Abstract
In the present work we aim to clarify the role of the microstructure and the crystalline distribution from the thermo-oxidation of solid isotactic PP (iPP) and ethylene-propylene (EP) copolymers. The effects of the content and quality of the isotacticity interruptions, together with the associated average isotactic length, on the induction time (ti) as well as on the activation energy (Eact) of the thermo-oxidation are analysed. Both parameters have been found to change markedly at an average isotactic length (n1) of 30 propylene units. While ti reaches a minimum when n1 is approximately 30 units, Eact increases quasi-exponentially as the number of units decreases from 30. This variation can be explained in terms of changes induced in the crystalline interphase, i.e. local molecular dynamics, which are closely linked to the initiation of the thermo-oxidation of isotactic PP-based polyolefins.
Introduction
The improved performance of propylene-based polyolefins requires a deep understanding of the effects of different polypropylene structure modifications on the thermal stability of these materials in the solid state. Actually, it is well-known that the iPP part of block-copolymers or blends is selectively thermo-oxidised, which limits the lifetime of these thermoplastics [Citation1–3].
A general consideration is that factors that play a role in the thermo-oxidative degradation of pure iPP are expected to apply in some way when the typical iPP semi-crystalline morphology is retained. In this sense, low-conversion copolymerisation may have a minor effect in the form of a chemical change, but it still has an important effect on the degradation behaviour because of discrete units acting as defective structures that can disturb the stereoregularity and related features, i.e. the content, quality and distribution of the crystalline phase. Consequently, in ethylene-propylene copolymers (EP) effects other than the stabilization caused by the introduction of more stable methylene units may be expected.
In this respect, Terano and coworkers [Citation2, Citation4–7] have shown that the PP primary structure plays a major role in the degradation behaviour of PP-based polyolefins. The authors found that not only mr and rr triads but also ethylene units confer a high thermal stability to iPP because these units act as interruptions of the isotacticity, which is the configuration in which degradation is particularly favoured.
These studies were performed on PP and EP copolymers at temperatures where the materials were about to melt or were in a completely molten state. Under these conditions, the induction time, measured as the moment at which either the oxygen uptake or the intensity of emitted light increases, reflects the ability of individual iPP chains to accumulate or decompose hydroperoxides, respectively. In both cases, the degradation behaviour is expected to be driven mostly by the chain microstructure. From this point of view, typical isotacticity interruptions in iPP homopolymers other than racemic units, such as 2,1-insertions, should also be proposed as stabilising units because they interrupt the stereoregularity.
Our results on iPP thermo-oxidation in the solid state indeed show a major role of the content of racemic insertions together with the molar mass [Citation8, Citation9] but, contrary to Terano's conclusion, a destabilising effect of such units was found. The apparently contradictory role of mr propylene units is thought to arise from their different aggregation state, because the effect of microstructure-derived features must be taken into account in the solid state rather than the sole microstructure of the isolated chain. In particular, the necessary accumulation of isotacticity interruptions in the crystalline interphase makes this region rich in overaverage-size local free volume, which has been suggested to control the induction time of iPP samples within a wide isotacticity range. According to this proposal, at temperatures where the oxygen accessibility is restricted to the disordered phase and the intrachain oxidation buildup is greatly hindered, hydroperoxides would concentrate in the interphase up to the content required to undergo decomposition by the widely recognised bimolecular Russell mechanism.
This hypothesis was tentatively verified by evaluating the effect of introducing a small number of isolated ethylene units (2.6 mol.%) on the iPP thermal stability [Citation9]. Actually, taking the interphase dynamics as a criterion to monitor changes in the local free volume, the induction time (ti) of the EP copolymer was found to fit the trend of pure iPP samples. On this basis, in the present work we aim to confirm, on one hand, the existence of a destabilising effect of the isotacticity interruptions in solid iPP thermo-oxidation and, on the other hand, that changes in the intensity of local dynamics linked to the interphase account for the variations of ti for solid-state iPP degradation. For this purpose, three families of metallocene polyolefins were synthesized. The first series correspond to iPP samples that differ in both molar mass and the content of interruptions, the second series corresponds to propylene-rich EP copolymers and the third series is composed of polyethylene samples and ethylene-rich EP copolymers with an extremely low propylene content.
Experimental
Samples
The samples used in this study are usually named by the acronyms PP, EP and PE if they are polyproylene, ethylene–propylene copolymer and polyethylene samples, respectively. The value of the intrinsic viscosity (ml g−1) is used to complete the name in the case of homopolymers (PP and PE) and the ethylene conversion (mol.%) is used for EP copolymers.
The three series of polyolefins were obtained by homogeneous metallocene catalysis using the conditions listed in tables 1, 2 and 3. Catalyst traces were removed by pouring the reaction bulk into a CH3OH/HCl (5 vol.%) mixture. Finally, the polymers were filtered, washed with CH3OH and dried under vacuum at room temperature.
Polymerisation conditions of PP samples.
Polymerisation conditions of EP copolymers.
Polymerisation conditions of PE samples.
Characterisation of samples
Molar mass
The molar mass was estimated using the intrinsic viscosity of polymers measured from decaline solutions at 135 °C in nitrogen (tables 4, 5 and 6).
Viscosimetry, DSC and 13C-NMR characterisation of PP samples.
Viscosimetry, DSC and 13C − NMR characterisation of EP copolymers.
Viscosimetry and DSC characterisation of PE samples.
Crystallinity and melting point
The DSC measurements were carried out in a Perkin-Elmer DSC-7 calorimeter with about 5 mg of powdered sample heated at 10 °C min−1 between 40 and 210 °C in a N2 flow. Only the first scan was considered in the determination of the melting temperature (Tm), the crystallinity degree (χ c) and the endotherm shape, which characterises the precise morphology of samples when they are degraded. The standard heat of melting for a 100% crystalline sample used in crystallinity calculations was 209 J g−1 [Citation10] for PP and propylene-rich EP copolymers and 293 J g−1 [Citation11] for PE and ethylene-rich EP copolymers. The results of DSC characterisation are shown in tables 4, 5 and 6.
