Abstract
The electronic structure of boron–hydrogen complex and boron pair in diamond are studied by first-principles density-functional calculations with supercell models. The electronic structure calculated for the B–H complexes with C2v or C3v symmetry and the nearest-neighbor B pair is used to interpret recent experimental results such as B 1s x-ray photoemission spectroscopy, 11B nuclear quadruple resonance and B K-edge x-ray absorption spectroscopy, which cannot be explained solely by the isolated substitutional boron.
Introduction
Previously, it has been shown by several first-principles calculations [Citation1–5] that substituting a few per cent of carbon atoms in diamond by boron introduces hole carriers to the valence band maximum (VBM), which might be responsible for superconductivity observed in B-doped diamond. Calculated electronic structure of the B-doped diamond is quite consistent with experimental findings by angle-resolved photoemission spectroscopy [Citation6], carbon 1s x-ray photoemission spectroscopy (XPES) [Citation6] and carbon K-edge x-ray absorption and emission spectroscopy [Citation7]. However, in film samples, the growth-orientation dependence of Tc has been found [Citation8, Citation9], indicating certain kind of passivation of carriers and a close relation to the existence of uniaxial strain [Citation10]. Furthermore, more than one B site in diamond has been observed by B K-edge x-ray absorption spectroscopy [Citation7], B 1s XPES [Citation11] and 11B nuclear quadruple resonance (NQR) [Citation12]. These results suggest possible existence of B–H complexes and/or B pairs in B-doped diamond. Calculated B K-edge x-ray absorption spectra (XAS) for different B concentrations doped at the substitutional sites in diamond show hole states at VBM but no in-gap states [Citation13]. In the measured XAS, on the other hand, there are clear in-gap states in addition to the hole states [Citation7]. Because the B K-edge XAS involve the electric dipole transitions from B 1s to 2p states, the in-gap states must be of B 2p origin associated with a different environment from the substitutional site. The experimental B 1s XPES reveals distinct features for different B concentrations [Citation11]. Samples with low B concentration show single peak whereas samples with high B concentration have multi-peak structure at higher binding energy, indicating different boron sites. The 11B NQR reveals at least two B sites; at one site the electric field gradient has a finite value and at another it is exactly zero [Citation12]. The former site is attributed to the B–H complex and the latter to the high-symmetry substitutional B. The NQR spectrum suggests more than one type of B–H complexes and/or B pairs.
The electronic structure and stability of B–H complex in diamond have been extensively studied by electronic structure calculations [Citation14–21]. It has been found that the most stable position of the H atom around the substitutional B in diamond is a site slightly shifted from the bridging point between the two B–C bonds. The calculated total energy is 0.6 eV lower than that of H at the B–C bond-center position [Citation16]. Theoretical studies of diamond doping, including B–H complex, have been concisely summarized in a recent review [Citation22]. As for B pairs, some first-principles calculations have been carried out to understand their stability and electronic structure [Citation17, Citation21, Citation23–25]. Boron pairs are definitely more stable than the isolated substitutional B. It has been shown that the formation energies of B pairs at the neighboring substitutional positions are between 0.3 and 0.8 eV [Citation23]. Although the electronic structure and stability of B–H complexes and B pairs in diamond are well understood from those electronic-structure calculations, the x-ray absorption spectra, the electric field gradients observed by NQR and the core levels addressed by recent experiments [Citation7, Citation11, Citation12] have not been studied in detail theoretically.
In this study, we investigate the electronic structure of some B–H complexes and B pair in diamond by calculating XAS at B K-edge, electric field gradients at the B site and core level of B in order to explain experimental results mentioned above. Our calculations are based on the first-principles density-functional theory within the local density approximation.
Method
One-electron Kohn–Sham equations are solved with the all-electron full-potential linear augmented-plane-wave method in a scalar relativistic manner. In order to simulate impurities in diamond, supercell models are adopted in the present calculations. Typical supercell is a 64-atom (2a×2a×2a) cubic cell of diamond structure. Relaxation of the internal coordinates by impurity doping is fully taken into account while the lattice constant is fixed at the calculated equilibrium value for pure diamond. Computational details in this study basically follow the previous work [Citation13].
