation of LA/methyl lactate from trioses.
Table 4. Representative examples of the heterogeneously catalyzed oxidation of HMF to FDCA with O2Footnotea.
Table 5. Representative examples of the heterogeneously catalyzed oxidation of glycerol to trioses with O2Footnotea.
2.1. Micro- and mesoporous materials
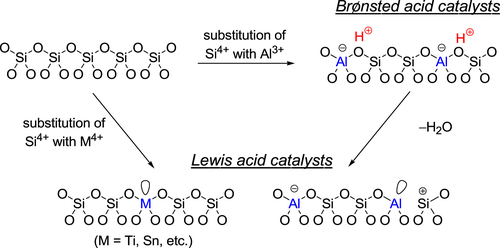
Zeolites are an important class of inorganic crystalline materials that have been widely used in petroleum refining, and in the petrochemical and fine chemical industries as catalysts, adsorbents, and ion-exchangers [Citation56–Citation85]. The zeolite framework has an ordered distribution of micropores with diameters typically less than 2 nm. In comparison with other microporous materials, the zeolite framework is built exclusively from TO4 tetrahedra (T denotes tetrahedrally coordinated Si, Al, or P). Each TO4 tetrahedron is connected with four neighbors by sharing their vertex O atoms, which forms the three-dimensional four-connected zeolite framework. Although all zeolites are constructed from TO4 tetrahedra, they can be connected in different ways, which leads to the rich variety of zeolite structures. Figure illustrates the mechanism for the formation of Br⊘nsted and Lewis acid sites within the zeolite framework. The substitution of Si atoms with trivalent Al atoms results in the formation of protons at the bridging O atoms (Si–OH–Al), and the evolved protons can act as Br⊘nsted acid sites. Acidic protons can be replaced by various metal cations (e.g., Na+, K+, Cs+, Mg2+, Ca2+, Ag+, Cu2+, Pd2+, Pt2+ etc) by ion-exchange. These metal ion-exchanged zeolites mainly work as base, oxidation, and hydrogenation catalysts depending on the nature of metal cations, and they are applied to a wide range of reactions including cracking of hydrocarbons, removal of nitrogen monoxide from emission of diesel engines, selective oxidation of methane and benzene, and synthesis of fine chemicals [Citation56–Citation85]. When Si atoms are replaced with high coordination number heteroatoms, the incorporated heteroatoms function as Lewis acid sites that can activate various nucleophiles. Some Lewis acid-doped zeolites exhibit remarkable activity, selectivity, and lifetime for multiple processes [Citation86–Citation88]. For instance, the development of TS-1 (a TiIV-doped MFI-type zeolite, figure (a)) is viewed as one of the greatest breakthroughs in sustainable chemistry in the last few decades, having resulted in a greener processes that include the epoxidation of propylene with hydrogen peroxide (H2O2) [Citation89]. In addition, Sn-β (SnIV-doped BEA-type zeolite, figure (a)) has shown unparalleled activity and selectivity for the isomerization of glucose to fructose and the Baeyer–Villiger oxidation of ketones to lactones with H2O2 as the green oxidant [Citation90–Citation92]. In this section, the acid-catalyzed (i) formation of lactate from trioses and (ii) conversion of hexoses over micro- and mesoporous materials are described.
Figure 5. Mechanism for the formation of Br⊘nsted and Lewis acid sites within the zeolite framework.
2.1.1. Micro- and mesoporous materials for the formation of lactate from trioses
LA is currently emerging as a building block for a new generation of materials such as biodegradable plastics and solvents [Citation93–Citation95]. These new materials can be produced from biomass-derived precursors and have the potential to replace existing petroleum-based materials due to comparable or even superior properties [Citation96]. LA also has the potential to become a central chemical feedstock in the chemical industry for the production of acrylic acid, propylene glycol, and various useful condensation products. Despite its high potential, the major obstacle to the wider implementation of LA-based materials and the use of an LA platform in the chemical industry is the high cost associated with the cumbersome manufacturing route of LA; large-scale production of LA relies on batch-wise fermentation of aqueous glucose under anaerobic conditions [Citation97–Citation102]. The fermentation reaction typically takes 2–4 days and requires continuous addition of calcium hydroxide to maintain a neutral pH level for optimal bacterial function, which results in the formation of calcium lactate. Crystallization of calcium lactate followed by acidification with sulfuric acid releases crude LA and gypsum. Typically, one ton of gypsum is formed for every ton of LA produced [Citation93]. Further purification of LA is performed by esterification to methyl lactate followed by distillation and hydrolysis to release pure LA. Therefore, alternative chemocatalytic approaches toward the production of LA and its derivatives from abundant carbohydrates are of significant interest.
The catalytic production of lactate from trioses (DHA and GLA) has been studied because the trioses can be obtained by aerobic oxidation of glycerol [Citation103–Citation106] or by fermentation using the Gluconobactor suboxydans strain [Citation107–Citation110]. Figure represents the generally accepted mechanism for the conversion of trioses into alkyl lactates or LA. The two triose isomers of DHA and GLA easily interconvert according to a ketose–aldose isomerization, which shifts the carbonyl between C1 and C2. This reaction is, in principle, performed via an acid-catalyzed hydride shift, via a base-catalyzed mechanism with a proton shift (and intermediate enol), or via a concerted proton-coupled hydride shift in neutral media. The latter isomerization was recently studied in the presence of heterogeneous Lewis acid (Sn) by Roman-Leshkov and co-workers for hexose monosaccharides [Citation90, Citation111, Citation112] and by Assary and Curtiss for trioses [Citation113]. While LA formation from trioses starts with a dehydration step, it is still controversial as to whether DHA or GLA is the ultimate dehydrating substrate. Although the reaction rates are different, depending on the use of DHA or ALD, the conversion generally works well from both substrates. This is probably due to the rapid equilibrium between the two triose isomers. The dehydration reaction forms pyruvaldehyde (PAL) via an enetriol intermediate and enol pyruvate. PAL is prone to nucleophilic attack by water or an alcohol at the carbonyl carbon atom of the aldehyde to form a hydrate or hemiacetal, respectively. In alcoholic media, the hemiacetal can further react in two ways; the formation of a dialkyl acetal (in equilibrium with the hemiacetal) by reaction with another alcohol molecule or isomerization into an alkyl lactate. In water, only the latter isomerization to LA is possible. The isomerization and nucleophilic attack may also be concerted without the formation of intermediates. The isomerization step assumes a formal 1,2-hydride shift, which has been explained as an internal Cannizzaro reaction from isotope labeling experiments [Citation114] or as a combination of Meerwein–Ponndorf–Verley (MPV) reduction and Oppenauer oxidation [Citation115]. Both are formally indistinguishable, but either way, it was confirmed that the H atom of the aldehyde in PAL shifts to the adjacent carbon atom.
Figure 6. Reaction pathways for the conversion of trioses into lactate (alkyl lactate or LA) catalyzed by a Lewis acid catalyst [Citation113].
![Figure 6. Reaction pathways for the conversion of trioses into lactate (alkyl lactate or LA) catalyzed by a Lewis acid catalyst [Citation113].](/cms/asset/95e10384-2121-4d4f-8d02-4568af3c4390/tsta_a_11661295_f0006_ob.jpg)
Two research groups independently investigated the use of H-USY zeolites (FAU-type structure, figure (a)) for the production of LA from trioses [Citation114, Citation116]. They performed a screening with H-USY, H-ZSM-5, H-BEA and H-MOR along with a sulfated zirconia and H-montmorillonite, both in water and methanol. The H-USY zeolite with a bulk Si/Al ratio of 6 (H-USY(6)) outperformed all the other catalysts and also the other H-USY with a Si/Al ratio of 30 (H-USY(30)). Methyl lactate was obtained in 96% yield. Fourier transform-infrared spectroscopy (FTIR), using pyridine as a probe for acidity, combined with NH3-temperature programmed desorption (TPD) measurements showed that the ratio of Br⊘nsted over Lewis acid sites were 1.8 and 5.6 for H-USY(6) and H-USY(30), respectively. Working in water generally entailed lower rates, lower yields and also lower lactate selectivity. In addition, the zeolite catalyst in a promising continuous flow reaction setup was studied to evaluate the stability of the zeolite and its deactivation process. It was determined that LA destroys the catalyst structure, even at a concentration as low as 0.3 M. As expected, this is not observed for methyl lactate in methanol. Another cause for the deactivation of zeolite in water is catalyst coking, which is mainly attributed to PAL decomposition. An additional kinetic study with H-USY(6) in water revealed activation energy barriers of 53 and 61 kJ mol−1 for DHA dehydration and PAL to LA reaction, respectively. These values are clearly lower than those obtained with soluble Al3+ salts [Citation115], while the rate-determining step is reversed, the hydride shift being the slowest step with the H-USY(6) zeolite.
