Abstract
In recent years, culture-independent methods based on polymerase chain reaction (PCR) have been developed for the study of environmental bacteria. Denaturing gradient gel electrophoresis (DGGE) is one of the most useful of these methods. However, in the DGGE method, the low resolution of gel electrophoresis compared with the high diversity of soil bacterial communities can be a problem. Soil communities contain several thousand species of bacteria, but the resolution of more than 20–50 bands on a gel is difficult. To overcome these problems, we designed a two-dimensional electrophoresis (2-DE) method that combines size separation and the DGGE methods. In the present study, bacterial DNA was first used to design a protocol for 2-DE using PCR products of bacterial 16S rDNA. After examining different 2-DE approaches, the bacterial 16S rDNA could be separated successfully using the 2-DE described in this study. This protocol was applied to an environmental sample (i.e. soil) and the results showed that one band of size separation or DGGE produced two or more spots after 2-DE, thereby providing additional useful information about the bacterial population in the environment. Our results suggest that the new 2-DE method may be useful to monitor soil microbial populations in detail.
INTRODUCTION
Bacteria are excellent indicators of soil health because they are involved in many soil processes, they respond quickly to changes in the soil ecosystem and they have tightly coupled relationships with their surroundings because of their high surface to volume ratio (CitationWinding 2004). Methods are being developed to better determine microbial abundance, diversity and distribution in natural environments in the hope of associating microbial diversity with ecosystem function. However, there are large numbers of organisms in soil, and their presence or absence has yet to be correlated to any microbial process or any chemical or physical properties of soil (CitationKent and Triplett 2002). The investigation of bacterial diversity in the soil is important because a number of bacterial species are related to many microbial processes. More definitive descriptions of those organisms that appear or disappear with a given treatment are required (CitationKent and Triplett 2002). Approaches to determine microbial diversity and community functions can be broadly divided into culture-dependent and culture-independent methods, both of which include genetic characterization techniques. Traditionally, culture-dependent methods have been used to classify the taxonomy of bacteria based on morphological, metabolic and physiological traits. Recent theoretical and empirical analyses of the microbial diversity of soils have indicated that soils harbor in the order of 7000 different taxa at an abundance of approximately 109 cells per cubic centimeter (CitationKassen and Rainey 2004). However, attempts to cultivate the bacteria in soils typically yield culturable cell numbers of less than 5% (usually less than 1%) of the total microscopically countable cell numbers (CitationHengstman et al. 1999). Therefore, culture-based methods do not represent in situ bacterial diversity in soil, suggesting the necessity for culture-independent methods.
Methods that extract bacterial DNA directly from environmental samples have allowed us to develop culture-independent methods to study bacterial diversity. For analysis of genetic diversity, polymerase chain reaction (PCR)-based methods, such as temperature gradient gel electrophoresis (TGGE), denaturing gradient gel electrophoresis (DGGE), terminal restriction fragment length polymorphism (T-RFLP), single strand conformation polymorphism (SSCP), restriction fragment length polymorphism (RFLP) and amplified ribosomal DNA restriction analysis (ARADA) using 16S rDNA in the DNA extracted from environmental samples, are often used (CitationWinding 2004). Among these methods, DGGE provides an immediate qualitative and semiquantitative display of the constituents of a population (CitationMuyzer et al. 1993) and specific phylogenetic information can be derived from excised bands (CitationDíez et al. 2001). However, for DGGE methods, the low resolution of gel electrophoresis compared with the high diversity of bacterial communities can be a problem. To improve the resolution, a two-dimensional electrophoresis (2-DE) method that combines size separation and DGGE is being proposed. Such 2-DE methods will be able to produce more detailed information regarding bacterial diversity in environmental samples, such as soil. This approach has been used in the field of medicine (CitationNyström-Lahti et al. 1996; CitationVan Orsouw et al. 1996, Citation1998) and for comparison of whole genomes between bacterial strains or species (CitationDullaghan et al. 2002; CitationMalloff et al. 2001, Citation2003). The 2-DE method described in the present study includes separation based on differences in chain length and GC contents of the fragments of bacterial 16S rDNA. That is, we developed a 2-DE method to analyze bacterial communities in the environment in the present study. High resolution was necessary, particularly for size separation.
