Abstract
A rapid simultaneous multi-element analysis method for soils and environmental samples has been established using polarizing energy dispersive X-ray fluorescence (EDXRF) spectrometry. The pressed powder pellet technique was adopted because it is simple and requires no specialized skills for sample preparation. The analytes examined were: Na2O, MgO, Al2O3, SiO2, P2O5, K2O, CaO, TiO2, MnO, Fe2O3, V, Cr, Co, Ni, Cu, Zn, Rb, Sr, Y, Zr, Nb, Sn, Sb, Cs, Ba, La, Ce, Pr, Nd, Pb and Th. Twenty-six reference samples were used for the calibrations. Compton scatter radiation was used as an internal standard to compensate for variations in the sample matrix, particle size, packing density and operating characteristics of the instrument. The correlation coefficients of the linear regression lines were >0.98, with the exception of Na2O, MgO, Al2O3, SiO2, P2O5, Co, Pr and Nd. The values obtained using the proposed EDXRF spectrometry were compared with the values obtained using other methods. Out of the 31 analytes examined, the results for 12 compounds (CaO, K2O, TiO2, MnO, Fe2O3, Ni, Zn, Rb, Sr, Cs, Ba and Ce) obtained by the proposed EDXRF spectrometry compared favorably with those determined through conventional wet chemical methods. In contrast, the results for eight compounds (Na2O, MgO, Cr, Co, Zr, Sn, Sb and Pr) obtained by the proposed EDXRF spectrometry exhibited poor agreements with those obtained using chemical methods. Among these analytes, the poor agreements of Cr, Zr and Sn were attributable to incomplete dissolution and/or volatilization losses during the chemical treatments based on an acid attack. It can be concluded, therefore, that there is a high possibility of underestimating the concentrations of these elements if wet chemical methods are used. Although the results of the other 11 analytes were not as good as those of the first group, they still appeared to be of practical use, considering the time consuming and potentially hazardous wet chemical pretreatment.
Introduction
In recent years, both the number of soil samples and the types of elements to be analyzed have been rapidly increasing. Although techniques such as inductively coupled plasma-mass spectrometry (ICP-MS), inductively coupled plasma-atomic emission spectrometry (ICP-AES) and atomic absorption spectrometry (AAS) are commonly used to meet the above requirements (CitationButler et al. 2006, 2009; CitationYamasaki 2000), all of these methods require time-consuming and potentially hazardous acid treatments for sample dissolution. Furthermore, a well-equipped chemical laboratory and the assistance of skillful analysts are indispensable. In contrast, X-ray fluorescence spectrometry (XRF) has many advantage in terms of: (1) simultaneous multi-element capability, (2) wider dynamic range, from parts per million (mg kg−1) to 100%, (3) simple sample preparation procedure, (4) avoidance of poor recovery for certain elements during acid digestion, (5) easier machine operation, (6) higher sample throughput, (7) good reproducibility, (8) low operating costs. Because of these inherent advantages, XRF has been widely adapted for the analysis of major and trace elements in foods and plants (CitationMargui et al. 2005; CitationNielson et al. 1991), farmyard manure (CitationMatsunami et al. 2009), and soil and environmental samples (CitationGunicheva et al. 1995; CitationJørgensen et al. 2005; CitationNakayama et al. 2007; CitationOrescanin et al. 2008; CitationYamasaki et al. 1980; CitationYu et al. 2002).
Despite these advantages, energy dispersive X-ray fluorescence (EDXRF) spectrometry is used to a lower extent comparatively than wavelength dispersive X-ray fluorescence (WDXRF) spectrometry owing to its lower energy resolution, particularly for light elements. However, technical advances have been made in EDXRF instrumentation. Because the conventional and widely adopted EDXRF uses unpolarized X-rays as its primary excitation radiation, background levels are higher because the unpolarized X-rays contain scattering from the X-ray tube and sample matrices. As a consequence, the detection limits are degraded for trace elements in light matrices, such as plant materials and high silicate rocks and soils (CitationHeckel et al. 1992; CitationHittipathirana 2004). Spectrum analysis programs have also been developed that allow multiple-peak X-ray profiles to be fitted to spectra; such a fitting compensates for problems attributable to peak overlapping.