Mechanical relaxations
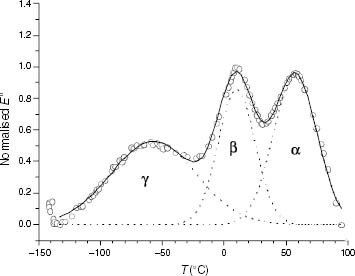
The gamma (γ) relaxation of PP and PP-like EP copolymers was measured by dynamic thermomechanical analysis using a Thermal Instrument DMA 983 analyser in flexion mode of samples processed under conditions reported elsewhere [Citation12]. The frequency of oscillation was 0.1 Hz, the temperature range was from − 150 to 130 °C, the heating rate was 5°C min−1 and the deformation amplitude was 0.8 mm. The relative intensity of the γ relaxation was calculated by measuring the relative sub-Tg area from the deconvoluted E″ spectra, as performed in earlier work [Citation13]. The criterion for performing the deconvolution follows the example given in figure , that is, each mechanical relaxation was fitted with one Gaussian component centred at the corresponding maximum.
Figure 1 Mechanodynamical loss modulus curve of PP92.4. (○) Experimental, (—) calculated, (····) single components.
Microstructure
The configuration and composition of samples were obtained by the 13C-NMR technique. Details corresponding to the calculus of the different tactic triads, regiodefects and ethylene contents can be found in a previous work [Citation12]. Tables 4 and 5 show the tactic and regio microstructures of iPP and propylene-rich EP copolymers, respectively.
Spectra were obtained using a Varian Unity 500 spectrometer operating at 125.708 MHz in d2-tetra-chloroethane (100 mg ml−1), at 125 or 90 °C depending on the solubility of the sample. The acquisition time was 1 s, the relaxation delay was 4 s, the pulse angle was 90° and at least 8000 scans were accumulated. Although proton broadband decoupling was used, the NOE effect on the different tactic methyl groups was assumed to be the same [Citation14] and a quantitative valuation of the configurational microstructure could be performed.
Content of chromophores
The relative content of chromophores was estimated by comparing the intensity of the photoluminescence emission spectra. The spectra were obtained from some selected samples in the form of films by irradiation at λ exc = 270 nm and − 130 °C and after a 0.5 s delay. The excitation wavelength and the temperature were chosen to produce the maximum intensity of the phosphorescence emission. Circular films (5 cm diameter, 130–150 μ m thick) were obtained by molding the powdered sample for 3 min. at 190 bar and a temperature that corresponded to the onset of the DSC melting endotherm of the virgin polymer. Photoluminescence was measured in an airtight chamber equipped with a UV-vis 150 W xenon source [Citation15]. This device allows the synchronisation between excitation and emission detection. The spectral light analysis was carried out using of a liquid-nitrogen-cooled CCD camera and a low-dispersivity monochromator (4.5 nm resolution).
Thermo-oxidation of samples
Chemiluminescence (CL) kinetics during the isothermal thermo-oxidation of samples was obtained using a Lumipol 2 device, which is produced at the Polymer Institute, Slovak Academy of Sciences, Bratislava, Slovakia. The high level of discrimination of this apparatus (2 count s−1 at 40 °C) allowed us to perform an accurate measurement of the induction time of the iPP thermo-oxidation (ti). About 3 mg of polymer in powder form was added to an aluminium pan, which was placed inside the oven of the CL device. A flow of oxygen at 50 ml h−1 passed through the reactor during the measurement.
In this work oxidation kinetics at temperatures ranging from 90 to 130 °C, at intervals of 10 °C, are presented. The CL curves were recorded throughout the induction period and the autoacceleration stage, and were stopped after the maximum intensity was reached. All kinetics were normalised to a mass of 3.0 mg. The intersection of the line that fits the autoacceleration period with the x-axis yields ti. These values of ti are in agreement with those reported in previous studies [Citation8, Citation16, Citation17] as well as with those usually found in the literature. In fact, the value of ti obtained from the CL measurements carried out at 100 °C on a single layer of powder particles of an unstabilized iPP in an oxygen gas flow rate of 50 ml h−1 [Citation18] is close to the range of ti found for our iPP samples at the same temperature (around 16 000 s compared with 18 000–42 300 s).
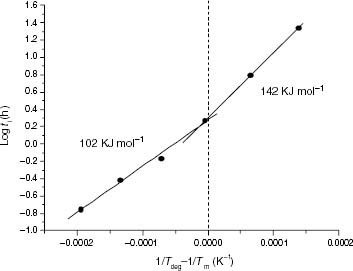
The apparent values of Eact for the degradation processes were calculated from Arrhenius plots of ti. As shown in figure for the case of EP6.2, this procedure was found to be sufficiently sensitive to detect kinetic transitions in the iPP initiation step and served to confirm that the CL kinetics arises from the thermo-oxidation of the iPP. In fact, the value of 103 kJ mol−1 found for the iPP samples with the highest molar mass corresponds to the typical value obtained for the degradation of an unstabilised iPP at nearly the same temperature range (80–150 °C) [Citation19].
Figure 2 Arrhenius plot of ti for EP6.2. The 1/ T scale is normalised to the melting point.
Results
Analysis of the isothermal CL curves
The results of evaluating the changes undergone by the CL kinetics with degradation temperature, molar mass and ethylene content are shown in figures to . A logarithmic time scale is used in graphs 3 to 5 to clearly show the early trends of the kinetics. Figure shows, as an example, the change in the CL buildup of EP1.1 with the degradation temperature. It is clear that an increase in Tdeg shortens the induction period and results in a higher maximum CL intensity.
Figure 3 ICL kinetics of EP1.1 at degradation temperatures between 90 and 130 °C.
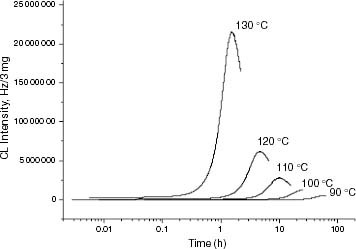
Figure 4 ICL kinetics of iPP samples at 120 °C: (○) PP178.1, (—) PP147.8, (····) PP131.9, (– – –) PP99.5, (–·–) PP92.4, (□) PP77.9.