CitationKang et al. (2001) showed that length polymorphism (LPM) analysis is effective for monitoring changes in bacterial communities. Moreover, LPM analysis is more convenient and less costly than conventional T-RFLP. Thus, in the present study, we combined LPM (size separation) and DGGE. High resolution is indispensable for the study of bacterial diversity in environmental samples by LPM (size separation) analysis because DNA fragments have only small ranges of chain length in the variable region of bacterial 16S rDNA (e.g. 30 bp for V1 region). Hence, the proposed 2-DE method should have higher resolution than the 2-DE methods mentioned previously (CitationDullaghan et al. 2002; CitationMalloff et al. 2001, Citation2003; CitationNyström-Lahti et al. 1996; CitationVan Orsouw et al. 1996, 1998) because there are a number of species of soil bacterial DNA fragments in narrow range of chain length, the structural difference of variable region of bacterial 16S rDNA is not remarkable. To demonstrate the possibility of direct scanning of bacterial diversity in environmental samples, a 2-DE method has been proposed. One of the objectives of the present study was to confirm the separation of PCR products extracted from different species known to be present in the environment using 2-DE. These PCR products have a narrow range of chain
Table 1 GC content and chain length of the 16S rDNA V1–V2 region of the bacterial species used in this study
MATERIALS AND METHODS
Bacteria
The bacterial strains Bacillus subtillis (ATCC 9466), Mydroides odoratus (ATCC 4651), Acinetobacter baumannii (ATCC 19606) and Flavobacterium aquatile (ATCC 11947) from the Institute of Applied Microbiology, the University of Tokyo, Tokyo, Japan, were used as markers (chain length and GC content are shown in ) in this study. Bacillus subtillis, M. odoratus and F. aquatile were cultured using nutrient medium (5 g peptone, 3 g beef extract, 3 g NaCl in 1 L of distilled water, pH 7.0) at 30°C. Acinetobacter baumannii was also cultured in the same nutrient medium, but the incubation temperature was 37°C. These bacterial strains were first cultured in liquid culture medium. After observing sufficient growth in liquid medium, the spread plate method was used to obtain pure cultures of the bacterial strains, and a single colony was again inoculated into liquid medium and incubated for 24 h. This culture medium was centrifuged at 3,000 g for 5 min at 4°C. The supernatant was discarded and the pellet was stored at −20°C until further analysis.
Soil
Subsurface soil samples were collected from grassland at the campus of Yokohama National University, Yokohama, Japan. After removing all visible roots and other materials from the soil samples, the samples were sieved (2 mm mesh) and stored at −20°C for DNA analysis.
DNA extraction
Pellets of the bacterial samples were used to extract DNA using a FastDNA Kit (Q·BIOgene, Carlsbad, CA, USA) in accordance with the manufacturer's protocol. DNA extraction from the soil was carried out using the bead-beater method (CitationCullen and Hirsch 1998). Six milliliters of phosphoric acid sodium buffer (SPB, including 1% sodium dodecyl sulfate (SDS)) and 0.5 g of glass beads (equal amounts of 0.1 mm, 0.5 mm and 1 mm) were added to 0.5 g of the soil sample in a tube. Mechanical lysis (5,000 rpm for 30 s three times) was carried out with a mini-bead beater (BioSpec Products, Bartlesville, OK, USA). After centrifugation (3,000 g for 15 min at 10°C) the supernatant was collected and 0.25 volumes of Ethylene Diamine Tetra-acetic Acid (EDTA) (0.5 mol L−1, pH 8.0) and 1/10 volume of potassium acetate solution were added, followed by incubation on ice for 20 min. Centrifugation (9,000 g for 15 min at 4°C) was carried out to remove organic matter and protein. One volume of isopropanol was added to the supernatant for DNA precipitation. Agarose gel (2%) electrophoresis was carried out to check the DNA extracted from the bacteria and the soil samples. After staining with ethidium bromide, the presence of bands was checked under ultraviolet (UV).