Taking all of these factors into consideration, we decided in the present study to examine the applicability of the recently developed EDXRF spectrometer with 3-D optics, in the hope that it will realize the efficient analysis of a large variety of elements in numerous soil and environmental samples with acceptable speed and accuracy.
Materials and methods
Instrumentation
The EDXRF spectrometer (Epsilon 5; PANalytical B. V., Lelyweg, The Netherlands) used in the present study was equipped with a Sc/W anode X-ray tube with the goal of increased sensitivity for lighter as well as heavier elements. The instrument has a series of user selectable secondary targets and is equipped with a liquid-nitrogen-cooled Ge solid-state high-resolution detector with a Be (8 μm) window. A 3-D design (or Cartesian geometry) was adopted for the instrument to eliminate the X-ray tube spectrum by polarization. Consequently, the background can be an order of magnitude lower than the traditional 2-D optics, resulting in much lower detection limits. The overlapping of the characteristic L lines of heavier elements with the K lines of lighter elements (a typical example is the overlap of Cd L lines on K K lines) is a major drawback in determining heavy elements using the EDXRF method. However, because the instrument used in the present study is equipped with a 100 kV Sc/W tube, even heavy rare earth elements (from Eu to Lu) can be analyzed using the more sensitive K lines, and hence the instrument is less susceptible to spectral overlapping with the K lines of lighter elements.
Construction of the calibration curves
The instrument was calibrated for 10 major constituents and 21 selected trace elements using 26 geological and environmental standard reference materials. Although calibration can be done using “homemade” (secondary) standards, including the method of standard additions, the most widely used and well-established method of utilizing standard reference materials was adopted in the present study. The reference materials are listed in , together with their basic information and the producers. More detailed information has been presented elsewhere (CitationAndo et al. 1989; CitationGovindaraju 1994; CitationImai et al. 1995a,b, 1996; CitationNational Bureau of Standards and Technology 1993a,b, 2000, 2002; CitationTerashima et al. 1998). The selection of trace elements was based on previous information concerning the background concentration of each element in soils (CitationEgashira et al. 1996, 2004, 2007; CitationKimura et al. 2008; CitationNanzyo et al. 2002; CitationTakeda et al. 2004; CitationYamasaki et al. 2001, 2009). Although the existence of Cl and S was occasionally identified in the X-ray spectrum, these elements were not included because their occurrence rates were not very high. In contrast, although Br and I were observed in almost all of the samples, a lack of appropriate reference materials made it impossible to construct calibration curves for these elements.
The measurement conditions for the major constituents and trace elements are given in and , respectively. As for the trace elements, the choice of secondary targets was a compromise between sensitivity and analytical speed. For example, the emitted X-ray intensities from Rb and Sr are much stronger when Zr is used as a target rather than Mo. However, the use of a Zr target makes it impossible to analyze both Zr and Y. As a result, these elements must be analyzed separately from Rb and Sr.
Sample preparation for X-ray spectrometry
The samples were prepared in the form of pressed powder pellets, without adding binders. They were first pulverized for 10 min in a planetary ball mill (pulverisette 7; Fritsch GmbH, Idar-Oberstein, Germany) at 450 rpm (135 m s−2 or 13.8 G) and then pressed into 32-mm internal diameter pellets using a hydraulic press operated at a pressure of 20 tonnes (Specac, Kent, UK). To reduce errors resulting from any inhomogeneity caused by sample preparation, the spinner system in the spectrometer constantly rotated the samples during the measurements.