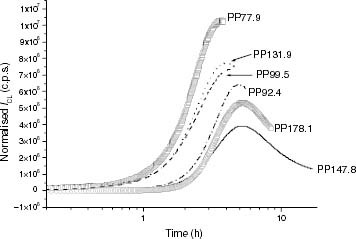
Figure 5 ICL kinetics of propylene-rich EP samples at 120 °C: (—–) EP0.6, (– – –) EP1.1, (····) EP3.3, (–·–) EP5.1, (—) EP6.1, (–··–) EP23.3.
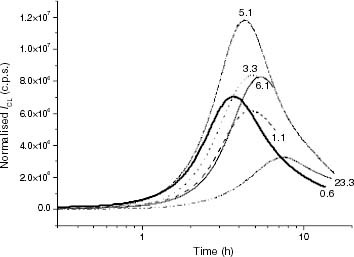
Figure 6 ICL kinetics of PE and ethylene-rich EP samples at 120 °C: (—–) PE242.5, (—) EP99.7, (– – –) EP99.0.
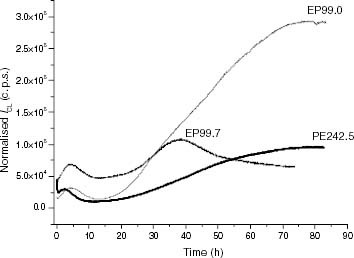
Figure shows the CL kinetics at 120 °C for some iPP samples with different molar masses. It was found that there is no a clear correlation between the induction time and the chain size. The two highest-molar-mass samples turn out to be the most stable, and the lowest-molar-mass iPP has the shortest ti but the intermediate values do not vary accordingly. As far as the maximum CL intensity is concerned, the less stable the iPP, the higher the CL level is observed.
When the CL kinetics of propylene-rich EP copolymers at 120 °C are inspected (figure ), one can observe that the shapes and maximum CL levels are similar to those exhibited for iPP materials in figure ; however, in this case, molar mass does not vary as much as in the iPP family (tables 4 and 5). It would thus be reasonable to conclude that differences in the CL buildup features are related to the whole microstructure rather than to only the ethylene content, because no coherent variation of both ti and the maximum CL intensity with composition is apparent up to an ethylene conversion of about 6 mol.%. It is only EP23.3 that is clearly the most stable sample.
Figure shows some examples corresponding to the thermo-oxidative CL kinetics of PE and ethylene-rich EP samples at 120 °C. In these materials, the maximum CL intensities are about one order of magnitude lower than those yielded by PP or propylene-rich EP samples. In addition, a small CL peak appears before the autoacceleration stage revealing the existence of hydroperoxides into the virgin material. As expected, the values of ti for the PE and ethylene-rich EP samples are greater than the values obtained for PP and propylene-rich EP samples.
Analysis of the photoluminescence
The phosphorescence emission of iPP with λmax between 445 and 488 nm is known to originate from α β-unsaturated ketones (enones) and polyconjugated enones [Citation20, Citation21]. This is the wavelength range where all polyolefin samples tested were found to exhibit the maximum intensity in the emission spectrum (figure ). Thus, the phosphorescence intensity was taken as being directly correlated with the content of such chromophores.
Figure 7 Photoluminescence emission spectra: (a) PP and propylene-rich EP samples; (b) PE and ethylene-rich EP samples obtained under irradiation at λ exc = 270 nm and T = - 130 °C after a 0.5 s delay. Samples are identified in the graphs.
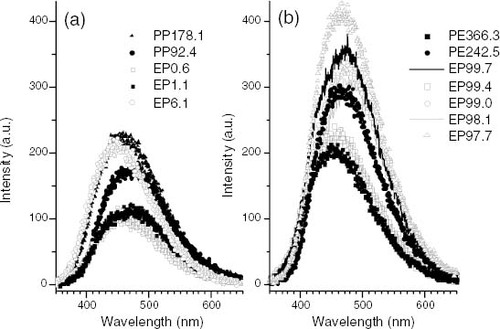
Figure shows that while the phosphorescence intensity for some PP and propylene-rich EP copolymers reaches a maximum of about 225 a.u. (figure (a)), PE and ethylene-rich EP copolymers exhibit a maximum intensity of approximately this value for two samples and significantly larger maximum values for the other samples (figure (b)). Consequently, the content of enones in PE and ethylene-rich EP samples is either similar to or higher than that in PP and propylene-rich EP samples.
Variation of ti
Molar mass
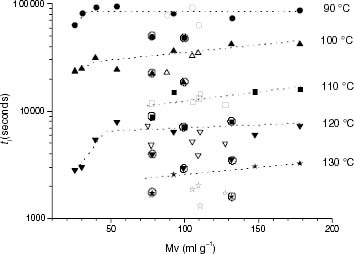
The thermal performance of iPP samples between 90 and 130 °C is partially related to their molar masses, as shown in figure . The induction time increases steeply for molar masses up to about 45 g ml−1 and smoothly above this value. This trend seems to occur at all temperatures, although some samples give a value of ti that is less than the value predicted from the molar mass (circled points). This molar mass dependence of ti is reasonably strong for propylene-rich EP copolymers between 90 and 110 °C but is weak at 120 and 130 °C. At 120 °C the degradation temperature is still considerably less than the melting point, between 12 and 25 °C less than Tm in EP5.1 and EP0.6, respectively (table 5).
Figure 8 Variation of ti with the molar mass for PP samples (black symbols) and propylene-rich EP copolymers (white symbols).
Stereo-defect content
The variation of ti with the content of isotacticity interruptions caused by stereo defects is shown in figure . From 90 to 120 °C, ti for pure iPP polymers has a minimum value at an mr content of 5–6 mol.%. At 130 °C, ti decreases continuously and linearly.
Figure 9 Variation of ti with the content of stereo defects for PP samples (black symbols) and propylene-rich EP copolymers (white symbols).
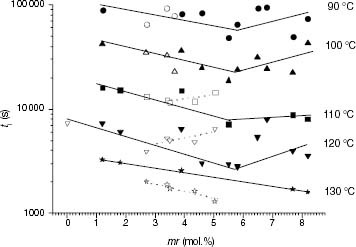
For the propylene-rich EP copolymers, tacticity reveals a distinct dependence on ti. Although the stability of the less defective propylene-rich EP samples fits the iPP trend at each temperature between 90 and 120 °C, the more complete series at 110 and 120 °C also exhibit minima at concentrations of mr triads of about 3–4 mol.%. In this propylene-rich EP family, thermo-oxidation at 130 °C has not only a negative linear dependence of ti on the mr content but also a different dependence to that displayed by iPP samples.