Results and discussion
The first B–H complex in diamond to be investigated is substitutional B and interstitial H situated near and simulated with a C63BH supercell model. Optimized B–H complex in diamond has local C3v symmetry with the B-H bond length of 1.17 Å, called H at the back-bond (BB) site.
Calculated electronic structure shows that an in-gap empty s state of H is hybridized with the neighboring carbon p states but not with the boron states. Holes introduced at VBM by B doping at the substitutional sites are compensated by electrons from H, resulting in B−–H+ pairs. Energy lowering of occupied B p states is due to attractive potential by H and hybridization with the hydrogen s states.
The second B–H complex is substitutional B and H at the bond-center (BC) site between B and C; this complex has local C3v symmetry. Insertion of H introduces large relaxation of the nearest B and C, leading to the bond lengths of 1.18 Å for B–H and of 1.03 Å for C–H whereas displacements of further sites are rather minor. In this C3v bond-center case, the in-gap state has the largest amplitude at boron p states and on its nearest neighbor carbon p states but almost no contribution at hydrogen. Again, holes introduced to the VBM by substitutional B doping are compensated by electrons from H (B−–H+ pair formation).
The third B–H complex is substitutional B and interstitial H at the bridging site; the complex has C2v symmetry. Optimized B–H bond length is 1.20 Å. Similar to the C3v back-bond situation, an in-gap state consists of H s states and neighboring C p states but not B states. Hole compensation and energy lowering of the B p states are also revealed for this configuration.
Let us discuss the energetics and stability of B–H complexes. Calculated total energy differences are:
(1)
(2)
The B–H complex with C2v symmetry is the most stable among the three studied complexes, in agreement with the previous total-energy calculation [Citation16].
In addition to the B–H complexes in diamond, the electronic structure of boron pairs is investigated from the first principles. Two B atoms are fixed at the nearest-neighbor substitutional sites, forming B dimers. Optimized bond length of B dimer is significantly longer (by 0.4 Å) than the C–C bond in diamond. The stability of the nearest-neighbor B pair relative to the isolated substitutional B in diamond is analyzed by a total-energy calculation. Calculated total energy shows that the B pair is more stable, by about 1 eV per pair, which is slightly larger compared with the previous first-principles calculation [Citation23].
The most striking feature in the electronic structure of the nearest-neighbor B pair in diamond is an empty split-off state located just above VBM and composed mostly of the B p orbital (say pz) pointing to the B–B bond direction with bonding character. The other B p components (px and py) perpendicular to the B–B bond with bonding character are mostly located inside the valence band. Because boron has three valence electrons, one of the four bonding states associated with the B s and p orbitals should be empty. The elongation of the B–B bond makes the px and py (pz) bonding states lower (higher) in energy, resulting in the stability of the B dimer in diamond. The existence of this empty split-off state means that B doping in a dimer form introduces no hole carriers at VBM.
Let us compare the electronic structure of B–H complexes and B dimer in diamond with some recent experimental results. We consider first the C 1s core state spectra. In the previous study, the shoulder structure at lower binding energies was successfully explained by the 1s core shift of C surrounding the substitutional B site [Citation13]. It is found in this study that the shoulder structure in the theoretical spectra for all B–H complexes and B dimer in diamond becomes slightly weaker than that in the isolated substitutional B case but is still present in the lower binding energy region. Experimental B 1s XPES shows multiple structure for heavily B-doped samples at higher binding energies [Citation11]. Calculated B 1s core levels for the B–H complexes and nearest-neighbor B pair in diamond are lower by 1.4–1.8 eV than that of the isolated substitutional B, being consistent with the experimental findings. Broad structures observed experimentally at much higher binding energies may be of surface origin. These features of the C and B 1s spectra do not rule out any B–H complex or B pair in diamond.