Taarning and co-workers proved that particular Lewis acidic β-zeolites (BEA structure, figure (a)) were very active for DHA conversion [Citation115]. A comparison of a series of zeolites with Ti-, Zr-, and Sn incorporated within the framework revealed a correlation between the catalytic activity and Lewis acid strength, with Sn being the strongest among the series. From a comparison of Al-β with a Br⊘nsted acidic ion-exchange resin yielding only dialkyl acetals, the authors also postulated that strong Br⊘nsted acidic zeolites are selective towards acetals, whereas Lewis acidic zeolites are selective towards alkyl lactates. The initial turnover rate (per Sn site) of methyl lactate formation with the Sn-β zeolite (45 mol Sn mol–1 h–1) was higher than that of homogeneous SnCl4·5H2O (4.2 mol Sn mol–1 h–1). Interestingly, steaming a parent Al-β zeolite to produce extra-framework Al enhanced the lactate yield with respect to the pristine H-Al-β [Citation115]. However, the elaborate and time consuming synthesis procedure for Sn-β zeolite could hamper its large scale application. Alternative Sn-based catalysts or alternative synthesis routes to Sn-β, as very recently reported by Hermans and co-workers, may offer advantages in this respect [Citation117].
Bifunctional catalysis for alkyl lactate synthesis from trioses has recently been reported by de Clippel et al [Citation118]. Based on their previous work [Citation119–Citation121], they proposed a simple carbon–silica composite design to independently alter the number of Br⊘nsted acid sites from that of the Lewis acid sites (figure ) [Citation118]. Lewis acidity was imparted by grafting a mesoporous silica (MCM-41) with isolated SnIV. Br⊘nsted acidity was introduced by polymerizing furfuryl alcohol in the mesopores followed by pyrolysis of the polymer to an active carbon phase. Oxygen-containing functional groups such as carboxylic acids and phenols acted as weak Br⊘nsted acid sites, as ascertained by the solid-state 31P magic angle spinning nuclear magnetic resonance (MAS-NMR) spectroscopy. The density of acid sites was controlled by the carbon loading, the degree of oxidation (dependent on the pyrolysis temperature) and an optional post-synthesis oxidation. Quantitative DHA conversion into ethyl lactate was achieved in ethanol with a composite containing 15% carbon that was pyrolyzed at 773 K and post-treated at 573 K under oxygen flow. This composite achieved a 7-fold increase of the initial turnover frequency (TOF) with respect to the parent Sn-MCM-41. The Sn atom is crucial for the selectivity by facilitating the 1, 2-hydride shift of PAL; however, it does not determine the reaction rate. The acid sites at the carbon surface accelerate the rate-determining dehydration step of DHA to PAL. Therefore, the catalytic potential of Sn in this reaction is evident when sufficient Br⊘nsted acidity is available in the closed environment. Catalyst regeneration for these composites was demonstrated by the observation of constant conversion and selectivity over three consecutive runs in ethanol. However, the importance of the spatial organization of the two catalytic sites was not examined in this study. The conversion of trioses into LA in water was also successful and a similar bifunctional catalysis explains the observed chemistry. However, the use of water entailed a decrease of activity in consecutive runs, due to deactivation (by carbonaceous deposits) of the catalyst, which is consistent with earlier reports for other catalyst types in water [Citation116]. The composite was even capable of working in long-chain alcoholic media to afford long chain lactates in one step with high yields and interesting (e.g., solvent) properties [Citation118].
Figure 7. Schematic representation of the structure of carbon-deposited and Sn-incorporated mesoporous silica catalyst at various scale lengths [Citation118].
![Figure 7. Schematic representation of the structure of carbon-deposited and Sn-incorporated mesoporous silica catalyst at various scale lengths [Citation118].](/cms/asset/97d1f28c-0ab6-476f-9f52-6702eb37ece3/tsta_a_11661295_f0007_oc.jpg)
2.1.2. Micro- and mesoporous materials for conversion of hexoses
The isomerization of glucose into fructose is an important industrial reaction used mainly for the production of high-fructose corn syrup [Citation122]. In recent years, glucose isomerization has played a crucial role in the synthesis of biomass-derived chemical platforms used for the production of fuels and chemicals [Citation123]. The reaction is equilibrium-limited (Keq ≈ 1 at 298 K), slightly endothermic (ΔHr = 3 kJ mol–1), and typically catalyzed by an immobilized enzyme (xylose isomerase) [Citation124]. Equilibrium mixtures often yield a product distribution of approximately 42% (w/w) fructose, 50% (w/w) glucose, and 8% (w/w) other saccharides [Citation122]. Although fructose yields are high, the window of operation for this enzyme is very narrow and requires strict control over reactant purity, reaction temperature, and solvent pH. This significantly limits the cost-effective coupling of glucose isomerization with upstream and downstream biomass processing schemes such as cellulose hydrolysis and carbohydrate dehydration. In this respect, a robust heterogeneous inorganic catalyst would have clear advantages over the biological system.
Zeolites with the beta topology that contain tin (Sn-β) or titanium (Ti-β) metal centers in the framework are highly active catalysts for the glucose isomerization reaction in aqueous media. A 10 wt% glucose solution contacted with a catalytic amount of Sn-β at 383 K generated glucose, fructose, and mannose with respective yields of 46, 31, and 9% [Citation90]. Similar product yields were achieved for glucose solutions with concentrations up to 45 wt%. Notably, the Sn-β catalyst exhibited high stability in that it did not show signs of deactivation after multiple cycles or after calcination, there was no leaching of Sn detected by elemental analysis; and a hot filtration test showed that the catalysis occurred heterogeneously. Most importantly, the Sn-β catalyst was able to promote the isomerization reaction in highly acidic aqueous environments with retention of activity and product distribution as in media without added acid. This feature enables Sn-β to couple isomerization with other acid-catalyzed reactions. The zeolite topology and the nature of the tin site were shown to significantly influence the catalytic activity. The isomerization reaction did not proceed with a medium pore zeolite (MFI structure, figure (a)), most likely because glucose molecules are not able to enter the smaller pores. Mesoporous stannosilicates (e.g., Sn-MCM-41) were active, while their activities were considerably lower than that of Sn-β. The reaction did not proceed when using SnO2, SnCl4, or SnO2-β (created by the incorporation of SnO2 nanoparticles into the pores of zeolite beta) as catalysts [Citation90, Citation111].
These results indicate that isolated tin sites tetrahedrally coordinated to the crystalline zeolite framework are necessary for catalyzing the isomerization of glucose in aqueous media and that the degree of hydrophobicity surrounding the active sites is likely an important parameter to achieve the desired reactivity. A detailed NMR study revealed that Sn-β acts as a true Lewis acid during the isomerization of glucose in water [Citation111]. Isotopically labeled glucose molecules were used to show that glucose isomerization with Sn-β proceeds via intramolecular hydride shift. This hydride shift pathway is similar to that observed in MPV reactions mediated by Lewis acids involving a six-membered transition state between the metal center, the carbonyl group, and the hydroxyl group in the sugar (figure ) [Citation111, Citation125]. In contrast, a similar spectroscopic study performed with labeled glucose and NaOH showed that the reaction proceeds via proton abstraction and an enolization pathway that is typically observed in base-mediated isomerization (figure ) [Citation111]. Replacing water with methanol as the solvent resulted in no isomerization activity for Sn-β, which is unexpected given that most Lewis acid catalysts perform better in the absence of water.
Figure 8. Possible reaction mechanism for Lewis acid (Sn-β) or Br⊘nsted base (NaOH) catalyzed transformation of glucose into fructose [Citation111, Citation125].
![Figure 8. Possible reaction mechanism for Lewis acid (Sn-β) or Br⊘nsted base (NaOH) catalyzed transformation of glucose into fructose [Citation111, Citation125].](/cms/asset/a9a72a90-8aef-4790-9548-ce7766c8a30e/tsta_a_11661295_f0008_ob.jpg)
Nikolla et al published promising results for glucose dehydration with a bifunctional catalyst system of tin and titanium β-zeolites in combination with HCl [Citation126]. An optimum HMF yield of 57% at 79% conversion was obtained from a 10 wt% glucose solution in 26 wt% aqueous NaCl solution at pH = 1 and in the presence of 0.5 wt% Sn-β and 3 equivalents (v/v) of tetrahydrofuran (THF) as an extraction solvent for 70 min at 453 K. When Ti-β was used, the yield was 53% at 76% conversion for 105 min. Sn-β maintained its activity, even in the presence of aqueous phases saturated with chloride salts, such as NaCl. In this case, the specific combination of Lewis and Br⊘nsted acid catalysts was shown to benefit glucose conversion toward HMF by efficiently performing a cascade reaction without excessive by-product formation. Dealuminated beta zeolite also could act as an effective bifunctional catalyst for direct transformation of glucose to HMF [Citation127].