Ethylene content
The effect of the ethylene content on ti for EP copolymers is qualitatively similar to that described for the mr interruptions (figure ). In fact the stability of EP copolymers exhibit a minimum at very low ethylene concentrations (between about 3 and 4 mol.%). The ti of PP92.4 was taken as the reference value for pure iPP. This sample has both a molar mass and an isotacticity similar to those of EP copolymers that exhibit the decrease in thermal stability in figure , up to a conversion of about 3 mol.%.
Figure 10 Variation of ti with the ethylene content.
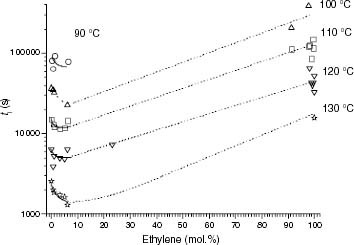
This particular variation suggests that, in addition to the well-known stabilising effect of introducing ethylene units in EP copolymers [Citation22], other factors should be considered to account for such a negative effect.
Discussion
Effect of molar mass
Figure shows the expected general relation between stability and molar mass for iPP samples, that is, the larger the chain size, the more stable the behaviour of the material; however, the relation is not as simple as it would be if the only effect was the content of chain ends. Actually, the chain size can explain the observed rapid increase in ti up to molar masses of about 45 g ml−1 but, for higher values, other parameters must play a role because considerable deviations from the average trends are found. On the other hand, the stability of iPP samples at 90 °C does not change anymore above 45 g ml−1.
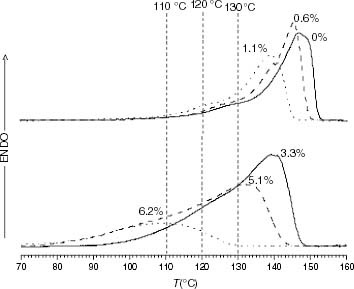
In the case of propylene-rich EP copolymers, ti roughly fits the molar mass variation of iPP samples between 90 and 110 °C. In contrast, the stabilities of the propylene-rich EP copolymers are markedly less than those corresponding to iPP with equivalent molar masses at 120 and 130 °C. This divergence may be associated with the fact that the introduction of a low number of ethylene units causes a decrease in Tm, which becomes considerable at 1.1 mol.% (see table 5). Furthermore, the melting endotherm broadens with the ethylene conversion as depicted in figure . The consequence is that EP thermo-oxidation occurs not only at temperatures closer to the melting point than pure iPP thermo-oxidation, but also in a partially melted state, as inferred from the superposition of the degradation temperatures on the DSC curves in figure . It is clear that above 120 °C the low-Tm crystalline fraction is increasingly involved in the melting process when the ethylene content increases, and the enhanced diffusion of oxygen may contribute to the lower stability.
Figure 11 DSC melting endotherms of PP and propylene-rich EP samples (the ethylene content is indicated on each curve).
The ti-molar mass correlation observed agrees with the reported effect of molar mass on the thermal stability of propylene-based polyolefins [Citation23], but it also shows that other features, i.e. microstructure and/or crystallinity, must play an important role.
Effect of the chromophore content
Among the anomalous chemical functions known to depress the thermal stability of polyolefins, enones have a recognised role [Citation20, Citation21]; nevertheless, the values of ti for PE samples and ethylene-rich EP copolymers are clearly higher than those for PP samples and propylene-rich EP copolymers (figure ), in spite of their greater content in these chromophores (figure ). Moreover, EP0.6 and EP1.1, which are found to have the least stability in figure , exhibit the lowest phosphorescence intensity (figure (a)). Thus, enones are excluded as a main cause of thermal unstability, and other microstructure characteristics, i.e. configuration and composition, should be considered as essential factors determining the stability of propylene-based polyolefins, either intrinsically or through their effect on the iPP semicrystalline structure.
Effect of the ethylene content
It is accepted that the thermal stability of EP copolymers is improved when the ethylene content increases [Citation3, Citation5]. This fact is clear from the change in stability of the solid-state EP copolymers shown in figure ; however, ti is surprisingly low for very low ethylene contents (under 5 mol.%). This effect is more clearly shown in figure , which shows the variation of ti only for propylene-rich EP copolymers. The increase in ti over a certain ethylene content can be accounted for by the higher chemical stability of ethylene units, as well as the barrier effect of the comonomer in the intrachain hydroperoxide formation [Citation2–5], when the degradation temperature approaches the melting point.
Figure 12 Variation of ti with the ethylene content for the propylene-rich EP copolymers.
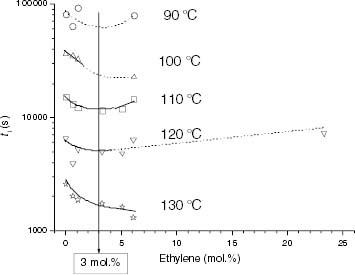
It is clear that at less than a 5 mol.% ethylene, there must be other factors that cause the stability to decrease. In this regard, both the decrease in Tm (table 5) and the broadening of the PP melting endotherm on the lower-temperature side, which is significant at low levels of ethylene insertion (figure ), can be claimed as destabilizing factors because they promote the partial melting of the crystalline phase and therefore the diffusion of oxygen through a higher fraction of the polymer bulk. The effect of the solid-melt transition phase on the stability trend is illustrated in figure . The complete melting of certain EP copolymers (points in red) at Tdeg between 110 and 130 °C reveals that the aggregation state is essential in the thermal stability of these polyolefins. In contrast to the upward tendency of ti exhibited by solid samples with over 5 mol.% ethylene conversion, a continuous decrease in ti with ethylene conversion is found in the melt. The results obtained by Achimsky et al [Citation24] show that iPP degradation at 140 °C is strongly controlled by diffusion and that the oxidation rate increases with the oxygen pressure. Therefore, chain accessibility to oxygen is further enhanced when Tdeg> Tm and is expected to play a prevailing role in the thermo-oxidation of EP copolymers, compared with the effect of the ethylene content, in the composition range studied (up to 11 mol.%). The negative effect of the ethylene content on the stability of propylene-rich EP copolymers, shown in figure , suggests that the degradation is not kinetically controlled. Moreover, the decrease in the calculated Eact for some propylene-rich EP copolymers when Tdeg> Tm, as indicated in the experimental section (figure ), shows that a change in the thermo-oxidation mechanism takes place associated with the solid-melt transition phase. On this basis, the discussion has been restricted in this study to the results on stability at temperatures within the Tα–Tm range (typically 80–150 °C), where no kinetic transitions occur.