The NQR quadruple coupling frequency provides information on electric field gradient at the studied nucleus. The electric field gradient tensor at a nuclear site is given as
(3)
where V is the crystal potential around the nuclear site. By diagonalizing the tensor, the principal axes X, Y, and Z, the electric field gradient q, and anisotropy parameter η can be evaluated as
(4)
The electric field gradient q is related to the quadruple coupling frequency in the NQR experiment
(5)
Here e is the unit charge, Q is the electric quadruple moment (Q=0.04e×10−28 m2 for 11B), I is the nuclear magnetic moment (I=3/2 for 11B) and h is the Planck constant. Mukuda et al have measured 11B NQR spectra for B-doped diamond and found a lower symmetry signal at νQ=1.8 MHz in addition to the high symmetry one [Citation12]. The electric field gradients are calculated for B–H complexes and B dimer in diamond and listed in table 1. The observed frequency is consistent with B-H complex with bond-center configuration or with the B dimer. Because the experimental data may include smaller but finite frequency components, B–H complexes with C2v and C3v back-bond structures can not be ruled out completely.
Calculated principal axis Z, electric field gradient q, quadruple coupling frequency νQ and anisotropy parameter η for B–H complexes and B dimer in diamond.
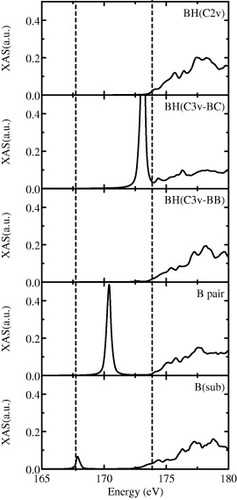
Finally, B K-edge XAS are discussed in detail. The experimental spectra have recently been reported by Nakamura et al [Citation7] for superconducting and non-superconducting B-doped diamond samples and have shown the existence of in-gap states. XAS are calculated with the Fermi's golden rule within the electric dipole approximation. Some detailed methodological points for XAS calculation have been given elsewhere [Citation13, Citation26]. Figure depicts calculated B K-edge XAS for B–H complexes and B dimer in diamond in addition to the isolated substitutional B. (Note that the absolute value of the photon energy for the transition (1s→2p) is underestimated because of the one-electron approximation.) The spectra for the B–H complex with C3v back-bond and the nearest-neighbor B pair are reasonably consistent with the experimental ones with significant in-gap states. The reason why the in-gap state appears so deep for the B dimer, despite the B pair impurity state located just above VBM, is mainly due to the deeper 1s core state of the nearest-neighbor B pair than that of the isolated substitutional B. Theoretical observation of the in-gap states and related hole compensation for the B–H complex and B pair in diamond may explain the experimental fact that the in-gap states are generally more prominent for heavier doped samples with relatively low Tc [Citation7].
Figure 1 Calculated B K-edge x-ray absorption spectra for B–H complexes (C2v, C3v back-bond and C3v bond-center), nearest-neighbor B pair and isolated substitutional B in diamond. Broken lines denote rough positions of the valence band maximum and conduction band minimum.
Concluding remarks
The electronic structure of the B–H complexes and the nearest-neighbor B pair in diamond are calculated from first principles and compared with recent B 1s XPES, 11B NQR and B K-edge XAS. It is shown that the B-H complex with C3v bond-center structure and the nearest-neighbor B pair may possibly exist in heavily B-doped film samples. The most stable H position near the substitutional B in diamond is known as an intermediate site between the C3v bond-center and C2v [Citation15, Citation16, Citation19]. A further investigation for the most stable B–H complex is important and is underway.
Acknowledgments
The author acknowledges T Yokoya, J Nakamura, and H Mukuda for invaluable discussions. This work is supported in part by a Grant-in-Aid for Scientific Research in Priority Area ‘Invention of Anomalous Quantum Materials’ of the Ministry of Education, Culture, Sports, Science and Technology, Japan. The computation in this work has been partly performed using the facilities of the Supercomputer Center, Institute for Solid State Physics, University of Tokyo, and Information Media Center, Hiroshima University.