2.2. Metal oxides
Some metal oxides and phosphates have both Br⊘nsted and/or Lewis acid sites on the surface that can be catalytically active for various organic reactions [Citation128, Citation129]. Representative examples are metal oxides and phosphates consisting of group IV and V elements. Niobic acid (Nb2O5·nH2O) is the most convenient precursor for the preparation of niobium pentoxide (Nb2O5, figure (b)), which is obtained upon complete dehydroxylation [Citation130–Citation133]. The presence of labile protons in Nb2O5·nH2O is responsible for its strong acidity, which corresponds to a 70% H2SO4 solution. Contrary to that observed with most acidic metal oxides, its acidity decreases with increasing pretreatment temperature [Citation131, Citation132]. The dehydration of Nb2O5·nH2O occurs around 363–453 K, as revealed by thermogravimetric measurements in combination with differential thermal analysis [Citation131]. Upon complete dehydroxylation (calcination above 773 K), niobium pentoxide exhibits very weak acidity, accompanied by a significant loss in surface area and a change from the amorphous to crystalline form. The Br⊘nsted acidity of Nb2O5·nH2O is very high, comparable to that of protonic zeolites according to 1H NMR results, and turns toward Lewis acidity following water elimination [Citation131, Citation132]. Metal phosphates are well known for their acid properties, thus making them promising acid catalysts [Citation128]. They are characterized by polymeric metal(IV) phosphates with layered structures, each layer consisting in a plane of tetravalent metal atoms sandwiched between planes of different hydrogen phosphate/phosphate species [Citation134, Citation135], which contain both Br⊘nsted and Lewis acid sites [Citation136, Citation137], the nature and concentration of which result is strictly related to the phase structures and thermal treatment history. In particular, zirconium and titanium phosphates belonging to this class of compounds have been used as acid catalysts for the dehydration of alcohols [Citation138, Citation139], isomerization of olefins [Citation140–Citation143] and the rearrangement of terpenes [Citation144].
Layered cubic-zirconium- and γ-titanium-phosphates [Citation145–Citation147] were used as acid catalysts in the dehydration of fructose to HMF in water [Citation148]. When cubic zirconium pyrophosphate was employed in fructose dehydration, 86% HMF selectivity at 52% fructose conversion was obtained at 373 K for 1 h. Under analogous conditions, γ-titanium phosphate also exhibited promising performance (89% HMF selectivity at 89% fructose conversion). When the reaction products were extracted and the catalysts recycled, the performance was retained up to very high conversion.
Phosphate-immobilized niobium oxide (phosphate/Nb2O5, figure (a)) has been reported to be an effective catalyst for direct HMF formation from glucose in water [Citation149]. Phosphate/Nb2O5 produced HMF with 92% conversion and 52% selectivity, and no decrease in activity was observed even after several reuses of the catalyst. There is no difference in HMF formation between Nb2O5 and sodium-immobilized niobium oxide (Na+/Nb2O5) without Br⊘nsted acid sites, which indicates that HMF formation over Nb2O5 does not proceed on the Br⊘nsted acid sites. FTIR and Raman spectroscopies were used to investigate the Lewis acid sites on Nb2O5 in the presence of water, which indicated that NbO4 tetrahedra (Lewis acid sites) on Nb2O5 particle surfaces immediately form NbO4–H2O adducts in the presence of water. However, some adducts can still function as effective Lewis acid sites that catalyze the glucose to HMF transformation. Thus, some NbO4 tetrahedra present in insoluble Nb2O5 can act as Lewis acids, even in water (figure (a)).
Figure 9. Representative structure of Lewis acid center on (a) phosphate/Nb2O5·nH2O and (b) phosphate/TiO2 [Citation149, Citation152].
![Figure 9. Representative structure of Lewis acid center on (a) phosphate/Nb2O5·nH2O and (b) phosphate/TiO2 [Citation149, Citation152].](/cms/asset/7d3bd556-c6af-4b1b-bee1-0ff1f79d738f/tsta_a_11661295_f0009_oc.jpg)
Many metal oxides of the group 4 and 5 elements, including Nb2O5, are composed mainly of octahedral MO6 (M: metal) units with saturated coordination spheres (figure (a)), and polyhedral MOx with unsaturated coordination spheres, such as tetrahedral MO4, also present on the surface. Unsaturated coordination MO4 tetrahedra act as Lewis acids, although MO4 species are considered to not function as effectively in water as other Lewis acids. NbO4 species in Nb2O5 acting as water-tolerant Lewis acid sites suggests that ubiquitous anatase TiO2 (figure (b)) with TiO4 species on the surface would also function as an insoluble, easily separable, and water-tolerant Lewis acid catalyst [Citation149–Citation152]. However, bare anatase TiO2 cannot act as an efficient heterogeneous catalyst for the selective transformation of glucose into HMF, which requires selective isomerization of glucose into fructose and intramolecular dehydration of fructose, because of intermolecular side reactions [Citation152]. In contrast, TiO2 modified with H3PO4 (phosphate/TiO2, figure (b)), where OH groups on TiO2 are esterified into O-PO(OH)2 by phosphoric acid, exhibits high HMF yield (ca. 80%) in a THF–water mixture [Citation152]. Such a high HMF yield can be achieved even with a diluted glucose solution (ca. 1 wt%) and high catalyst/glucose ratio (50/20 wt%). The original activity was maintained for subsequent reactions, which demonstrates that phosphate/TiO2 can function as a stable and reusable heterogeneous catalyst for HMF production. Self-assembled mesoporous TiO2 nanoparticulate material with well-defined nanospherical morphologies also efficiently catalyzed the dehydration of D-fructose and D-glucose into HMF in dimethylacetamide-LiCl solvent under microwave assisted heating [Citation153].
2.3. Supported metal catalysts
Supported metal catalysts are one of the most important catalysts that have been utilized for many large-scale processes, including petroleum refining, vehicle exhaust gas scrubbing, and chemicals syntheses such as hydrogenation and oxidation [Citation154–Citation156]. Recently, catalysis by metal nanoparticles (especially Au) has attracted much global attention from researchers after reports by the Haruta group on the exceptional performance of Au nanoparticles supported on TiO2 for CO oxidation [Citation157–Citation163]. Control of the nanoparticle size, shape, and dispersity generally has a major effect on the reactivity and selectivity of these catalysts. In addition, the immobilization of metallic nanoparticles on a catalytically inert metal-oxide support often results in a dominant impact on the activity due to strong metal–support interactions that influence the electronic charge of the metallic catalysts. Not only monometallic nanoparticles, but also bi- and multi-metallic nanoparticles, have well investigated for many catalytic reactions, due to their beneficial catalytic properties that are typically different from those of the constituent metals, which result from electronic and/or geometric synergistic effects [Citation164–Citation167].
Much attention has been paid to the development of catalytic oxidation systems, and the choice of oxidant determines their practicability and efficiency. While many oxidants have been extensively investigated for catalytic liquid-phase oxidation processes, H2O2 and molecular oxygen (O2) are regarded as green oxidants due to their high content of active oxygen species, high atom efficiency, and production of only water as a by-product [Citation89, Citation168–Citation170]. The ideal system for greener and cleaner oxidation employs O2 together with a recyclable solid catalyst in a nontoxic solvent. In this section, liquid-phase oxidation (mainly in water) of (i) HMF to FDCA and (ii) glycerol to trioses, three-carbon acids, and two-carbon acids with O2 over supported metal catalysts are described.
Both oxidation processes consist of two important reactions steps, (i) the oxidation of alcohols to carbonyl compounds such as aldehydes and/or ketones and (ii) subsequent oxidation of aldehydes to carboxylic acids. The proposed reaction mechanism for the oxidation of alcohols into carbonyl compounds over supported metal catalysts is shown in figure [Citation171]. The oxidation likely proceeds in three steps, the first of which involves adsorption of alcohol onto the metal surface to give an adsorbed metal alkoxide. Secondly, β-hydride elimination proceeds to produce a carbonyl species and a metal hydride. Lastly, the metal-hydride is oxidized by O2 to regenerate the metal surface. The oxidation of an aldehyde to carboxylic acid has been proposed to proceed through a geminal diol intermediate that is formed by the reaction of an aldehyde with water.
Figure 10. Proposed mechanism for the oxidation of a primary alcohol and successive oxidation of the corresponding aldehyde over a supported metal catalyst with O2 [Citation171].