DNA purification
The extracted DNA was further purified using spin columns packed with polyvinylpolypyrollidone (PVPP) (CitationCullen and Hirsch 1998) and Sepharose-4B (CitationJackson et al. 1997). Aliquots of 200 µL of DNA samples were placed onto the PVPP (0.15 g) columns, which were then centrifuged at 2,500 g for 5 min. This process was repeated. Further purification was carried out by placing the samples purified by passage through the PVPP columns onto a Sepharose-4B (3 mL) column, followed by centrifugation at 2,000 g for 5 min (CitationKang et al. 2001). The purity of DNA was again checked using agarose gel (2%) electrophoresis.
PCR
The PCR was carried out using DNA purified from the bacteria and soil samples with a thermal cycler (Perkin Elmer GeneAmp PCR system 2400; Perkin Elmer, Norwalk, CT, USA). The primers (Invitrogen, San Diego, CA, USA) used in the experiment are listed in (CitationLane 1991). A GC clamp was attached to the 5′ end of the forward primer. In the present study, the V1–V2 region was used for bacterial DNA from pure cultures and the V1 domain was used for the soil DNA. The PCR mixtures were prepared as follows: in a volume of 50 L, 45 µL of PCR SUPERMIX (Invitrogen) (22 mmol L−1 Tris-HCl, pH 8.4, 55 mmol L−1 KCl, 1.65 mmol L−1 MgCl2, 220 µmol L−1 dGTP, 220 µmol L−1 dATP, 220 µmol L−1 dTTP, 220 µmol L−1 dCTP, 22 U Taq DNA polymerase mL−1), 2 µL of each primer (0.25 µmol L−1) and 1 µL of DNA template were added. For bacterial DNA, the PCR thermocycling program was as follows: initial denaturation at 94°C for 1 min, followed by 30 cycles of denaturation at 94°C for 30 s, annealing at 59°C for 30 s and extension at 72°C for 30 s, followed by incubation at 72°C for 1 min. The bacterial DNA was also amplified by touchdown PCR as follows: 20 cycles consisting of denaturation at 94°C for 1 min, annealing at 65°C (the temperature was decreased by 2°C every four cycles until reaching the touchdown temperature of 57°C) for 1 min and primer extension at 72°C for 1 min. Five additional cycles were carried out at an annealing temperature of 55°C and then incubated for 10 min at 72°C.
Soil DNA was amplified using nested PCR to improve the poor amplification efficiency. The following thermocycling program was used to amplify the longer target part with primer set II (): 95°C for 2 min, 30 cycles consisting of 95°C for 1 min, 59°C for 30 s and 72°C for 40 s, followed by a final cycle consisting of 72°C for 7 min. The resultant PCR products were used as templates for touchdown PCR with primer set III and the temperature program for touch down PCR described above for bacterial DNA. The PCR products were checked by agarose gel (2%) electrophoresis for amplification of the target part.
Two-dimensional electrophoresis
Equal amounts of PCR fragments with a GC clamp obtained using DNA of the bacterial strains were mixed and used in the experiment to improve separation based on GC content and fragment size. The different approaches of 2-DE are shown schematically in and detailed descriptions are given below.
Two-dimensional electrophoresis carried out using the same gel
For the first dimension, the chamber was used for DGGE analysis of the PCR products obtained from pure cultures (). Electrophoresis was carried out on 6% polyacrylamide gels using an ascending gradient of 36–66% (100% denaturant was 7 mol L−1 urea plus
Table 2 Primer sequences used in this study†
Figure 1 Schematic representation of the different two-dimensional electrophoresis approaches. (a) Two-dimensional electrophoresis using the same gel (denaturing gradient gel electrophoresis [DGGE] and size separation); (b) two-dimensional electrophoresis using the same gel (size separation and DGGE); (c) two-dimensional electrophoresis using disk gels; (d) two-dimensional electrophoresis using specially prepared gels. C, chamber apparatus; S, stage apparatus; UF, urea and formamide.