Table 1 Standard reference materials used for calibration
Table 2 Measurement conditions for the major constituents
Table 3 Measurement conditions for the trace elements
Correction of matrix effects
The X-ray spectra were analyzed using AXIL software (Analysis of X-ray Spectra by Interactive Least Squares Fitting [CitationVan Espen et al. 1986; CitationVekemans et al. 1994]). The use of powder pellets pressed without a binder eliminates sample dilution and, therefore, increases the sensitivity and lowers the detection limits for trace elements. Additional advantages of the above technique are its simplicity and the fact that no specialized skills are required for sample preparation. Disadvantages include the difficulty of obtaining pellets with sufficient physical strength from soil samples rich in siliceous sand. Particle size effects and matrix effects are other negative factors with this approach. Therefore, to cope with these problems Compton scatter radiation was used as an internal standard to compensate for variations in the sample matrix, particle size, packing density and operating characteristics of the instrument (). The fundamental parameter method (FP) was also used for matrix correction for major constituents (). This method aims to establish a proper correction for the inter-elemental effect caused by other matrix elements in the standard reference materials by using several physical constants (fundamental parameters). Accordingly, information concerning all of the major constituents (including loss on ignition) in the reference materials should be registered in the data file, even when there is only one analyte of interest. As various specific parameters related to the instrument are also included in the software provided by the manufacturer, the details of the correction procedures are kept closed to the users. However, the effectiveness of this approach was shown by the fact that correlation coefficients obtained using FP were higher than those without FP.
Sample dissolution
The EDXRF spectrometry results were compared with the results obtained by chemical methods and the borate fusion-WDXRF method (SiO2 only), using three groups of samples. Group 1 contained the oldest samples collected nationwide from 1980 to 1982 as cooperative research work on “Ando Soils in Japan” (CitationWada 1986). The sampling sites were selected in such a way as to cover typical volcanic ash soils common to Japan. Group 2 consisted of part of the samples systematically collected from agricultural lands from 1979 to 1997, where the concentrations of more than 40 trace elements have already been determined (CitationKimura et al. 2008). Group 3 contained the newly sampled specimens and chemical analyses were carried out in parallel with the EDXRF measurements.
The sample dissolution techniques were based on an attack with mixed acids containing HF and were, therefore, essentially similar. However, the details of the procedures were considerably different between the three groups of samples because the experiments were carried out in different laboratories and at different times.
The most primitive technique was used for acid digestion for Group 1 samples. One gram of a finely ground sample was treated with 10 mL of HClO3 + HNO3 (1:1 mixture) to decompose the organic matter in the soils, followed by a double treatment with 15 mL of HClO3 + HF (1:2 mixture) in a polytetrafluoroethylene (PTFE) beaker on a hot plate manufactured for domestic use. The residue was heated with 5 mL of HNO3 and dissolved by the addition of 30–50 mL of H2O with gentle boiling and finally made up to a volume of 100 mL. The final solutions were stored in plastic bottles until the measurements were conducted (CitationYamasaki 1978; CitationYoshida et al. 1992).
For Group 2, a more efficient and less sample-consuming and acid-consuming microwave digestion technique was adopted. In brief, 0.1 g of finely ground sample was first treated with 2 mL of HNO3 in a tightly sealed PTFE container in a domestic microwave oven for 15 min, then with 0.5 mL of HF for another 15 min, and finally with 0.5 mL of HClO4 for 15 min. The digest was dried up on a hot plate after removing the sealed cup. The residue was heated with 2 mL of HNO3 and dissolved by adding 10–20 mL of H2O with gentle boiling and finally made up to a volume of 40 mL (CitationKimura et al. 2008; CitationMizushima et al. 1996).
For Group 3, the sample amounts were increased to avoid possible errors resulting from sample inhomogeneity. With this procedure, a specially designed heat block (DigiPREP MS; SCP SCIENCE, Bale-d' Urfé, Quebec, Canada) and digestion vessels made of PTFE were used. In this apparatus, it was possible to treat up to 48 samples at one time under a more precisely controlled temperature and heating period. The finely ground sample (0.500 g) was first treated with 4 mL of HClO4–HNO3 (1:1 mixture) for 45 min at 120°C and then with 4 mL of HClO4–HF (1:1 mixture) twice for 45 min at 170°C. The residue was heated with 5 mL of HNO3 and dissolved by adding 20 mL of H2O at 100°C for 3–4 h and finally made up to a volume of 100 mL (CitationYamasaki et al. 2009).