Figure 13 Variation of ti with the ethylene content for the propylene-rich EP samples at 110, 120 and 130 °C. Black points correspond to Tdeg < Tm and red points correspond to Tdeg > Tm.
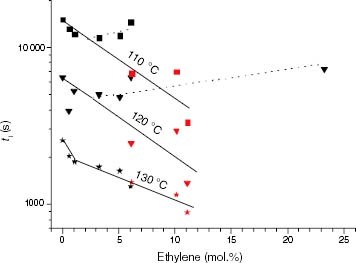
The main result shown by figures and is that the insertion of ethylene in polypropylene chains has not only a positive effect on the thermal stability of solid-state EP copolymers, but also a negative effect at very low ethylene conversions, either because of the introduction of isotacticity interruptions or because of the increased oxygen diffusion, or both.
Effect of the stereo-defect content
Similarly to ethylene insertion, a stereo-defect content of up to a 6 mol.% causes a decrease in stability of solid iPP samples between 90 and 120 °C (figure ). Above 6 mol.%, the stability recovers. In this case, because of the absence of a chemical change, the variation of ti is evidence that stereo defects have an equivalent effect to that found for ethylene units on thermal stability. Therefore, the composition dependence of ti found in propylene-rich EP copolymers can be attributed, at least partially, to the role played by ethylene units as interruptions.
Interruptions of the stereoregularity have a well-known effect on both chain flexibility and crystallinity, two aspects that are expected to markedly affect the thermal performance of polyolefins. As far as the crystallinity is concerned, the decrease in the iPP stability is related to the depletion of the crystalline content; however, this relationship exhibits very strong deviations. Straightforwardly, the bulk fraction accessible to oxygen is a prime parameter in thermo-oxidation as expected, but it cannot provide a reliable and accurate prediction for ti over the whole molar mass and microstructure ranges studied.
The variation of ti at 130 °C appears to change because ti decreases linearly in the whole mr range. At this temperature, the low-Tm shoulder of the iPP DSC endotherm is clearly involved and there is a change in both the quantity and the quality of the crystalline phase. At this stage, where incipient melting occurs, oxygen diffusion is promoted, while the effects from solid-state parameters, i.e. interphase features, are expected to vanish.
The minimum ti at an interruption content of 6 mol.% corresponds to an average isotactic length (n1) of approximately 30 propylene units according to the following expression taken from the analysis of propylene-based copolymers performed by Randall [Citation25].
Randall's equation considers the populations of dyads in a composition but it can be used in a equivalent form by taking the populations of dyads of CH2 groups, each of them being meso (m) or racemic (r), for the regioregular insertion of propylene units, or adjacent to an anomalous unit (x) in the case of the regioirregular insertion of propylene or ethylene.
The minimum ti at n1 = 30 suggests that the iPP stability is related to a microstructure-induced change in the average size of crystals which, together with the mr content, is thought to be critical in the quality of their associated interphases. Therefore, the content and quality of the crystalline interfacial region are thought to be the main criteria in ascertaining the iPP stability.
Figure shows that for the propylene-rich EP copolymers a minimum in ti is related to a change in the configurational microstructure; however, some EP copolymers are more stable than expected from their mr content. These samples turn out to be the richest in ethylene and they reflect the effect of composition on ti. Alternatively, it has been shown experimentally that the ti values of the richest propylene samples (up to 3 mol.%) do not agree with the above-mentioned general trend of the iPP-crystallinity. This fact supports the idea that, although the diffusion of oxygen is required, there must be other important aspects in the disordered fraction that make the initiation of thermo-oxidation more or less feasible.
The result of this analysis allows us to propose that the insertion of isotacticity interruptions has an effect on the thermal stability of iPP, irrespective of the chemical nature of the interruption.
Effect of the total content of interruptions
Since all types of isotacticity interruption seem to play a common role, isotactic interruptions can be used to test whether the mere fact of interrupting the stereoregularity is a major cause of the thermal stability with a greater effect than the chemical nature of the interruption. Figure shows that a distinctive variation of ti with the total content of interruptions, including a very small contribution of regiodefects (tables 4 and 5), develops between 90 and 130 °C for iPP and propylene-rich EP copolymers.
Figure 14 Variation of ti with the total of isotacticity interruptions for PP samples (black symbols) and propylene-rich EP copolymers (white symbols).
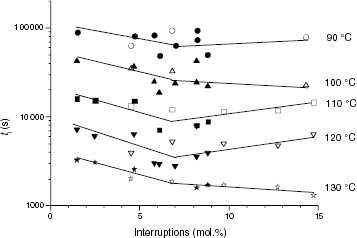
Although increased oxygen diffusion may be an explanation for the decrease of ti within the 0 to 7 mol.% range of interruptions, this reason is not consistent with the further stability increase above 7 mol.%. This is particularly evident in the pure iPP family, where the increase in isotacticity interruptions does not imply the introduction of more stable units, as is the case of EP copolymers.
The coincident effects of mr and a low ethylene contents on changes in ti for PP and propylene-rich EP copolymers indicate their common characteristics associated with their roles of stereoregularity interruptions. The determination of these parameters can be guided by a deeper insight into the microstructure of these propylene-based samples.