![Figure 10. Proposed mechanism for the oxidation of a primary alcohol and successive oxidation of the corresponding aldehyde over a supported metal catalyst with O2 [Citation171].](/cms/asset/d22576e2-ee2a-433b-a11b-172de017a4e1/tsta_a_11661295_f0010_ob.jpg)
2.3.1. Supported metal catalysts for oxidation of HMF to FDCA
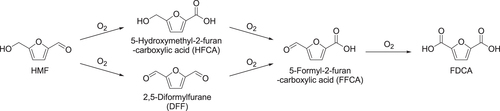
To achieve highly selective oxidation of HMF into FDCA, the oxidation of two functional groups (aldehyde and alcohol) is required, the reaction pathways of which are shown in figure . The conversion of HMF into 5-formyl-2-furan-carboxylic acid (FFCA) proceeds via two possible intermediates of 2,5-diformylfurane (DFF; alcohol oxidation product) and 5-hydroxymethyl-2-furan-carboxylic acid (HFCA; aldehyde oxidation product), and the pathways are dependent on reaction conditions such as the type of metal and additives and the additive concentration (table [Citation172–Citation184]). Supported Ru(OH)x species are active for the aerobic oxidation of alcohols to carbonyl compounds [Citation172]; thus, Riisager and co-workers reported HMF oxidation in water over Ru(OH)x on various supports and under base-free conditions [Citation173–Citation175]. Among these catalysts, Ru(OH)x/CeO2 and Ru(OH)x/MgAl2O4 gave moderate yields of FDCA (38–56%) and could be reused 3–4 times without a significant loss of activity. The competitive formation of two intermediates (DFF and HFCA) was proposed from the reaction profiles. In contrast with HMF oxidation in water, the reaction in ionic liquids over Ru(OH)x/La2O3 showed significant leaching of Ru species out of the support and into the reaction solution [Citation175].
Figure 11. Reaction pathways for the catalytic oxidation of HMF to FDCA with O2.
The oxidation of HMF in water over supported monometallic nanoparticles such as Au, Pt, and Pd in the presence of 2–4 equivalents of NaOH with respect to HMF has been investigated [Citation176, Citation177]. The base additive is required because (i) the base can facilitate dehydrogenation of hydroxyl groups and (ii) the amount of carboxylic acid adsorbed on the metal (especially Au) surface can be reduced. The reaction pathway under basic conditions has been proposed via HFCA and it is generally accepted that the alcohol oxidation step is slow in this reaction [Citation171, Citation176]. Davis et al reported that the catalytic activity of Au/C was much higher than that for Pd/C and Pt/C, while the selectivity toward FDCA with Au/C (8%) was much lower than that with Pd/C and Pt/C (71–79%) [Citation177]. The selectivity of supported Au catalysts toward FDCA can be increased to >99% by changing the carbon support to CeO2, TiO2, or HY zeolite [Citation176, Citation180]. Corma and co-workers reported the efficient oxidation of HMF to FDCA over Au nanoparticles (3.5 nm) supported on CeO2; however, the catalytic activity of the recovered catalysts decreased under identical conditions [Citation176]. Xu and co-workers reported that Au nanoclusters (1 nm) encapsulated within a HY supercage had higher catalytic activity than Au on other supports (TiO2, CeO2, Mg(OH)2, ZSM-5, and H-MOR) for the selective oxidation of HMF into FDCA [Citation180]. The acidic OH groups inside the supercage can stabilize the Au nanoclusters and promote reaction activity by strong interaction between gold and the hydroxyl groups.
The types of additives, metal species, and supports have a strong influence on the selectivity for HMF oxidation. In the case of Au/TiO2, FFCA could be selectively obtained with 78% yield using trifluoroacetic acid (HTFA) as an additive instead of NaOH [Citation179]. In contrast with supported Au, Pt, and Pd catalysts, the Ag-OMS-2 catalyst selectively gave DFF in 99% yield [Citation182]. Investigation on the effect of Ag substitution in K-OMS-2 by temperature-programmed reduction and NH3/CO2-TPD analyses showed that the addition of Ag decreased the reduction temperature of K-OMS-2 and acidic sites that cause side reactions of HMF to undesired products resulted in high catalytic activity and selectivity toward DFF.
Bimetallic catalysts such as Au–Cu, Au–Pd, and Pt–Bi exhibit superior activity than the corresponding monometallic counterparts [Citation183–Citation185]. Hutchings and co-workers reported that well defined Au–Cu alloy nanoparticles (4.4 nm) supported on anatase TiO2 exhibited much higher activity and stability for HMF oxidation than their monometallic counterparts (i.e., Au/TiO2 and Cu/TiO2), possibly due to Au site isolation effects caused by alloying [Citation183]. In addition, the Au–Cu/TiO2 catalyst could be easily recovered and reused without significant leaching or agglomeration of the metal nanoparticles. Similar effects of alloying on the activities and stabilities of Au8Pd2/AC and Pt–Bi/C have been reported [Citation184, Citation185].
The base-free oxidation of HMF to FDCA has attracted considerable attention because HMF is not stable under basic conditions. However, most catalytic systems showed very low FDCA yields in the absence of a base, and thus highly efficient oxidation of HMF to FDCA under base-free conditions is a challenging research subject. Ebitani and co-workers reported that Au nanoparticles (3.2 nm) supported on basic hydrotalcite catalysts gave an excellent yield of FDCA without the addition of a homogeneous base such as NaOH and NaHCO3 [Citation178]. A subsequent study by Riisager and co-workers showed that Mg2+ ions leached out of hydrotalcite (i.e., hydrotalcite acted as a stoichiometric base) during the oxidation reaction [Citation174]. Recently, Wang and co-workers reported the stability and efficiency of Au–Pd nanoparticles (2–3 nm) supported on carbon nanotubes (CNTs) for the aerobic oxidation of HMF to FDCA in water without the addition of a base [Citation186]. The functionalization of CNT surfaces plays an important role in the formation of FDCA, and CNTs that contain more carbonyl/quinone and less carboxyl groups favor FDCA formation by enhancing the adsorption of reactants and reaction intermediates.
2.3.2. Supported metal catalysts for formation of trioses from glycerol
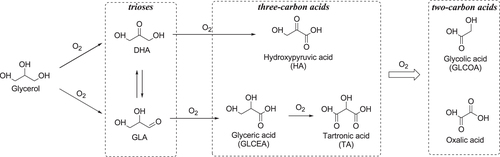
Glycerol is a major by-product of biodiesel synthesis through the transesterification of triglycerides with methanol, as discussed in section 1.2 [Citation43–Citation51]. Glycerol oxidation to produce high value-added chemicals including trioses (DHA and GLA) and three-carbon acids (glyceric acid (GLCEA) and tartronic acid (TA)) could help biodiesel economics become more competitive with those of diesel produced from non-renewable resources. The possible reaction pathways for the oxidation of glycerol are shown in figure . Glycerol has two primary alcohols and one secondary alcohol; therefore, several oxygenated products are formed via multiple pathways. Oxidation of a primary or secondary alcohol in glycerol produces GLA or DHA, respectively, and these two products are in equilibrium when in aqueous solution, depending on the pH value. The sequential oxidation of GLA and DHA gives three-carbon acids such as hydroxypyruvic acid (HA), GLCEA, and TA, with successive oxidation of the three-carbon acids to C–C bond cleaved two-carbon acids such as glycolic acid (GLCOA) and oxalic acid. While many research groups have reported the oxidation of glycerol to carboxylic acids (e.g., HA, GLCEA, GLCOA) with O2 over heterogeneous catalysts, the selective oxidation of glycerol to carbonyl compounds (especially for GLA) is still limited (table [Citation187–Citation199]). In this section, several examples of the selective oxidation of glycerol to trioses with O2 over supported metal catalysts are described.
Figure 12. Reaction pathways for the catalytic oxidation of glycerol with O2.
In the presence of 1–2 equivalents of NaOH with respect to glycerol, selective oxidation of glycerol over supported Pd, Pt, and Au nanoparticles was investigated. Hutchings and co-workers investigated the oxidation of glycerol at 333 K with 0.3 MPa of O2 over Pd, Pt, and Au nanoparticles supported on graphite and carbon [Citation187]. In all cases, GLCEA was the main product with very little GLA formed. The Pd/C and Pt/C catalysts gave respective GLA yields of 4 and 13%. Among the catalysts, Au/graphite or Au/activated carbon showed 100% selectivity to GLCEA with higher conversion than the Pt and Pd catalysts (70–97% selectivity to GLCEA). It has been proposed that NaOH aids initial dehydrogenation via H-abstraction of one of the primary OH groups of glycerol. An investigation on the effect of the support on glycerol oxidation over supported Au catalysts showed that a carbon support was the best [Citation188] and that the oxygen-free carbon supports would influence electron mobility to and from the gold surface, which probably promotes both adsorption and the regeneration of hydroxide ions to result in high catalytic performance.