Reference methods used for comparison
Inductively coupled plasma-mass spectrometry
After a 10–100-fold dilution, the acid digests were analyzed by ICP-MS for all of the trace and ultra-trace elements. Indium (In) was used as an internal standard element. The validity of the proposed method was examined by analyzing several reference materials, as shown in . The working standards were prepared from a series of SPEX Multi-Element Plasma Standards (XSTC-1, XSTC-7, XSTC-8 and XSTC-13) supplied by SPEX Industries (Edison, NJ, USA). It was possible to obtain working standard solutions containing up to 70 elements in this way.
For Group 2 samples, a quadrupole type ICP-MS, HP-4500 (Hewlett Packard [now Agilent Technologies], Palo Alto, CA, USA), was used for most of the trace elements (>0.1 mg kg−1 soil). However, for several ultra-trace elements (<0.1 mg kg−1) and/or trace elements susceptible to interferences resulting from spectral overlaps, it was necessary to use a high-resolution ICP-MS, ELEMENT (Finnigan MAT [now Thermo Scientific], Bremen, Germany). This type of ICP-MS is equipped with a double focusing mass spectrometer to separate the analyte ions from the overlapping polyatomic ions originating from gaseous components and/or matrix elements (CitationYamasaki 2000). The commonly used quadrupole type ICP-MS instruments are incapable of separating chemically different ions at the same nominal mass values and are, therefore, more prone to inaccurate results, particularly when the concentrations of the analytes become low. For the latter instrument, a special attachment (capacitive decoupling device) was installed to enhance the sensitivity.
For Group 3, an Elan DRC II ICP-MS (Perkin Elmer SCIEX, Concord, Ontario, Canada) was used. This instrument is equipped with a dynamic reaction cell (DRC) to suppress any Ar-originated interferences. However, it was not necessary to activate this device because the concentration levels of the elements susceptible to the above interferences were sufficient enough in all samples analyzed. The detection limits attained by this new type of instrument were approximately three orders of magnitude lower than those obtained using HP-4500. Therefore, it was possible to obtain acceptable results without using a high-resolution ICP-MS. The operational conditions and analytical settings for all of the instruments described above were basically those recommended by the manufacturers.
Inductively coupled plasma-atomic emission spectrometry
The instrument used for this part of the study was Vista-PRO (Varian, Palo Alto, CA, USA). Preliminary experiments showed that matrix effects were not negligible when acid digests were aspirated without dilution. Accordingly, all of the major constituents (Na2O, MgO, Al2O3, K2O, CaO, TiO2, MnO and Fe2O3) and trace elements (V, Sr and Ba) were analyzed after a 10-fold dilution, although the concentration levels of Cu and Zn became too low to obtain reliable results. First, the emission intensities from each element were measured at the positions of 4–8 different prominent lines using the simultaneous multi-wavelength capability of the instrument. Then, the most suitable analytical lines for each element, shown in , were selected after taking into consideration such factors as signal intensities, background levels and the reproducibility of the check standard solutions analyzed after every 10–15 real samples.
The mixed working standards solutions were prepared to cover the concentration ranges of most of the samples, while at the same time keeping the total concentrations of each solution approximately equal. The analytical results for V, Sr and Ba by ICP-AES were within ±5% of those obtained by ICP-MS in most cases.
Atomic absorption spectrometry
Manganese, Fe, Cu and Zn were also determined by AAS using a Varian AA240FS Fast Sequential AAS (Varian Australia, Mulgrave, Victoria, Australia). All of the elements except Fe were determined by direct aspiration of the acid digests. For Fe, however, samples were analyzed after a 10-fold dilution, using the less sensitive line of 372.0 nm rather than the commonly used 248.3 nm. The values obtained by AAS were in good agreement with those determined by ICP-AES and ICP-MS (Cu and Zn).