![Figure 1 Schematic representation of the different two-dimensional electrophoresis approaches. (a) Two-dimensional electrophoresis using the same gel (denaturing gradient gel electrophoresis [DGGE] and size separation); (b) two-dimensional electrophoresis using the same gel (size separation and DGGE); (c) two-dimensional electrophoresis using disk gels; (d) two-dimensional electrophoresis using specially prepared gels. C, chamber apparatus; S, stage apparatus; UF, urea and formamide.](/cms/asset/33e0fdf5-db5e-4e44-a51e-f3bb59e0e506/tssp_a_10382434_o_f0001g.gif)
Two-dimensional electrophoresis using disk gels
This type of 2-DE method has often been used for protein separation (CitationGillette et al. 2005; CitationMiyoshi et al. 1999; CitationO’Farrell 1975). Electrophoresis in the first dimension was carried out using the gel in a glass tube (Nihon Eido, Tokyo, Japan). In this method, samples were first separated by fragment size on a 10% non-denaturing polyacrylamide gel (3 mm × 130 mm) for 2 h at 200 V. Subsequently, the gel was placed on top of a 6.5% polyacrylamide denaturing gradient gel containing an ascending gradient of 36–66% as shown in , and then DGGE was carried out in a chamber at a constant voltage of 100 V for 3 h at 60°C.
Two-dimensional electrophoresis using specially prepared gels
In this method, samples were first separated by fragment size on a 8% non-denaturing polyacrylamide gel for 14 h at 100 V using D the DCode universal mutation detection system (Bio-Rad, Hercules, CA, USA). This gel contained Spreadex Polymer NAB (10×: Elchrom Scientific, Cham, Switzerland) to provide high resolution. Next, one of the lanes containing the sample was cut out and removed. This piece of gel was placed between two glass plates at the time of the preparation of the denaturing gradient gel (). Then, a 10% polyacrylamide denaturing gradient gel (25–60%) was prepared. The remaining region at the top was filled with 6.5% non-denaturing polyacrylamide gel (). DGGE was then carried out at 60°C for 10 h at 200 V. A similar experiment was also carried out for PCR products of soil DNA. Samples were first separated by fragment size on a 10% polyacrylamide gel containing Spreadex Polymer NAB (10×: Elchrom Scientific) for 13 h at 100 V. The samples were then separated by sequence composition on 10% polyacrylamide gels containing a 30–60% denaturant gradient for 13.5 h at 130 V at 60°C.
RESULTS AND DISCUSSION
PCR
The extracted bacterial DNA was first amplified by PCR using the primer set I (27fGC and 342r). The result of size separation revealed that in addition to the target band, smaller spurious bands were also noted after 10% polyacrylamide gel electrophoresis by using regular PCR condition. Then, touchdown PCR was carried out to eliminate the formation of non-target bands (CitationDon et al. 1991).
In the present study, the annealing temperature of PCR was decreased by 2°C every fourth cycle from 65°C to touchdown at 55°C. Under these PCR conditions, only one band was produced. Using touchdown PCR, the imbalance between correct and spurious annealing was automatically redressed and allowed amplification of sufficient amounts of product of the correct length. In the present study, touchdown PCR was used for bacterial samples, whereas touchdown PCR was used as the second round of nested PCR for soil samples.
Two-dimensional electrophoresis using the same gel
The 2-DE was first carried out with DGGE as the first dimension followed by size separation in the second dimension (). In this method, after DGGE in the chamber, the gel was taken out of the glass plates and rotated through 90° for size separation. Subsequently, electrophoresis was carried out in the second dimension using a stage-type apparatus. Using this apparatus, it was possible to carry out electrophoresis in the second dimension in the same gel after removal from the glass plates. After DGGE, the sample DNA becomes branched molecules (CitationSheffield et al. 1989). The gel used for DGGE was cooled to room temperature to allowing the branched DNA to become double-stranded as size separation in the second dimension must be carried out using double-stranded sample DNA. In this method, the results of size separation (second dimension) were found to be poor (data not shown). This may have been because the DNA samples had not become completely double-stranded. From these results, it was understood that the order of 2-DE (DGGE followed by size separation) was not suitable to obtain good separation.
Consequently, the order of electrophoresis was changed to size separation followed by DGGE for the second dimension with the same stage-type apparatus. Although size separation in the first dimension was found to be satisfactory, DGGE analysis failed to properly separate the bands (results are not shown). DGGE could not be carried out properly because of the possible variation in temperature. It was assumed that the temperature of the stage might be lower than the required temperature of 60°C, resulting in poor separation of the bands by DGGE. Therefore, this approach was not followed for further analysis and the use of the disk gels was attempted.