Effect of the nature of the interruption
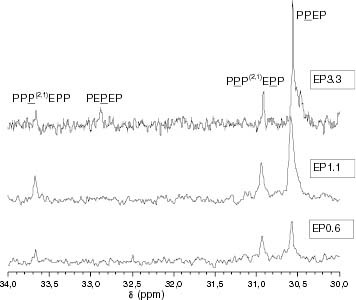
The 13C-NMR analysis showed that iPP samples contain mostly short interruptions of the mrrm type. Actually, with the exception of PP131.9, 13C-NMR characterisation yields mr/rr ratios of approximately 2 (table 4). In the case of propylene-rich EP copolymers, the samples with the lowest ethylene conversion contain only short interruptions of either mrrm (mr/rr value approximately 2, table 5) or isolated ethylene units. Figure displays the methine window of the 13C-NMR spectra corresponding to EP samples with an ethylene conversion of up to 3.3 mol.%. It is clear that 0.6 and 1.1 mol.% samples have only isolated ethylene units that are either normal (PEP) or associated with a propylene misinsertion (P(2,1)EP) [Citation26], but in the 3.3 mol.% sample longer interruptions are present. From this conversion, PEPEP appears and interruptions become longer and richer in ethylene as the ethylene conversion increases.
Figure 15 Methine window of the 13C-NMR spectra for EP0.6, EP1.1 and EP3.3.
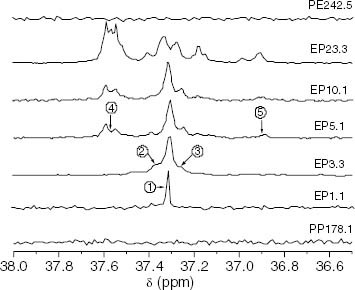
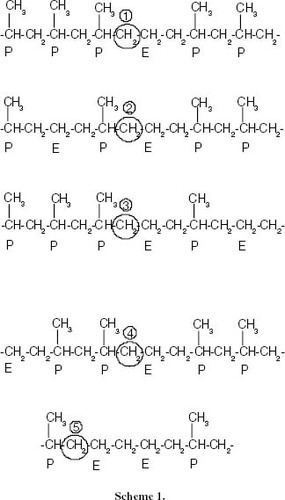
The change in the nature of the interruption is particularly noticeable above a conversion of 5.1 mol.%, where ethylene sequences appear and the content of PEP structures associated with short propylene sequences increases conspicuously. This evolution is inferred from the analysis of the 13C signals of methylene units of propylene-ethylene bonds in figure . Scheme 1 attached to figure displays the different CH2 groups resolved in the spectra according to the assignment reported in the literature [Citation27].
Figure 16 Changes in the 13C-NMR spectrum with the ethylene content showing the region corresponding to propylene-ethylene insertions of methylene, which are contiguous to methines. The chemical environment of the CH2 nuclei is shown in scheme 1.
The minimum value of ti in figure occurs at an interruption content equivalent to an average isotactic length of approximately 30 units, which, in the case of propylene-rich EP copolymers, coincides with a change in the quality of the interruptions. The correlation between this stability change and the microstructure is supported by the analysis of the variation of Eact, which is calculated from the temperature dependence of ti. Figure confirms that there is a unique Eact variation with n1 resulting from the ensemble of iPP and propylene-rich EP copolymers. Eact for the thermo-oxidation is roughly constant at about 103 kJ mol−1 for samples with n1 of over 30 units, but it increases quasi-exponentially for n1 shorter than 30. The fact that for the microstructure, the Eact values of iPP and propylene-rich EP copolymers are in a unique function suggests that n1 controls the oxidation mechanism irrespective of the chemical nature of the isotacticity interruptions, at least in the range of iPP samples considered (20 to 130 propylene units). In the case of EP copolymers, a further increase in Eact as the number of propylene units decreases from 20, may also reflect a degradation process taking place in increasingly stable ethylene-rich material. Figure shows a direct correlation between Eact and the ethylene content in the solid and melt states.
Figure 17 Variation of Eact of the thermo-oxidation in the solid state with the average isotactic length (n1) for PP samples (black symbols) and propylene-rich EP copolymers (white symbols). Red points correspond to the Eact values of EP samples degraded in the melt.
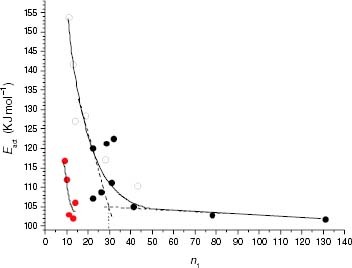
Figure 18 Variation of Eact with the ethylene content for the thermo-oxidation of propylene-rich EP copolymers in the solid state (black symbols) and molten state (red symbols). The range of Eact corresponding to PP samples is indicated on the Y-axis.
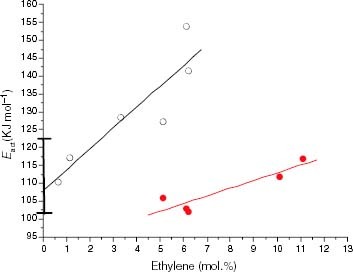
The variation of Eact in the melt is also shown in figure for some EP samples. It can be seen that the typical value of approximately 103 kJ mol−1, corresponding to the degradation of the most isotactic PP, is found for propylene-rich EP copolymers with n1 of approximately or slightly over 10 propylene units. The main effect of the transition phase is thus to allow the most isotactic crystallisable propylenic fraction to be oxidised. For n1 under 10 propylene units, the increase in the apparent Eact reveals that the ethylene content is sufficiently high for the chains to be more stable than those in PP.
The increase in Eact in iPP thermo-oxidation as the isotacticity decreases has been attributed to an increasing predominance of the unimolecular mechanism in the POOH decomposition, which is more energy-consuming than the bimolecular mechanism [Citation6]. Without disregarding a minor contribution of the former to the apparent Eact of the iPP thermo-oxidation, the CL emission is recognised to mostly originate from the bimolecular process, which is the main source of alcoxi radicals and, thus, of light through Russell's mechanism [Citation28]. Many studies on iPP thermo-oxidation support the occurrence of a unimolecular POOH decomposition process restricted to the induction period, while the CL buildup is essentially associated with the bimolecular process [Citation29–31]. Consequently, a minimum concentration of POOH is required for the decomposition to take place. Such a POOH threshold can be reached via intra- or interchain oxidation depending on whether regular conformations of the iPP chain are accessible to a backbiting POOH buildup or not. The aggregation state is thought to have a strong effect on the balance between intra- and interchain oxidation. The oxidation of dissolved and molten iPP is expected to result in mostly intrachain POOH interactions. In contrast, oxidation in the solid state is expected to yield a high content of interchain POOH associations. However, the decomposition process remains essentially bimolecular and the change in Eact found at n1 = 30 should be related to a change in the quality of the ‘weakest points’ initiating the oxidation.