Table 4 Analytes and analytical lines for inductively coupled plasma-atomic emission spectrometry
Wavelength dispersive X-ray fluorescence
For the samples in Group 1, SiO2 was determined by WDXRF using an X-ray fluorescence spectrometer (model AFV 777; Tokyo Shibaura Electric Company, Tokyo, Japan). A finely powdered sample (0.500 g) and 4.5 g of Na2B4O7 were fused using a high-frequency furnace at approximately 1100°C to make a glass bead. Calibration curves were constructed using 12 rock standards provided by the US Geological Survey (AGV-1, BCR-1, G-2, GSP-1 and PCC-1 [CitationFlanagan 1973]), the Geological Survey of Japan (JB-1 and JG-1 [CitationAndo et al. 1974]) and the Centre de Rechereches Pétrographique et Geochimigues, France (BR, GA, GH, Mica Fe and Mica Mg [CitationRoubault et al. 1970]). A mathematical approach was used to correct errors resulting from matrix effects. More details of these procedures have been presented elsewhere (CitationYamasaki et al. 1980).
Spectrophotometry
The phosphorus (P2O5) content of the samples in Group 1 was measured photometrically as molybdenum blue. A 5 mL aliquot of the acid digest containing less than 0.1 mg of P was transferred to a 50 mL measuring flask. After adding 20–30 mL of water, 8 mL of the mixed reagent (30 mL of ammonium molybdate solution [40 g L−1], 60 mL of ascorbic acid solution [17.6 g L−1], 10 mL of potassium antimony tartrate solution [2.7 g L−1] and 100 mL of 2.5 mol L−1 H2SO4) was added and finally made up to a volume of 50 mL. The absorbance of the developed blue color was measured at 880 nm (CitationMurphy and Riley 1962).
Results and discussion
Calibration curves
and list the correlation coefficients, the root mean square error (RMSE) and concentration ranges of the calibration curves for the major constituents and trace elements examined. The numbers of samples used for the construction of the calibration lines differed from analyte to analyte and were less than the total number of reference materials (26) because the chemical values of certain elements were not available. When the concentration level of a certain element was extremely high compared with the level of the other samples, such reference materials were also excluded from the statistical calculations.
Among the 31 examined analytes, the correlation coefficients of only eight analytes were less than 0.98. Lower values for correlation coefficients were observed in low atomic number major constituents, between Na2O and P2O5. The lower correlation coefficients of these components apparently result from the lower total counts originating from the lower detection efficiency of the Ge detector at lower energy ranges ().
As the weight percentages of Na2O and MgO in soil samples are generally lower than those of geological reference materials (,), it can be concluded that there appears to be little possibility of successfully using EDXRF for the determination of these two components.
The smaller value obtained for P2O5 can obviously be attributed to the combined effects of the lower weight percentage of this component in the reference materials used to construct the calibration lines and the lower detection efficiency, as can be observed from the values listed in and . The differences between the certified values and the calculated values using the regression equation were within the range of ±10% when the contents of P2O5 were more than 0.2%. This may suggest a higher possibility of using the EDXRF approach for analyzing surface soils obtained from intensive agricultural lands because, in general, such samples contain much higher amounts of P2O5.
Table 5 Analytical figures of merit for the major constituent calibrations
Table 6 Analytical figures of merit for the trace element calibrations
Table 7 Instrument stability of the major constituents†
Table 8 Instrument stability of the trace elements†
In the case of Co, the overlapping of the Fe Kβ line is responsible for the lower correlation coefficient. Because the Fe content is three to four orders of magnitude higher than that of Co in most soil and environmental samples, it is common for the signal from the Co Kα line to be completely swallowed up by the huge peak in the Fe Kβ line. The use of the Co Kβ line in place of the Co Kα line was also impractical because the intensity of the Co Kβ line was strongly weakened owing to the absorption of Fe. Accordingly, it should be concluded that the EDXRF approach does not appear to be feasible for the analysis of Co in soils and related samples.