While the Au/C catalyst was inactive for glycerol oxidation in the absence of NaOH [Citation187], the supported Pt catalysts were active under base-free conditions. Zhaoyin and co-workers reported that Pd nanoparticles (3.1 nm) supported on carbon achieved a GLCEA yield of 47% and that the catalyst was reused six times without significant loss of catalytic activity [Citation190]. The GLCEA yield could be increased to 56–62% by changing the carbon support to multi-wall carbon nanotubes (MWCNTs) [Citation191], sulfur-treated multi-wall carbon nanotubes (S-MWCNTs) [Citation192], and gas-phase sulfonated mesoporous polydivinylbenzene (SPDVB) [Citation193], while the catalytic activity or leaching of the Pt species out of the support could be decreased.
In a similar way to HMF oxidation to FDAC, bi- and multi-metallic catalysts such as Pt–Cu [Citation196] and Au–Pd–Pt [Citation199] exhibit superior activity than the corresponding monometallic counterparts. Hutchings and co-workers reported that the trimetallic Au–Pd–Pt/TiO2 was formed by the addition of Au to the best bimetallic catalyst, Pd–Pt/TiO2. [Citation199] The TOF of the trimetallic Au–Pd–Pt/TiO2 was 378 h–1 and this value was 1.8 times larger than that for the bimetallic Pd–Pt/TiO2 catalyst (210 h–1) with retention of the selectivity towards C3 products. However, the Au–Pd–Pt/TiO2 catalyst was not reusable and small amounts of metal were leached, and significant particle agglomeration was observed.
Not only catalytic activity but also chemoselectivity could be controlled by alloying. Bimetallic catalysts of Pt–Bi/C [Citation195], PtSb/MWCNTs [Citation197], and Pd–Ag/C [Citation198] showed much higher chemoselectivity toward DHA than Pt/C and Pd/C. Kimura et al reported that the Pt–Bi catalysts had high selectivity (ca. 50% at 20% glycerol conversion) toward DHA in a semibatch reactor [Citation103], and the highest DHA yield of 48% at 80% conversion was reported by Varma and co-workers [Citation195]. Hirasawa et al reported that Pd–Ag/C showed higher selectivity toward DHA and higher activity than Pd/C, where the DHA yield reached 44% at 52% glycerol conversion over Pd–Ag/C (Ag/Pd = 1) [Citation198]. The Pd–Ag alloy phase is required to achieve high activity and selectivity toward DHA. The mechanistic investigation suggested that the terminal OH group of glycerol is adsorbed on the Ag site and the neighboring secondary OH group (CH–OH) is attacked by an oxygen species that is dissociatively adsorbed on the Pd site [Citation200].
2.4. Sulfonated polymers
Commercially available polymeric ion-exchange resins have been used for a range of industrially important transformations [Citation201]. The two main classes of ion-exchange resins are based on styrene-based sulfonic acids (Amberlyst® and Dow type resins, figure (d)), which exhibit very high activity for esterification and etherification, and the perfluorosulfonic acid-based catalysts, including the recently developed Nafion® resin/silica nanocomposites [Citation202–Citation206]. Nafion® resin is a perfluorinated resin-sulfonic acid and is a copolymer derived from tetrafluoroethylene and perfluoro-2-(fluorosulfonylethoxy)propyl vinyl ether. After hydrolysis of the sulfonyl fluoride, it yields the strongly acidic terminal -CF2CF2SO3H group (figure (d)) [Citation201, Citation202]. These resins have very high activity for the formation of linear alkyl benzene, isomerization, and some select acylation type reactions [Citation201–Citation206]. Typical Br⊘nsted acids including these resin catalysts are not applicable to various sugar conversion reactions, because Lewis acid catalysts exhibit much higher catalytic performance for hydride transfer (glucose–fructose and PAL–LA isomerization step) in water than the Br⊘nsted acids [Citation207–Citation209]. In addition, Br⊘nsted acids can decompose HMF into two organic acids (levulinic acid and formic acid) in water, thus requiring continuous extraction of HMF from the reaction solution by an organic solvent or the stabilization of HMF by an appropriate solvent toward subsequent acid-catalyzed decomposition [Citation207–Citation209]. In this section, two representative examples of Br⊘nsted-acid catalyzed HMF formation from fructose are briefly described.
The catalytic dehydration of fructose into HMF by microwave heating was studied in acetone–water mixtures in the presence of a cation-exchange resin (Dowex 50wx8-100) catalyst [Citation210]. The reaction in acetone–water reaction media at 423 K gave HMF in 73% yield at 94% conversion. No decrease of catalytic activity or selectivity was observed for five reuses of the resin, as determined by elemental analysis, which showed that sulfonic acid groups attached to the resin were stable under the experimental conditions. The comparison between conventional heating and microwave heating revealed that the latter has a significant acceleration effect, not only on fructose conversion, but also on the HMF yield. Fructose conversion and HMF yields (92 and 70%, respectively) by microwave heating were higher than those by sand bath heating (22 and 14% respectively). Shimizu et al developed a simple method for the effective production of HMF from fructose with various solid acid catalysts in dimethyl sulfoxide (DMSO), that was based on the continuous removal of water from the reaction mixture under reduced pressure at 0.97 × 105 Pa [Citation211]. The removal of water was effective for suppression of two undesired reactions; (i) the hydrolysis of HMF to levulinic acid and (ii) the reaction of partially dehydrated intermediates to condensation products. Nafion® resin and Amberlyst-15 gave high HMF selectivity with complete fructose conversion by the continuous removal of water from the reaction system.
3. Conclusions and future opportunities
Renewable biomass has attracted significant attention as a sustainable feedstock, and solid catalysts play an important role in the achievement of chemocatalytic processes for the manufacture of high value-added products such as biofuels, commodity chemicals, and new bio-based materials such as bioplastics. Various types of solid catalysts including micro- and mesoporous materials, metal oxides, supported metal catalysts, and sulfonated polymers can catalyze the conversion of biomass feedstocks into value-added chemicals and fuels. Heterogeneous Br⊘nsted and Lewis acid catalysts such as metal-substituted zeolites, surface-modified metal oxides, and cation-exchange resins are intrinsically effective for the hydrolytic conversion of cellulose and hemicellulose into C5 and C6 monosaccharides and their subsequent transformation into chemicals such as polyols, furans, and acids. Supported metal catalysts are also very useful and are most frequently studied for O2-based oxidation of HMF to FDCA and glycerol to trioses. In particular, bi- and multi-metallic catalysts exhibit superior activity and stability compared to the corresponding monometallic counterparts.
Although most solid catalysts can be easily recovered by simple filtration or centrifugation after the reaction and recycled without appreciable loss of their catalytic performance, successful examples of completely recovered and recycled catalysts are still limited. In addition, severe reaction conditions, such as hydrothermal conditions and high oxygen pressure, require a high ratio of additives, and microwave irradiation can sometimes cause instability and ion leaching of the solid catalysts. Therefore, future targets in this area will require novel catalyst design strategies to overcome these problems. Such concepts will open up a new avenue for the development of solid catalysts that are workable under mild reaction conditions for practical biomass conversion including challenging reactions such as high-yield direct syntheses of value-added chemicals from waste biomass as raw materials, and selective conversion of lignin into useful aromatic components. The density (i.e., quantity in a certain place) of lignocellulose is much lower than that of crude oil and natural gas, and the properties of biomass feedstocks are very much dependent on their sources; therefore, biorefinery processes cannot replace petroleum refinery processes at present. Our own impression is that other technological aspects, including collection and transport, should also be discussed to establish truly efficient biorefinery processes.
Acknowledgments
This work was supported in part by the ALCA, CREST, and PREST programs of the Japan Science and Technology Agency (JST), the Novel Cheap and Abundant Materials for Catalytic Biomass Conversion (NOVACAM) program of JST and the European Commission Directorate-General for Research and Innovation, and Grants-in-Aid for Japan Society for the Promotion of Science (JSPS) Fellows and Scientific Research from the Ministry of Education, Culture, Science, Sports, and Technology of Japan.