The values of Eact are within the typical range reported for a typical hydroperoxide decomposition process [Citation32], but the change is so abrupt that we think that the qualities of structures involved in thermo-oxidation are markedly different; short interruptions are associated with n1 over 30 units for low Eact values, and short or long interruptions are associated with n1 under 30 units for high Eact values.
The average isotactic length and the content of interruptions determine the average size of crystals and the interphase characteristics that are thought to be involved in the initiation of oxidation. As a tentative approach, the local free volume at the interphase is proposed to be the common parameter involved in all interruptions, and is able to govern the conditions, i.e., the accessibility of oxygen and local dynamics, under which oxidation takes place.
Effect of the sub-Tg local dynamics
The greater importance of the interphase in the oxidative degradation of semicrystalline polymers has been observed, for example, in polyethyleneoxide and polyethylene [Citation33]. In the particular case of the themo-oxidation of unstabilised polypropylene, this fact is beyond doubt because Lacey and Dudler [Citation34] succeeded in picturing the initial degradation states by the chemiluminescence imaging. Various reasons have been put forward to account for the ability of the interphase to initiate oxidation. Usually, the effects of catalyst residues [Citation35] and low-molar-mass atactic fractions [Citation36], which are rejected at the crystal boundaries, have been suggested as the main reasons.
In previous studies by our group we have tried to determine whether the local dynamics of the iPP crystalline interphase makes this region behave as a ‘weak’ zone in thermo-oxidation. It has been found that the α relaxation is associated with an abrupt change in the fraction of the polymer involved in the initiation [Citation16]. On the other hand, the γ relaxation is related to the crystalline phase. Actually, the initial annealing undergone by a metallocene iPP degraded at 75 °C promotes the growth of low-Tm crystals together with the simultaneous increase and disappearance of the γ and α relaxations, respectively [Citation13].
On the basis of these works, the sub-Tg dynamics of iPP is thought to occur at high temperatures unless the crystalline morphology to which it is associated changes or the destruction of the structures responsible for the dynamics takes place. It is reasonable to expect that such dynamics will operate at high frequency at temperatures well above Tg and thus, will promote the cleavage of chain bonds and the subsequent reaction of radicals with the available oxygen. The vanishing of the γ relaxation in the very early stages of thermo- and photodegradation [Citation37, Citation38] indeed confirms the dominant role of the sub-Tg dynamics in the selective degradation of the iPP interphase.
The correlation of ti with the sub-Tg local dynamics has been found to occur in iPP samples [Citation9]. In this study we attempted to verify the validity of this correlation for the thermo-oxidation of solid iPP-based polyolefins. Figure shows evidence that ti for the thermo-oxidation of solid-state iPP-based samples is proportional to the relative intensity of the γ relaxation in the temperature range studied. This correlation is evidently valid when the crystalline phase is retained and typical iPP γ dynamics is active. In cases where the degradation temperature implies that a significant fraction of the low-Tm crystalline phase melts, local motions associated with the crystalline distribution of iPP disappear. This happens at 130 °C where no unique dependence of ti on the intensity of γ is apparent.
Figure 19 Variation of ti with the relative intensity of sub-Tg DMA relaxation.
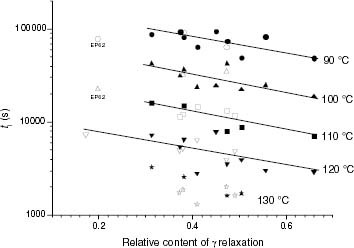
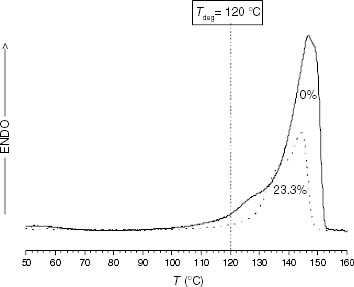
The deviation from the ti-γ relaxation correlation of EP6.2 at temperatures below its melting point (90 and 100 °C) can be ascribed to the fact that this copolymer lacks a typical iPP crystalline phase. Figure shows that this EP copolymer has an almost single low-temperature melting endotherm, and it is plausible that the local motions associated with a double-melting crystalline distribution disappear. In contrast, the EP23.3 sample unexpectedly fits the ti-γ relaxation trend at 120 °C, in spite of its high ethylene conversion. Further characterisation is required to determine whether this sample is a block copolymer or a blend. However, the γ relaxation, although small, corresponds to the local sub-Tg dynamics associated with a true iPP crystalline phase, as inferred from the double-melting endotherm shown in figure . Consequently, the value of ti at 120 °C appears to be mainly determined by the γ relaxation associated with the small iPP crystalline fraction.
Figure 20 DSC melting endotherms of PP92.4 (—) and EP23.3 (····).
The correlation between ti and the relative intensity of the γ relaxation is evidence that the local dynamics of localized regions in the iPP assist the oxidation chemistry involved in the initial formation of POOH and further oxidation products.
Conclusions
The chain microstructure has been found to play a crucial role in the thermal stability of iPP and propylene-rich EP copolymers in the solid state. In particular, average isotactic lengths of over 30 propylene units, associated with short isotacticity interruptions, exhibit thermo-oxidation with a low Eact value of about 103 kJ mol−1. Provided that these microstructure requirements are fulfilled, the insertion of isotacticity interruptions depresses the thermal stability of the material. In contrast, if the average isotactic length is under 30 units, the insertion of defects either in the configuration or in the composition, causes both Eact and the stability of the polyolefin to increase. The role of the chain microstructure can be explained in terms of the induced effects on the crystalline interphase features, i.e. local molecular dynamics, which are suggested to be closely linked to the initiation of thermo-oxidation. The relative intensity of the iPP sub-Tg relaxation can be used to predict the induction time of the degradation process in the solid state.
Acknowledgments
The authors acknowledge the financial support from Comisión Interministerial de Ciencia y Tecnología (CICYT MAT2005-05648-C02-01). The corresponding author also acknowledges the financial support from Comisión Interministerial de Ciencia y Tecnología (CICYT MAT2006-03831).