References
- BoeriLKortusJAndersenO K 2004 Phys. Rev. Lett. 93 237002 http://dx.doi.org/10.1103/PhysRevLett.93.237002
- LeeK WPickettW E 2004 Phys. Rev. Lett. 93 237003 http://dx.doi.org/10.1103/PhysRevLett.93.237003
- BlaseXAdessiCConnétableD 2004 Phys. Rev. Lett. 93 237004 http://dx.doi.org/10.1103/PhysRevLett.93.237004
- XiangH JLiZYangJHouJ GZhuQ 2004 Phys. Rev. B 70 212504 http://dx.doi.org/10.1103/PhysRevB.70.212504
- TseJ SMaYTutuncuH M 2005 J. Phys.: Condens. Matter 17 S911 http://dx.doi.org/10.1088/0953-8984/17/11/023
- YokoyaTNakamuraTMatsushitaTMuroTTakanoYNagaoMTakenouchiTKawaradaHOguchiT 2005 Nature 43 647 http://dx.doi.org/10.1038/nature04278
- NakamuraJ et al 2008 J. Phys. Soc. Japan 77 054711 http://dx.doi.org/10.1143/JPSJ.77.054711
- TakanoY 2006 Sci. Technol. Adv. Mater. 7 S1 http://dx.doi.org/10.1016/j.stam.2006.06.003
- TakanoYTakenouchiTIshiiSUedaSOkutsuTSakaguchiIUmezawaHKawaradaHTachikiM 2007 Diam. Relat. Mater. 16 911 http://dx.doi.org/10.1016/j.diamond.2007.01.027
- TakenouchiT et al private communications
- YokoyaT et al 2007 Phys. Rev. B 75 205117 http://dx.doi.org/10.1103/PhysRevB.75.205117
- MukudaHTsuchidaTHaradaAKitaokaYTakenouchiTTakanoYNagaoMSakaguchiIKawaradaH 2006 Sci. Technol. Adv. Mater. 7 S37 http://dx.doi.org/10.1016/j.stam.2006.03.010
- OguchiT 2006 Sci. Technol. Adv. Mater. 7 S67 http://dx.doi.org/10.1016/j.stam.2006.02.011
- GossJ PBriddonP RSqueS JJonesR 2004 Phys. Rev. B 69 165215 http://dx.doi.org/10.1103/PhysRevB.69.165215
- BreuerS JBriddonP R 1994 Phys. Rev. B 49 10332 http://dx.doi.org/10.1103/PhysRevB.49.10332
- GossJ PJonesRHeggieM IEwelsC PBriddonP RÖbergS 2002 Phys. Rev. B 65 115207 http://dx.doi.org/10.1103/PhysRevB.65.115207
- GossJ PBriddonP RJonesRTeukamZBallutaudDJomardEChevallierJBernardMDeneuvilleA 2003 Phys. Rev. B 68 235209 http://dx.doi.org/10.1103/PhysRevB.68.235209
- DaiYDaiDLiuDHanSHuangB 2004 Appl. Phys. Lett. 84 1895 http://dx.doi.org/10.1063/1.1650909
- LombardiE BMainwoodAOsuchK 2004 Phys. Rev. B 70 205201 http://dx.doi.org/10.1103/PhysRevB.70.205201
- KenmochiKSatoKYanaseAKatayama-YoshidaH 2005 Japan. J. Appl. Phys. 44 L51 http://dx.doi.org/10.1143/JJAP.44.L51
- CaiYZhangTAndersenA BAngusJ CKostadinovL NAlbuT V 2006 Diam. Relat. Mater. 15 1868 http://dx.doi.org/10.1016/j.diamond.2006.08.029
- GossJ PEyreR JBriddonP R 2008 Phys. Status. Solidi 245 1679 http://dx.doi.org/10.1002/pssb.200744115
- GossJ PBriddonP R 2006 Phys. Rev. B 73 085204 http://dx.doi.org/10.1103/PhysRevB.73.085204
- BourgeoisEBustarretEAchatzPOmnésFBlaseX 2006 Phys. Rev. B 74 094509 http://dx.doi.org/10.1103/PhysRevB.74.094509
- GossJ PEyreR JBriddonP R 2006 J. Phys.: Condens. Matter 20 085217 http://dx.doi.org/10.1088/0953-8984/20/8/085217
- TsumurayaTShishidouTOguchiT 2008 Phys. Rev. B 77 235114 http://dx.doi.org/10.1103/PhysRevB.77.235114