Two-dimensional electrophoresis using disk gels
The above two approaches did not provide good results, but did suggest a few points to be considered. One was that DGGE should be carried out after size separation in 2-DE. Another was that DGGE should be carried out in the chamber, although the stage had the advantage that the gel could be rotated by simple manipulation. Thus, the 2-DE was designed as follows: size separation for the first dimension was carried out using a disk gel that was then attached to the top of the gel to be used for DGGE (). This type of approach has often been used for the separation of proteins by 2-DE (CitationGillette et al. 2005; CitationMiyoshi et al. 1999; CitationO’Farrel 1975).
The results revealed that the bands were separated properly when compared with the results of either size separation or DGGE analysis. These results revealed that 2-DE could be carried out properly by combining the two methods (size separation and DGGE), as carried out in this approach. However, there were still some limitations in this type of 2-DE electrophoresis. One was that the disk gel does not have high resolution. Although a long disk gel (230 mm) was used in the present experiment to improve the resolution, satisfying separation ability could not be obtained. Another problem was that proper fixing of the disk gel on top of the DGGE gel was difficult ().
Two-dimensional electrophoresis using specially prepared gels
To overcome the problems mentioned previously, size separation was carried out using a 1 mm polyacrylamide gel as an alternative to a disk gel to improve resolution. Moreover, Spreadex Polymer NAB was added to the gel. This gel had better separation ability than ordinary polyacrylamide gels (CitationJanatováet al. 2003). This is an important point in the present study, which used environmental samples, because high resolution was necessary for the efficient size separation of 16S rDNA. The gel used for size separation in the first dimension was kept between the two glass plates at the time of DGGE in the second dimension (). Size separation in the first dimension was carried out using the same two samples and size markers. After electrophoresis, the gel was cut into two pieces, thereby providing one of the gel pieces with one sample. The gel sample with the size marker was stained to confirm size separation. The area where the target bands were expected to be present was cut from the non-stained gel and placed between the two glass plates as shown in . Subsequently, the denaturing gradient gel was made below the target gel. After polymerization of the denaturing gradient gel, the non-denaturing gel was used to fill the surroundings of the target gel. Thus, the target gel was now in a fixed position and DGGE in the second dimension was carried out using this gel.
The results of size separation and DGGE of bacterial DNA are shown in . Size separation of the mixture of bacterial PCR products produced two bands (). The upper band was B. subtillis (396 bp) and the lower band represented M. odoratus (385 bp), A. baumannii (386 bp) and F. aquatile (383 bp) in lane 5 (). In the result of DGGE, four bands that represented the four bacterial species were observed for the mixed PCR products (Lane 5) by DGGE ().
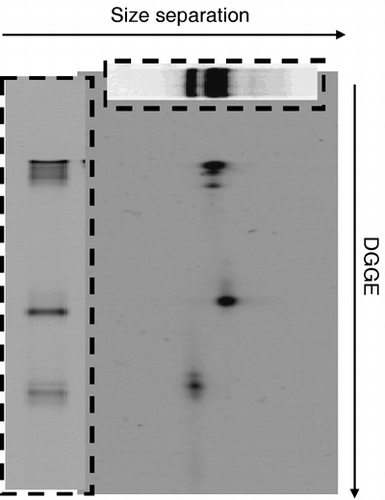
The results of 2-DE using the same PCR products are presented in along with the results of size separation and DGGE to show separation by 2-DE. Four spots
Figure 2 (a) Size separation and (b) denaturing gradient gel electrophoresis (DGGE) patterns of the bacterial 16S rDNA amplified with primer set I. Lane 1 B. subtillis, lane 2 M. odoratus, lane 3 A. baumannii, lane 4 F. aquatile and lane 5 bacterial mix. Lane M in (a) is the size marker.
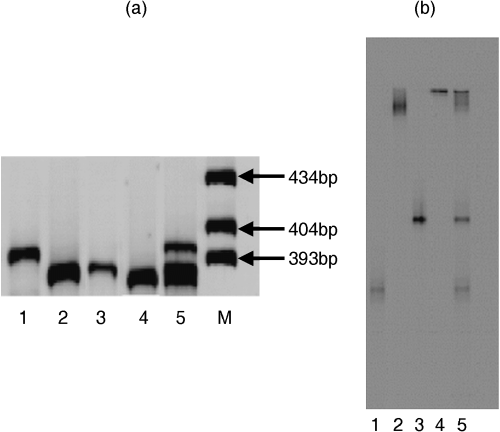
Figure 3 Two-dimensional electrophoresis patterns of bacterial 16S rDNA amplified with primer set I. The top boxed figure shows the results of size separation and the left boxed figure shows the results of denaturing gradient gel electrophoresis (DGGE).