References
- AnastasP TWarnerJ C 1998 Green Chemistry: Theory and Practice New York Oxford University Press
- SheldonR A 2012 Chem. Soc. Rev. 41 1437 10.1039/C1CS15219J
- ClarkJ H 1999 Green Chem. 1 1 10.1039/a807961g
- ThomasJ MRajaT 2005 Annu. Rev. Mater. Res. 35 315 10.1146/annurev.matsci.35.102003.140852
- HovárthI T 2002 ‘Green chemistry’ Acc. Chem. Res. 35 685 (special issue) 10.1021/ar020160a
- BullJ R 2000 ‘Green chemistry’ Pure Appl. Chem. 72 1207 (special issue)
- SheldonR A 2000 Pure Appl. Chem. 72 1233 10.1351/pac200072071233
- HagenJ 1999 Industrial Catalysis: A Practical Approach Weinheim Wiley
- ErtlGKnözingerHWeitkampJ 2008 Handbook of Heterogeneous Catalysis 2nd edn Weinheim Wiley
- AnKSomorjaiG A 2012 Chem. Cat. Chem. 4 1512 10.1002/cctc.201200229
- ThomasJ M 2014 Phys. Chem. Chem. Phys. 16 7647 10.1039/c4cp00513a
- ClimentM JCormaAIborraSSabaterM J 2014 ACS Catal. 4 870 10.1021/cs401052k
- MizunoN 2009 Modern Heterogeneous Oxidation Catalysis Weinheim Wiley
- CormaAIborraSVeltyA 2007 Chem. Rev. 107 2411 10.1021/cr050989d
- Mäki-ArvelaPHolmbomBSalmiTMurzinD Y 2007 Catal. Rev. 49 197 10.1080/01614940701313127
- GallezotP 2007 Catal. Today 121 76 10.1016/j.cattod.2006.11.019
- PalkovitsRTajvidiKProcelewskaJRinaldiRRuppertA 2010 Green Chem. 12 972 10.1039/c000075b
- VigierK D OJérômeF 2010 Top. Curr. Chem. 295 63
- Serrano-RuizJ CLuqueRSepúlveda-EscribanoA 2011 Chem. Soc. Rev. 40 5266 10.1039/c1cs15131b
- ClimentM JCormaAIborraS 2011 Green Chem. 13 520 10.1039/c0gc00639d
- CentiGLanzafamePPerathonerS 2011 Catal. Today 167 14 10.1016/j.cattod.2010.10.099
- ZhouC-HXiaXLinC-XTongD-SBeltraminiJ 2011 Chem. Soc. Rev. 40 5588 10.1039/c1cs15124j
- RuppertA MWeinbergKPalkovitsR 2012 Angew. Chem. Int. Edn 51 2564 10.1002/anie.201105125
- KobayashiHOhtaHFukuokaA 2012 Catal. Sci. Technol. 2 869 10.1039/c2cy00500j
- HuLZhaoGHaoWTangXSunYLinYLiuS 2012 RSC Adv. 2 11184 10.1039/C2RA21811A
- KuznetsovB NChesnokovN VYatsenkovaO VSharypoV I 2013 Russ. Chem. Bull. Int. Edn 62 1493 10.1007/s11172-013-0213-z
- BessonMGallezotPPinelC 2014 Chem. Rev. 114 1827 10.1021/cr4002269
- GallezotP 2012 Chem. Soc. Rev. 41 1538 10.1039/C1CS15147A
- ClimentM JCormaAIborraS 2014 Green Chem. 16 516 10.1039/c3gc41492b
- SchwartzT JO’NeillB JShanksB HDumesicJ A 2014 ACS Catal. 4 2060 10.1021/cs500364y
- SheldonR A 2014 Green Chem. 16 950 10.1039/C3GC41935E
- XiongHPhamH NDatyeA K 2014 Green Chem. 16 4627 10.1039/C4GC01152J
- SpiveyJEgbebiA 2007 Chem. Soc. Rev. 36 1514 10.1039/b414039g
- YueHMaHGongJ 2014 Acc. Chem. Res. 47 1483 10.1021/ar4002697
- IribarrenDSusmozasAPetrakopoulouFDufourJ 2014 J. Clean. Prod. 69 165 10.1016/j.jclepro.2014.01.068
- HeCChenC-LGiannisAYangYWangJ-Y 2014 Renew. Sustain. Energy Rev. 39 1127 10.1016/j.rser.2014.07.141
- GeboersJ AVan de VyverSOomsRde BeeckB OJacobsP ASelsB F 2011 Catal. Sci. Technol. 1 714 10.1039/c1cy00093d
- MartelFEstrineBPlantier-RoyonRHoffmannNPortellaC 2010 Top. Curr. Chem. 294 79 10.1007/128_2010_54
- Mäki-ArvelaPSalmiTHolmbomBWillfȯrSMurzinD Y 2011 Chem. Rev. 111 5638 10.1021/cr2000042
- ZakzeskiJBruijnincxP C AJongeriusA LWeckhuysenB M 2010 Chem. Rev. 110 3552 10.1021/cr900354u
- PandeyM PKimC S 2011 Chem. Eng. Technol. 34 29 10.1002/ceat.201000270
- BartaKFordP C 2014 Acc. Chem. Res. 47 1503 10.1021/ar4002894
- MeierM A RMetzgerJ OSchubertU S 2007 Chem. Soc. Rev. 36 1788 10.1039/b703294c
- ZhouC-HBeltraminiJ NFanY-XLuG Q 2008 Chem. Soc. Rev. 37 527 10.1039/B707343G
- RybakAFokouP AMeierM A R 2008 Eur. J. Lipid Sci. Technol. 110 797 10.1002/ejlt.200800027
- BarraultJJeromeF 2008 Eur. J. Lipid Sci. Technol. 110 825 10.1002/ejlt.200800061
- BehrAEiltingJIrawadiKLeschinskiJLindnerF 2008 Green Chem. 10 13 10.1039/B710561D
- MetzgerJ O 2009 Eur. J. Lipid Sci. Technol. 111 865 10.1002/ejlt.200900130
- BehrAGomesJ P 2010 Eur. J. Lipid Sci. Technol. 112 31 10.1002/ejlt.200900091
- KatryniokBKimuraHSkrzyńskaEGirardonJ-SFongarlandPCapronMDucoulombierRMimuraNPaulSDumeignilF 2011 Green Chem. 13 1960 10.1039/c1gc15320j
- ten DamJHanefeldU 2011 ChemSusChem 4 1017 10.1002/cssc.201100162
- RosatellaASimeonovS PFradeR F MAfonsoC A M 2011 Green Chem. 13 754 10.1039/c0gc00401d
- van PuttenR-Jvan der WaalJ Cde JongERasrendraC BHeeresH Jde VriesJ G 2013 Chem. Rev. 113 1499 10.1021/cr300182k
- ArendsI W C ESheldonR AWallauMSchuchardtU 1997 Angew. Chem. Int. Edn 36 1144 10.1002/anie.199711441
- SheldonR AWallauMArendsI W C ESchuchardtU 1998 Acc. Chem. Res. 31 485 10.1021/ar9700163
- AuerbachS MCarradoK ADuttaP K 2003 Handbook of Zeolite Science and Technology New York Dekker
- XuRPangWYuJHuoQChenJ 2007 Chemistry of Zeolites and Related Porous Materials: Synthesis and Structure Singapore Wiley
- ČejkaJCormaAZonesS 2010 Zeolites and Catalysis: Synthesis, Reactions and Applications Weinheim Wiley
- NiwaMKatadaNOkumuraK 2010 Characterization and Design of Zeolite Catalysts: Solid Acidity, Shape Selectivity and Loading Properties 1st edn Springer Series in Materials Science Berlin Springer
- van SantenR AKramerG J 1995 Chem. Rev. 95 637 10.1021/cr00035a008
- CormaA 1997 Chem. Rev. 97 2373 10.1021/cr960406n
- DemontisPSuffrittiG B 1997 Chem. Rev. 97 2845 10.1021/cr950253o
- HartmannMKevanL 1999 Chem. Rev. 99 635 10.1021/cr9600971
- KiricsiIFörsterHTasiGNagyJ B 1999 Chem. Rev. 99 2085 10.1021/cr9600767
- FrickeRKosslickHLischkeGRichterM 2010 Chem. Rev. 100 2303 10.1021/cr9411637
- GarcíaHRothH D 2002 Chem. Rev. 102 3947 10.1021/cr980026x
- CundyC SCoxP A 2003 Chem. Rev. 103 663 10.1021/cr020060i
- SmitBMaesenT L M 2008 Chem. Rev. 108 4125 10.1021/cr8002642
- NavrotskyATrofymlukOLevchenkoA 2009 Chem. Rev. 109 3885 10.1021/cr800495t
- KrishnaRSmitBCaleroS 2002 Chem. Soc. Rev. 31 185 10.1039/b101267n
- KrishnaR 2012 Chem. Soc. Rev. 41 3099 10.1039/c2cs15284c
- DavisM E 2002 Nature 417 813 10.1038/nature00785