Although it was not possible to specify the causes of the lower correlation coefficients for Pr and Nd, particularly for Pr in the present study, it might be attributable to the difficulty of obtaining accurate values for these elements in the reference materials.
Instrument stability
Instrument stability was examined using two reference materials, JLk-1 for the major constituents and JSO-2 for the trace elements, under the conditions listed in and , respectively. The X-ray intensity was measured 24 times consecutively at intervals of 1 h. The X-ray intensity results for JLk-1 and JSO-2 are summarized in and , respectively.
As for the major constituents, the relative standard deviation (RSD) was less than 1%, with the exceptions of Na2O and P2O5. The lower reproducibility of these two analytes can be attributed to the lower total counts, as mentioned in the section on the calibration curve.
Table 9 Results of the linear regression analysis
In the case of the trace elements, the RSDs were much lower than 1% for all of the elements measured. JSO-2 is a unique reference material in that the concentrations of a considerable number of trace elements are adjusted to be approximately 1000 or 100 mg kg−1 by spiking chemicals to the original volcanic ash soil (CitationTerashima et al. 2000, 2002). Accordingly, it can be expected that the RSDs of real world samples would not be as low as those listed in . However, as the RSDs of the non-spiked elements (Sr) were approximately 0.5%, it appears reasonable to assume that the RSDs would be better than 1.0%, even with real samples.
Comparison of the results
The values obtained by the EDXRF method were compared with those obtained by the reference methods (). The number of samples examined was approximately 70 for SiO2 and P2O5, 100 for all of the other major constituents, and more than 230 for the trace elements. The soil and environmental samples were selected in such a way as to cover a wide range of chemical as well as mineralogical compositions. The best-fit lines were calculated in such a way as to force the equation to cross the Y-axis (EDXRF value) at zero when the X values (chemical results) are zero. The examined analytes were conveniently classified into three groups based on the correlation coefficients of their linear regression lines.
Out of 31 analytes examined, the number of analytes classified into Group 1 (R > 0.98) was 12, Sr was the highest among them, with the decreasing order of Ni > MnO > TiO2 > Cs > Ba > Rb > Fe2O3 > CaO > Ce > Zn > K2O. shows the results for Sr as a typical example of the analytes in this group. The regression lines were practically superimposed on the 1:1 lines (the lines of perfect fitness). As for the other analytes, the X-coefficients of the regression equation were also very close to unity (1.0), with the exception of Ni and Rb. Therefore, it can be concluded for the analytes of Group 1 that the results obtained in the present study compared favorably with those determined through time-consuming wet chemical pre-treatments.
The analytes belonging to Group 2 were those having correlation coefficients between 0.98 and 0.92, and were in the order of Al2O3 > Nd > P2O5 > SiO2 > Cr > Cu > La > Th > Y > V > Nb > Pb. Good agreements were observed for Al2O3, SiO2, P2O5 and Nd, although the correlation coefficients of the calibration lines of these analytes were lower (,,). For these analytes, considerably greater scattering around the regression lines was observed, as manifested by the lower correlation coefficients. However, it can be concluded that the proposed EDXRF method is also acceptable for the analysis of these analytes because the X coefficients remained between 1.0 and 1.3, except for Cr. However, in the case of Cr, the slope was much higher than the other analytes, indicating that the values obtained by the proposed technique were significantly higher than those analyzed by the spectrophotometric methods. A similar tendency was also observed for Sn, and particularly for Zr, which were categorized as Group 3. One of the authors has already reported that the Cr and Zr values in certain samples containing considerable amounts of chromite and zircon, respectively, resulted in an underestimation when analyzed by ICP-MS (CitationYamasaki 1996; CitationYamasaki et al. 1990), presumably because of incomplete dissolution of these minerals. However, it has become evident in this study that the above underestimation is a rather universal phenomenon that is not limited to chromite-rich and zircon-rich specific samples, but is generally observed in a wide variety of soil samples. In fact, even though the total number of samples examined was more than 230, it was not possible to find a sample whose value of Zr using the proposed EDXRF method was lower than the value recorded using ICP-MS, as can be seen in . In the case of Cr, volatilization of Cr compound during acid digestion would be an additional factor resulting in lower values in a chemical analysis. In contrast, the lower value for Sn could be attributed to volatilization losses. The above results clearly indicate the superiority of EDXRF over chemical methods based on an acid attack. In fact, the excellent calibration curve for Zr can be attributed to the fact that the contents of Zr in most of the reference materials were obtained by XRF, although the details of the analytical procedures differed from sample to sample.