References
- SokolovskiiA ABorisovaN NAngertL G 1975 Visokomol. Soyed. A 17 1107
- ManabeNYokotaYNakataniHSuzukiSLiuBTeranoM 2006 J. Appl. Polym. Sci. 100 1831 http://dx.doi.org/10.1002/app.22058
- ManabeNYokotaYNakataniHSuzukiSLiuBTeranoM 2005 Polym. Bull. 54 141 http://dx.doi.org/10.1007/s00289-005-0370-9
- TeranoMLiuBNakataniH 2004 Macromol. Symp. 214 299 http://dx.doi.org/10.1002/masy.200451022
- SuzukiSLiuBTeranoMManabeNKawamuraKIshikawaMNakataniH 2005 Polym. Bull. 55 141 http://dx.doi.org/10.1007/s00289-005-0413-2
- NakataniHSuzukiSTanakaTTeranoM 2005 Polymer 46 12366 http://dx.doi.org/10.1016/j.polymer.2005.10.131
- SuzukiSNakamuraYHasanATMLiuBTeranoMNakataniH 2005 Polym. Bull. 54 311 http://dx.doi.org/10.1007/s00289-005-0389-y
- Gómez-ElviraJ MTiembloPElviraMRychláLRychlýJ 2004 Polym. Deg. Stab. 85 873 http://dx.doi.org/10.1016/j.polymdegradstab.2004.04.003
- HoyosMTiembloPGómez-ElviraJ MRychláLRychlýJ 2006 Polym. Deg. Stab. 91 1433 http://dx.doi.org/10.1016/j.polymdegradstab.2005.10.013
- WunderlichB 1976 Macromolecular Physics vol II New York Academic
- WunderlichB 1973 Macromolecular Physics vol I New York Academic
- HoyosMTiembloPGómez-ElviraJ M 2007 Polymer 48 183 http://dx.doi.org/10.1016/j.polymer.2006.11.034
- GuisándezJTiembloPGómez-ElviraJ M 2005 Polym. Deg. Stab. 87 543 http://dx.doi.org/10.1016/j.polymdegradstab.2004.10.014
- BusicoVCipulloRCorradiniPLandrianiLVacatelloM 1995 Macromolecules 28 1887 http://dx.doi.org/10.1021/ma00110a024
- TiembloPGómez-ElviraJ MTeyssedreGMassinesFLaurentC 1998 Polym. Int. 46 33 http://dx.doi.org/10.1002/(SICI)1097-0126(199805)46:1<33::AID-PI964>3.0.CO;2-M
- TiembloPGómez-ElviraJ MGarcíaSRychláLRychlýJ 2002 Macromolecules 35 5922 http://dx.doi.org/10.1021/ma0119373
- RychlýJRychláLTiembloPGómez-ElviraJ M 2001 Polym. Deg. Stab. 71 253 http://dx.doi.org/10.1016/S0141-3910(00)00169-5
- BillinghamN CGriggM N 2004 Polym. Deg. Stab. 83 441 http://dx.doi.org/10.1016/j.polymdegradstab.2003.06.001
- AchimskyLAudouinLVerduJMatisova-RychláL 1997 Polym. Deg. Stab. 58 283 http://dx.doi.org/10.1016/S0141-3910(97)00059-1
- LaceyD JDudlerV 1996 Polym. Deg. Stab. 51 109 http://dx.doi.org/10.1016/0141-3910(95)00192-1
- AllenN SHomerJMcKellarJ F 1977 J. Appl. Polym. Sci. 21 2261 http://dx.doi.org/10.1002/app.1977.070210823
- Alam MdSNakataniHLiuBTeranoM 2002 Recent Res. Devel. Macromol. 6 213
- Matisova-RychláLRychlýJTiembloPGómez-ElviraJ MMarcosM 2004 Macromol. Symp. 214 261 http://dx.doi.org/10.1002/masy.200451019
- AchimskyLAudouinLVerduJRychláLRychlýJ 1999 Eur. Polym. J. 35 557 http://dx.doi.org/10.1016/S0014-3057(98)00174-8
- RandallJ C 1978 Macromolecules 11 592 http://dx.doi.org/10.1021/ma60063a032
- BussicoVCipulloRRoncaS 2002 Macromolecules 35 1537 http://dx.doi.org/10.1021/ma011400p
- HayashiTInoueYChÛjoRAsakuraT 1988 Polymer 29 2208 http://dx.doi.org/10.1016/0032-3861(88)90113-9
- GeorgeG A 1981 Developments in Polymer Degradation vol 3 NGrassie London Applied Science Publishers p 173
- Matisova-RychláLRychlýJVerduJAudouinLCsomorovaK 1995 Polym. Deg. Stab. 49 51 http://dx.doi.org/10.1016/0141-3910(95)00048-Q
- StannettVMesrobianR B 1953 Discuss. Faraday Soc. 14 9 http://dx.doi.org/10.1039/df9531400009
- Matisova-RychláLRychlýJVavrekovaM 1978 Eur. Polym. J. 14 1033 http://dx.doi.org/10.1016/0014-3057(78)90163-5
- ZolotovaN VDenisovE T 1971 J. Polym. Sci. A-1: Polym. Chem. 9 3311 http://dx.doi.org/10.1002/pol.1971.150091117
- DeckerC 1977 Makromol. Chem. 178 2969 http://dx.doi.org/10.1002/macp.1977.021781022
- LaceyD JDudlerV 1996 Polym. Deg. Stab. 51 101 http://dx.doi.org/10.1016/0141-3910(95)00151-4
- GijsmanPHennekensJVincentJ 1993 Polym. Deg. Stab. 39 271 http://dx.doi.org/10.1016/0141-3910(93)90001-Y
- BillinghamN C 1989 Makromol. Chem. Macromol. Symp. 28 145
- OlivaresNTiembloPGómez-ElviraJ M 1999 Polym. Deg. Stab. 65 297 http://dx.doi.org/10.1016/S0141-3910(99)00019-1
- CastejónM LTiembloPGómez-ElviraJ M 2001 Polym. Deg. Stab. 71 99 http://dx.doi.org/10.1016/S0141-3910(00)00159-2