- CormaAGarciaH 2004 Chem. Commun. 1443 10.1039/b400147h
- MedinaM EPlatero-PratsA ESnejkoNRojasAMongeAGa´ndaraFGutie´rrez-PueblaECamblorM A 2011 Adv. Mater. 23 5283 10.1002/adma.201101852
- ČejkaJCentiGPerez-ParienteJRothW J 2012 Catal. Today 179 2 10.1016/j.cattod.2011.10.006
- KimH SPhamT C TYoonK B 2012 Chem. Commun. 48 4659 10.1039/c2cc30919j
- SchoonheydtR AGeerlingsPPidkoE Avan SantenR A 2012 J. Mater. Chem. 22 18705 10.1039/C2JM31366A
- BellussiGCaratiARizzoCMilliniR 2013 Catal. Sci. Technol. 3 833 10.1039/C2CY20510F
- ChatterjeeSHarishRSchützG M 2013 Chem. Ing. Tech. 85 1671 10.1002/cite.201300030
- HemelsoetKvan der MynsbruggeJDe WispelaereKWaroquierMvan SpeybroeckV 2013 ChemPhysChem. 14 1526 10.1002/cphc.201201023
- MintovaSGilsonJ-PValtchevV 2013 Nanoscale 5 6693 10.1039/c3nr01629c
- BhanAIglesiaE 2008 Acc. Chem. Res. 41 559 10.1021/ar700181t
- ThomasJ MRajaR 2008 Acc. Chem. Res. 41 708 10.1021/ar700217y
- MartínezCCormaA 2011 Coord. Chem. Rev. 255 1558 10.1016/j.ccr.2011.03.014
- Ristic´ALogarN ZHenningerS KKaučičV 2012 Adv. Funct. Mater. 22 1952 10.1002/adfm.201102734
- HammondC 2012 Angew. Chem. Int. Edn 51 5129 10.1002/anie.201108706
- WuPTatsumiTKomatsuTYashimaT 2001 J. Catal. 202 245 10.1006/jcat.2001.3278
- CormaADomineM ENemethLValenciaS 2002 J. Am. Chem. Soc. 124 3194 10.1021/ja012297m
- CavaniFTelesJ H 2009 ChemSusChem. 2 508 10.1002/cssc.200900020
- MolinerMRoman-LeshkovYDavisM E 2010 Proc. Natl Acad. Sci. USA 107 6164 10.1073/pnas.1002358107
- CormaANemethL TRenzMValenciaS 2001 Nature 412 423 10.1038/35086546
- RenzMBlascoTCormaAFornesVJensenRNemethL 2012 Chem. Eur. J. 8 4708 10.1002/1521-3765(20021018)8:20<4708::AID-CHEM4708>3.0.CO;2-U
- DhattaRHenryM 2006 J. Chem. Technol. Biotechnol. 81 1119 10.1002/jctb.1486
- SheldonR A 2005 Green Chem. 7 267 10.1039/b418069k
- WeeY-JKimJ-NRyuH-W 2006 Food Technol. Biotechnol. 44 163
- LeaversuchR 2002 Plast. Technol. 48 50
- VaradarajanSMillerD J 1999 Biotechnol. Prog. 15 845 10.1021/bp9900965
- ShiH FHuY CWangYHuangH 2007 Chin. Chem. Lett. 18 476 10.1016/j.cclet.2007.01.043
- CortrightR DSanchez-CastilloMDumesicJ A 2002 Appl. Catal. B 39 353 10.1016/S0926-3373(02)00121-2
- Serrano-RuizJ CDumesicJ A 2009 ChemSusChem. 2 581 10.1002/cssc.200900004
- GarlottaD 2001 J. Polym. Environ. 9 63 10.1023/A:1020200822435
- JohnR PNampoothiriK MPandeyA 2007 Appl. Microbiol. Biotechnol. 74 524 10.1007/s00253-006-0779-6
- KimuraHTsutoKWakisakaTKazumiYInayaY 1993 Appl. Catal. A 96 217 10.1016/0926-860X(90)80011-3
- KimuraH 1993 Appl. Catal. A 105 147 10.1016/0926-860X(93)80245-L
- GarciaRBessonMGallezotP 1995 Appl. Catal. A 127 165 10.1016/0926-860X(95)00048-8
- FarnettiEKasparJCrottiC 2009 Green Chem. 11 704 10.1039/b819870e
- HekmatDBauerRNeffV 2007 Process Biochem. 42 71 10.1016/j.procbio.2006.07.026
- YamadaSNabeKIzuoNWadaMChibataI 1979 J. Ferment. Technol. 57 215
- NabeKIzuoNYamadaSChibataI 1979 Appl. Environ. Microbiol. 38 1056
- FlickingerM CPerlmanD 1977 Appl. Environ. Microbiol. 33 706
- Roman-LeshkovYMolinerMLabingerJ ADavisM E 2010 Angew. Chem. Int. Edn 49 8954 10.1002/anie.201004689
- Bermejo-DevalR 2012 Proc. Natl Acad. Sci. USA 109 9727 10.1073/pnas.1206708109
- AssaryR SCurtissL A 2011 J. Phys. Chem. A 115 8754 10.1021/jp204371g
- PescarmonaP PJanssenK P FDelaetCStroobantsCHouthoofdKPhilippaertsADe JongheCPaulJ SJacobsP ASelsB F 2010 Green Chem. 12 1083 10.1039/b921284a
- TaarningESaravanamuruganSSpangsbergH MXiongJWestR MChristensenC H 2009 ChemSusChem 2 625 10.1002/cssc.200900099
- WestR MHolmM SSaravanamuruganSXiongJBeversdorfZTaarningEChristensenC H 2010 J. Catal. 269 122 10.1016/j.jcat.2009.10.023
- HammondCConradSHermansI 2012 Angew. Chem. Int. Edn 51 11736 10.1002/anie.201206193
- de ClippelF 2012 J. Am. Chem. Soc. 134 10089 10.1021/ja301678w
- de ClippelFHarkiolakisAKeXVoschTvan TendelooGBaronG VJacobsP ADenayerJ F MSelsB F 2010 Chem. Commun. 46 928 10.1039/B918864A
- de ClippelF 2011 Microporous Mesoporous Mater. 144 120 10.1016/j.micromeso.2011.04.003
- de ClippelF 2013 J. Mater. Chem. A 1 945 10.1039/C2TA00098A
- BhosaleSRaoMDeshpandeV 1996 Microbiol. Rev. 60 280
- VigierKJerômeF 2010 Carbohydrates in Sustainable Development II Topics in Current Chemistry vol 295 RauterA PVogelPQueneauY Berlin Springer p 63
- TewariY 1990 Appl. Biochem. Biotechnol. 23 187 10.1007/BF02942054
- BoronatMCormaARenzM 2006 J. Phys. Chem. B 110 21168 10.1021/jp063249x
- NikollaERoman-LeshkovYMolinerMDavisM E 2011 ACS Catal. 1 408 10.1021/cs2000544
- OtomoRYokoiTKondoJ NTatsumiT 2014 Appl. Catal. A 470 316 10.1016/j.apcata.2013.11.012
- TanabeKMisonoMOnoYHattoriH 1989 New Solid Acid and Bases Amsterdam Elsevier
- OhkuharaT 2002 Chem. Rev. 102 3641 10.1021/cr0103569
- MoraesMPintoW de S FGonzalezW ACarmoL M P MPasturaN M RLachterE R 1996 Appl. Catal. A 138 L7 10.1016/0926-860X(96)00044-0
- GuoCQianZ 1993 Catal. Today 16 379 10.1016/0920-5861(93)80077-E
- UshikuboTIizukaTHattoriHTanabeK 1993 Catal. Today 16 291 10.1016/0920-5861(93)80068-C
- NowakIZiolekM 1999 Chem. Rev. 99 3603 10.1021/cr9800208
- ClearfieldATroupJ M 1973 J. Phys. Chem. 77 243 10.1021/j100621a022
- ChristensenA NAndersenE KAndersenI G KAlbertiGNielsenMLehmannM S 1990 Acta Chem. Scand. 44 865 10.3891/acta.chem.scand.44-0865
- BuscaGLorenzelliVGalliPLa GinestraAPatronoA 1987 J. Chem. Soc. Faraday Trans. I 83 853 10.1039/f19878300853
- AlbertiGCostantinoU 1984 J. Mol. Catal. 27 235 10.1016/0304-5102(84)85083-X
- La GinestraAPatronoPBerardelliM LGalliPMassucciM AFerraginaCCiambelliP 1984 Proc. 17th Congr. Naz. Chim. Inorg. p 369
- ClearfieldATakurT S 1980 J. Catal. 65 185 10.1016/0021-9517(80)90293-6
- La GinestraAPatronoPMassucciM AGalliPFerraginaC 1986 Proc. 19th Congr. Naz. Chim. Inorg. p 439
- SegawaKKurusuYNakajimaYKinoshitaM 1985 J. Catal. 94 491 10.1016/0021-9517(85)90213-1
- La GinestraAPatronoPBerardelliM LGalliPFerraginaCMassucciM A 1987 J. Catal. 103 346 10.1016/0021-9517(87)90126-6