Figure 1 Energy dispersive X-ray fluorescence concentration data plotted against the values obtained by inductively coupled plasma-mass spectrometry or inductively coupled plasma-atomic emission spectrometry. The superimposed line of slope = 1.0 does not represent a line of “best fit”.
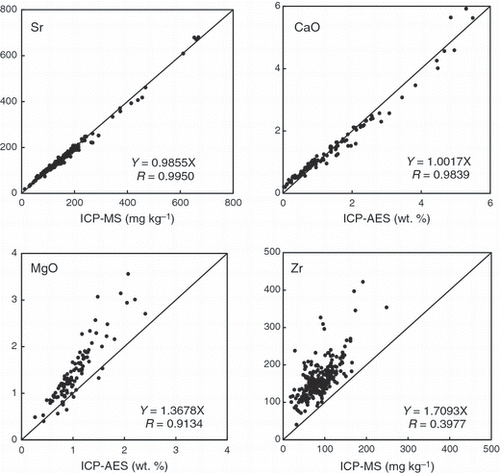
Group 3 was the worst class (R < 0.92). Except for Sn and Zr, the scatter plots were roughly within the range of RMSE. The causes of the poor results for these analytes were undoubtedly inherited from the lower correlation coefficients of the calibration lines (,) and/or the lower contents of these components in the samples (,,). Therefore, it can be concluded that chemical methods are more preferable for these five analytes.
Conclusions
We thoroughly examined the feasibility of using polarizing EDXRF spectrometry with pressed powder pellets to establish a rapid simultaneous multi-element analysis technique for the major constituents and trace elements in soil and environmental samples. The analytes examined were 10 major constituents (NaO, MgO, Al2O3, SiO2, P2O5, K2O, CaO, TiO2, MnO and Fe2O3) and 21 trace elements (V, Cr, Co, Ni, Cu, Zn, Rb, Sr, Y, Zr, Nb, Sn, Sb, Cs, Ba, La, Ce, Pr, Nd, Pb and Th). Twenty-six reference materials encompassing a wide range of rock, sediment and soil compositions were used for the calibrations. Compton scatter radiation was used as an internal standard to compensate for variations in the sample matrix, particle size, packing density and operating characteristics of the instrument. The correlation coefficients of the linear regression lines were >0.98, with the exception of Na2O, MgO, Al2O3, SiO2, P2O5, Co, Pr and Nd.
The values obtained using the proposed EDXRF method were compared with values obtained using conventional chemical methods for all of the elements except SiO2, the value for which was determined by borate fusion-WDXRF. The number of samples examined was approximately 70 for SiO2 and P2O5, 100 for all the other major constituents, and more than 230 for the trace elements. The soils and geological samples were selected in such a way as to cover a wide range of chemical as well as mineralogical compositions.