Two-dimensional electrophoresis using environmental samples
This 2-DE protocol was then used for analysis of soil DNA. Soil communities contain at least 7,000 different prokaryote taxa (CitationTorsvik et al. 1990), but the resolution of more than 20–50 bands on a gel is difficult. Thus 1 band on DGGE and size separation may contain DNA from multiple bacterial species. DGGE and size separation prevent the bacterial diversity especially the change of major population in environmental samples (e.g. soil) for this low resolution. Sequencing and identification of the visible bands on the gel following DGGE may further improve the resolution, but analysis is time consuming (CitationWinding 2004). For these reasons, 2-DE may be useful for analysis of environmental samples. Consequently, for soil DNA, the V1 region of 16S rDNA was amplified and analyzed as we concluded that this region was suitable for 2-DE. The sequence data of approximately 200 bacterial 16S rDNAs from the ribosomal database project (RDP) were used to subsequently calculate chain length and GC content in some variable regions. The data of V1 and V3 are shown in . The V1 region of bacterial 16S rDNA had more extensive differences in chain length and GC content than observed in other regions. With regard to DGGE, a recent study indicated that the V3 region produced more bands than the V1 region, although theoretically the V1 region with the highest sequence diversity should produce the greatest number of resolved bands (CitationYu and Morrison 2004). Thus, the V3 region has been one of the most useful regions in DGGE. However, as shown in , the V1 region had wide ranges of both chain length (70–130 bp) and GC content (40–60%). In the V3 region, there were many DNA fragments from bacteria with the same chain length, such as 169, 194 and 195 bp (). These results suggest that the V1 region is suitable for size separation. To improve the resolution of samples containing many DNA fragments, 2-DE should have detailed size separation of the V1 region. For this reason, the V1 region was found to be suitable for 2-DE using DNA extracted from environmental samples containing various species of bacteria.
The results of size separation and DGGE of soil DNA are shown in . Size separation in the first dimension was carried out using the Spreadex Polymer NAB. There were eight confirmed bands around the range of
Figure 4 Chain length and GC content of bacterial DNA fragments in the V1 (•) and V3 (◊) regions.
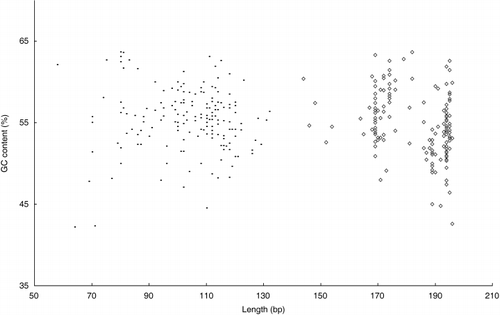
Figure 5 (a) Size separation and (b) denaturing gradient gel electrophoresis (DGGE) patterns of soil bacterial 16S rDNA amplified with primer set III. Lane M in (a) is the size marker.
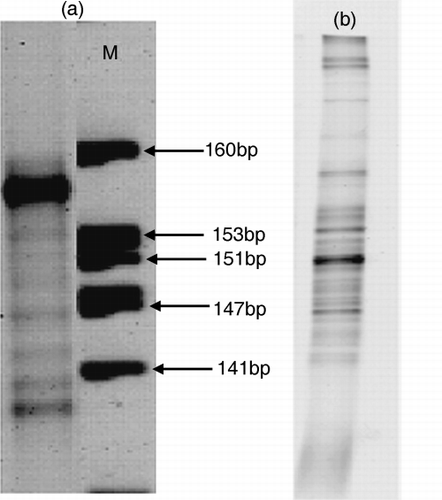
Figure 6 Two-dimensional electrophoresis patterns of soil bacterial 16S rDNA amplified with primer set III. The top boxed figure shows the results of size separation and the left boxed figure shows the results of denaturing gradient gel electrophoresis (DGGE).