- RamisGRossiP FBuscaGLorenzelliVLa GinestraAPatronoP 1989 Langmuir 5 917 10.1021/la00088a006
- CostaM C CJohnstoneR A WWhittakerD 1998 J. Mol. Catal. A 129 79 10.1016/S1381-1169(97)00135-0
- CostantinoULa GinestraA 1982 Thermochim. Acta 58 179 10.1016/0040-6031(82)87080-9
- YamanakaSTanakaM 1979 J. Inorg. Nucl. Chem. 41 45 10.1016/0022-1902(79)80391-7
- AllulliSFerraginaCLa GinestraAMassucciM ATomassiniN 1977 J. Inorg. Nucl. Chem. 39 1043 10.1016/0022-1902(77)80261-3
- BenvenutiFCarliniCPatronPGallettiA M RSbranaGMassucciM AGalliP 2000 Appl. Catal. A 193 147 10.1016/S0926-860X(99)00424-X
- NakajimaKBabaYNomaRKitanoMHayashiSHaraM 2011 J. Am. Chem. Soc. 133 4224 10.1021/ja110482r
- ShintakuHNakajimaKKitanoMIchikuniNHaraM 2014 ACS Catal. 4 1198 10.1021/cs401149n
- ShintakuHNakajimaKKitanoMHaraM 2014 Chem. Commun. 50 13473 10.1039/C4CC05711B
- NakajimaKNomaRKitanoMHaraM 2014 J. Mol. Catal. A 388–389 100 10.1016/j.molcata.2013.09.012
- DeSDuttaSPatraA KBhaumikASahaB 2011 J. Mater. Chem. 21 17505 10.1039/c1jm13229f
- GatesB C 1995 Chem. Rev. 95 511 10.1021/cr00035a003
- GrossESomorjaiG A 2013 Top. Catal. 56 1049 10.1007/s11244-013-0069-3
- QiS-CWeiX-YZongZ-MWangY-K 2013 RSC Adv. 3 14219 10.1039/C1JM13229F
- TaketoshiAHarutaM 2014 Chem. Lett. 43 380 10.1246/cl.131232
- CormaAGarciaH 2008 Chem. Soc. Rev. 37 2096 10.1039/b707314n
- WhiteR JLuqueRBudarinV LClarkJ HMacquarrieD J 2009 Chem. Soc. Rev. 38 481 10.1039/B802654H
- LiuXHeLLiuY-MCaoY 2014 Acc. Chem. Res. 47 793 10.1021/ar400165j
- IdeM SDavisR J 2014 Acc. Chem. Res. 47 825 10.1021/ar4001907
- Taladriz-BlancoPHervésPPérez-JusteJ 2013 Top. Catal. 56 1154 10.1007/s11244-013-0082-6
- ShuttleworthP SDe bruynMParkerH LHuntA JBudarinV LMatharuA SClarkJ H 2014 Green Chem. 16 573 10.1039/C3GC41555D
- HutchingsG J 2014 Catal. Today 238 69 10.1016/j.cattod.2014.01.033
- PaalanenPWeckhuysenB MMeenakshisundaramS 2013 Catal. Sci. Technol. 3 2869 10.1039/c3cy00341h
- FöttingerKRupprechterG 2014 Acc. Chem. Res. 47 3071 10.1021/ar500220v
- AlonsoD MWettsteinS GDumesicJ A 2012 Chem. Soc. Rev. 41 8075 10.1039/c2cs35188a
- SheldonR AKochiJ K 1981 Metal Catalyzed Oxidations of Organic Compounds New York Academic
- BäckvallJ-E 2004 Modern Oxidation Methods Weinheim Wiley
- PunniyamurthyTVelusamySIqbalJ 2005 Chem. Rev. 105 2329 10.1021/cr050523v
- DavisS EIdeM SDavisR J 2013 Green Chem. 15 17 10.1039/C2GC36441G
- YamaguchiKMizunoN 2002 Angew. Chem. Int. Edn 41 4538 10.1002/1521-3773(20021202)41:23<4538::AID-ANIE4538>3.0.CO;2-6
- GorbanevY YKegnæsSRiisagerA 2011 Top. Catal. 54 1318 10.1007/s11244-011-9754-2
- GorbanevY YKegnæsSRiisagerA 2011 Catal. Lett. 141 1752 10.1007/s10562-011-0707-y
- StåhlbergTEyjólfsdóttirEGorbanevY YSádabaIRiisagerA 2012 Catal. Lett. 142 1089 10.1007/s10562-012-0858-5
- CasanovaOIborraSCormaA 2009 ChemSusChem 2 1138 10.1002/cssc.200900137
- DavisS EHoukL RTamargoE CDatyeA KDavisR J 2011 Catal. Today 160 55 10.1016/j.cattod.2010.06.004
- GuptaN KNishimuraSTakagakiAEbitaniK 2011 Green Chem. 13 824 10.1039/c0gc00911c
- SahaBDuttaaSAbu-OmarM M 2012 Catal. Sci. Technol. 2 79 10.1039/C1CY00321F
- CaiJMaHZhangJSongQDuZHuangYXuJ 2013 Chem. Eur. J. 19 14215 10.1002/chem.201301735
- NieJXieJLiuH 2013 Chin. J. Catal. 34 871 10.1016/S1872-2067(12)60551-8
- YadavG DSharmaR V 2014 Appl. Catal. B 147 293 10.1016/j.apcatb.2013.09.004
- PasiniT 2011 Green Chem. 13 2091 10.1039/c1gc15355b
- RassH AEssayemNBessonM 2013 Green Chem. 15 2240 10.1039/c3gc40727f
- VillaASchiavoniMCampisiSVeithG MPratiL 2013 Chem. Sus. Chem. 6 609 10.1002/cssc.201200778
- WanXZhouCChenJDengWZhangQYangYWangY 2014 ACS Catal. 4 2175 10.1021/cs5003096
- CarrettinSMcMornPJohnstonPGriffinKKielyC JHutchingsG J 2003 Phys. Chem. Chem. Phys. 5 1329 10.1039/b212047j
- DemirelSKernPLucasMClausP 2007 Catal. Today 122 292 10.1016/j.cattod.2006.12.002
- RodriguesE GPereiraM F RChenXDelgadoJ JÓrfãoJ J M 2011 J. Catal. 281 119 10.1016/j.jcat.2011.04.008
- MengyuanZDanLRenfengNXiuyangLPingCZhaoyinH 2012 Chin. J. Catal. 33 1340 10.1016/S1872-2067(11)60411-7
- GaoJLiangDChenPHouZZhengX 2009 Catal. Lett. 130 185 10.1007/s10562-009-9849-6
- LiangDGaoJSunHChenPHouZZhengX 2011 Appl. Catal. B 106 423 10.1016/j.apcatb.2011.05.050
- RichterF HMengYKlasenTSahraouiLSchüthF 2013 J. Catal. 300 341 10.1016/j.jcat.2013.08.014
- DemirelSLehnertKLucasMClausP 2007 Appl. Catal. B 70 637 10.1016/j.apcatb.2005.11.036
- HuWKnightDLowryBVarmaA 2010 Ind. Eng. Chem. Res. 49 10876 10.1021/ie1005096
- LiangDGaoJWangJChenPWeiYHouZ 2011 Catal. Commun. 12 1059 10.1016/j.catcom.2011.03.019
- NieRLiangDShenLGaoJChenPHouZ 2012 Appl. Catal. B 127 212 10.1016/j.apcatb.2012.08.026
- HirasawaSNakagawaYTomishigeK 2012 Catal. Sci. Technol. 2 1150 10.1039/c2cy20062g
- KondratS A 2014 ChemSusChem 7 1326 10.1002/cssc.201300834
- HirasawaSWatanabeHKizukaTNakagawaYTomishigeK 2013 J. Catal. 300 205 10.1016/j.jcat.2013.01.014
- HarmerM ASunQ 2001 Appl. Catal. A 221 45 10.1016/S0926-860X(01)00794-3
- TanabeK 1994 Acidity and Basicity of Solids: Theory, Assessment and Utillity FraissardJPetrkisL Dordrecht Kluwer pp 53 79 353–79
- SonheiemerS JBruceN JFyfeC A 1986 J. Macromol. Sci., Rev. Macromol. Chem. Phys. C26 353 10.1080/07366578608081976
- OlahG AIyerP SPrakasnG K S 1986 Synthesis 513 10.1055/s-1986-31692
- ChakrabartiASharmaM M 1989 Ind. Eng. Chem. Res. 28 1757 10.1021/ie00096a004
- ChakrabartiASharmaM M 1993 React. Polym. 20 1 10.1016/0923-1137(93)90064-M
- Román-LeshkovYChhedaJ NDumesicJ A 2006 Science 312 1933 10.1126/science.1126337
- ZhaoHHolladayJ EBrownHZhangZ C 2007 Science 316 1597 10.1126/science.1141199
- Román-LeshkovYDavisM E 2011 ACS Catal. 1 1566 10.1021/cs200411d
- QiXWatanabeMAidaT MSmithR L Jr 2008 Green Chem. 10 799 10.1039/b801641k
- ShimizuKUozumiRSatsumaA 2009 Catal. Commun. 10 1849 10.1016/j.catcom.2009.06.012