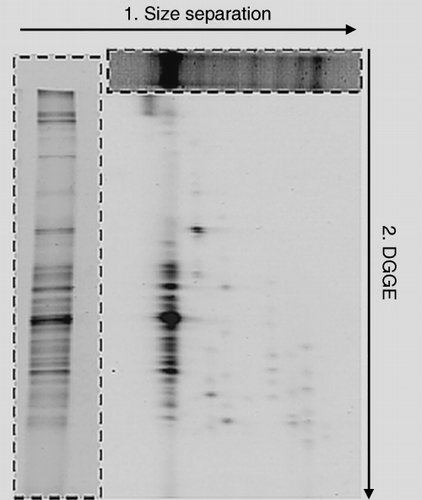
Typical results of 2-DE analysis are shown in . The results of size separation and DGGE are also shown in the figure for comparison. Approximately 70 spots were easily identified in the 2-DE gel, while only several bands were observed in the size separation gel and in the DGGE gel. For the 2-DE PCR product was loaded twice as much as DGGE, as some spots of the result of 2-DE were either invisible or not clearly seen for the high resolution. The bands of DGGE and size separation were separated to multiple bands by 2-DE. The bands observed in the first electrophoresis (size separation) were separated into a wide range by the second electrophoresis (DGGE). The results of 2-DE indicated that the PCR products had the same or similar lengths, but had different GC contents. It was confirmed that a single band observed on size separation was separated further into many bands after electrophoresis in the second dimension. Similarly, the PCR products that had the same or similar mobility associated with the GC content but had different lengths were also separated. showed that the spots derived from the strongest band in the size separation corresponded well to the DGGE bands. Moreover, the other bands of size separation also produced many spots by 2-DE. These results reveal that PCR products of environmental samples can be separated efficiently by 2-DE.
Thus, we successfully applied 2-DE to samples containing different bacterial DNA and the results indicated that the bacterial 16S rDNA from pure cultures and from soil samples could be separated well by 2-DE. The 2-DE method improved the low resolution of DGGE and allowed samples to be separated more distinctly than by size separation or DGGE analysis alone. Application of this method may allow us to effectively analyze bacterial populations in the environment and their roles in ecosystem function and soil health. As part of this research project, the analysis of 16S rDNA obtained from different environmental samples by 2-DE method is currently in progress in our laboratory.
ACKNOWLEDGMENTS
This work was supported by the 21st Century Center of Excellence (COE) program of the Ministry Education, Culture, Sports, Science and Technology of Japan, ‘Bio-Eco Environmental Risk Management.’
REFERENCES
- Winding , A . Indicators of soil bacterial diversity . Proc. of an OECD Expert Meeting on Soil Erosion and Soil Biodiversity Indicators . March 25–28 2003 , Rome, Italy. pp. 495 – 504 .
- Kent , AD and Triplett , EW . 2002 . “ Microbial communities and their interactions in soil and rhizosphere ecosystems ” . In AnnuRevMicrobiol Vol. 56 , 211 – 236 .
- Kassen , R and Rainey , PB . 2004 . “ The ecology and genetics of microbial diversity ” . In AnnuRevMicrobiol Vol. 58 , 207 – 231 .
- Hengstmann , U , Chin , K-J , Janssen , PH and Liesack , W . 1999 . “ Comparative phylogenetic assignment of environmental sequences of genes encoding 16S rRNA and numerically abundant culturable bacteria from an anoxic rice paddy soil ” . In ApplEnvironMicrobiol Vol. 65 , 5050 – 5058 .
- Muyzer , G , De Waal , EC and Uitterlinden , AG . 1993 . “ Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA ” . In ApplEnvironMicrobiol Vol. 59 , 695 – 700 .
- Díez , B , Pedrós-alió , C , Marsh , TL and Massana , R . 2001 . “ Application of denaturing gradient gel electrophoresis (DGGE) to study the diversity of marine picoeukaryotic assemblages and comparison of DGGE with other molecular techniques ” . In ApplEnvironMicrobiol Vol. 67 , 2942 – 2951 .
- Nyström-Lahti , M , Wu , Y Moisio , A-L . 1996 . “ DNA mismatch repair gene mutations in 55 kindreds with verified or putative hereditary non-polyposis colorectal cancer ” . In HumMolGenet Vol. 5 , 763 – 769 .
- Van Orsouw , NJ , Li , D Van der Vlies , P . 1996 . Mutational scanning of large genes by extensive PCR multiplexing and two-dimensional electrophoresis: application to the RB1gene . HumMolGenet , 5 : 755 – 761 .
- Van Orsouw , NJ , Dhanda , RK Rines , RD . 1998 . “ Rapid design of denaturing gradient-based two-dimensional electrophoretic gene mutational scanning tests ” . In Nucleic Acids Res Vol. 26 , 2398 – 2406 .
- Dullaghan , EM , Malloff , CA , Li , AH , Lam , WL and Stokes , RW . 2002 . Two-dimensional bacterial genome display: a method for the genomic analysis of mycobacteria . Microbiology , 148 : 3111 – 3117 .
- Malloff , CA , Fernandez , RC and Lam , WL . 2001 . “ Bacterial comparative genomic hybridization: A method for directly identifying lateral gene transfer ” . In JMolBiol Vol. 312 , 1 – 5 .
- Malloff , C , Dullaghan , E , Li , A , Stokes , R , Fernandez , R and Lam , W . 2003 . Two-dimensional DNA displays for comparisons of bacterial genomes . BiolProcedOnline , 5 : 143 – 152 .
- Kang , KS , Itoh , K , Murabayashi , M and Nakanishi , J . 2001 . “ Direct DNA extraction from soil and DNA purification ” . In JJpn SocWater Environ Vol. 24 , 175 – 179 .
- Cullen , DW and Hirsch , PR . 1998 . “ Simple and rapid method for direct extraction of microbial DNA from soil for PCR ” . In SoilBiolBiochem Vol. 30 , 983 – 993 .
- Jackson , CR , Harper , JP , Willoughby , D , Roden , EE and Churchill , PF . 1997 . “ A simple, efficient method for the separation of humic substances and DNA from environmental samples ” . In ApplEnvironMicrobiol Vol. 63 , 4993 – 4995 .
- Lane , DJ . 1991 . “ 16S/23S rRNA sequencing ” . In Nucleic Acid Techniques in Bacterial Systematics , Edited by: Stackebrandt , E and Goodfellow , M . 115 – 175 . Chichester : John Wiley & Sons .
- Gillette , WK , Esposito , D Frank , PH . 2005 . “ POET: Using proteomics to screen pools of open reading frames for protein expression MolCellProteomics ” . Vol. 4 , 1647 – 1652 .
- Miyoshi , E , Noda , K Ko , JH . 1999 . “ Overexpression of α1-6 fucosyltransferase in hepatoma cells suppresses intrahepatic metastasis after splenic injection in athymic mice ” . In Cancer Res Vol. 59 , 2237 – 2243 .
- O’Farrell , PH . 1975 . “ High resolution two-dimensionalel ectrophoresis of proteins ” . In JBiolChem Vol. 250 , 4007 – 4021 .
- Don , RH , Cox , PT , Wainwright , BJ , Baker , K and Mattick , JS . 1991 . “Touchdown” PCR to circumvent spurious priming during gene amplification ” . In NuclAcids Res Vol. 19 , 4008
- Sheffield , VC , Cox , DR , Lerman , LS and Myers , RM . 1989 . Attachment of a 40-base-pair G+C-rich sequence (GC-clamp) to genomic DNA fragments by the polymerase chain reaction results in improved detection of single-base changes . ProcNatlAcadSciUSA , 86 : 232 – 236 .
- Janatová , M , Pohlreich , P and Matouß , B . 2003 . Detection of the most frequent mutations in BRCA1gene on polyacrylamide gels containing Spreadex Polymer NAB . Neoplasma , 50 : 246 – 250 .
- Torsvik , V , Goksoyr , J and Daae , FL . 1990 . “ High diversity in DNA of soil bacteria ” . In ApplEnvironMicrobiol Vol. 56 , 782 – 787 .
- Yu , Z and Morrison , M . 2004 . Comparisons of different hypervariable regions of rrsgenes for use in fingerprinting of microbial communities by PCR-denaturing gradient gel electrophoresis . ApplEnvironMicrobiol , 70 : 4800 – 4806 .