Out of the 31 analytes examined, the results for CaO, K2O, TiO2, MnO, Fe2O3, Ni, Zn, Rb, Sr, Cs, Ba and Ce obtained by the proposed EDXRF method compared favorably with those determined through conventional wet chemical methods. In contrast, the results for eight analytes (Na2O, MgO, Cr, Co, Zr, Sn, Sb and Pr) obtained by the proposed EDXRF method exhibited poor agreements with the results obtained using the chemical methods. Among these analytes, the poor agreements of Cr, Zr and Sn were attributable to incomplete dissolution and/or volatilization losses during the chemical treatments based on an acid attack. It can be concluded, therefore, that there appears to be a high possibility of the underestimation of the concentrations of these elements if wet chemical methods are used. Although the results of the other 11 analytes were not as good as those of the first group, they still appeared to be of practical use, considering the time consuming and potentially hazardous wet chemical pretreatment.
Notes
Present address: National Institute of Livestock and Grassland Science, National Agriculture and Food Research Organization, Tochigi 329-2793, Japan
Related Research Data
References
- Ando , A , Kamioka , H , Terashima , S and Itoh , S . 1989 . 1988 values for GSJ rock reference samples, “Igneous rock series” . Geochem. J. , 23 : 143 – 148 .
- Ando , A , Kurasawa , H , Ohomori , T and Takeda , E . 1974 . 1974 compilation of date on the GSJ geochemical reference samples JG-1 granodiorite and JB-1 basalt . Geochem. J. , 8 : 175 – 192 .
- Butler , OT , Cook , JM , Davidson , CM , Harrington , CF and Miles , D . 2009 . Atomic spectrometry update. Environmental analysis . J. Anal. At. Spectrom. , 24 : 131 – 177 .
- Butler , OT , Cook , JM , Harrington , CF , Hill , SJ , Rieuwerts , J and Miles , D . 2006 . Atomic spectrometry update. Environmental analysis . J. Anal. At. Spectrom. , 21 : 217 – 243 .
- Egashira , K , Aramaki , K , Yoshimasa , M , Takeda , A and Yamasaki , S . 2004 . Rare earth elements and clay minerals of soils of the floodplains of three major rivers in Bangladesh . Geoderma , 120 : 7 – 15 .
- Egashira , K , Aramaki , K , Yoshimura , M , Takeda , A and Yamasaki , S . 2007 . Variation of rare earth element of soils in the progress of soil formation – a case study for terrace and hill soils in Bangladesh . Clay Sci. , 13 : 117 – 124 .
- Egashira , K , Yamasaki , S and Virakornphanich , P . 1996 . Trace elements in paddy soils from the Central Region of the Mekong River, Laos . Soil Sci. Plant Nutr. , 42 : 673 – 676 .
- Flanagan , FJ . 1973 . 1972 values for international reference samples . Geochim. Cosmochim. Acta , 37 : 1189 – 1200 .
- Govindaraju , K . 1994 . 1994 composition of working values and sample description for 383 geostandards . Geostandards Newsl. , 8 ( Special Issue ) : 2
- Gunicheva , TN , Aisueva , TS and Afonin , VP . 1995 . Non-destructive X-ray fluorescence analysis of soils and friable and marine sediments . X-Ray Spectrom. , 24 : 187 – 192 .
- Heckel , J , Haschke , M and Brumme , R . 1992 . Principles and application of energy-dispersive X-ray fluorescence analysis with polarised radiation . J. Anal. At. Spectrom. , 7 : 281 – 286 .
- Hittipathirana , TD . 2004 . Simultaneous determination of parts-par-million level Cr, As, Cd and Pr, and major elements in low level contaminated soils using borate fusion and energy dispersive X-ray fluorescence spectrometry with polarized excitation . Spectrochim. Acta , 59 : 223 – 229 .
- Imai , N , Terashima , S , Itoh , S and Ando , A . 1995a . 1994 compilation values for GSJ reference samples, “Igneous rock series” . Geochem. J. , 29 : 91 – 95 .
- Imai , N , Terashima , S , Itoh , S and Ando , A . 1995b . 1994 compilation of analytical data for minor and trace elements in seventeen GSJ geochemical reference samples, “Igneous rock series” . Geostandards Newsl. , 19 : 135 – 213 .
- Imai , N , Terashima , S , Itoh , S and Ando , A . 1996 . 1996 compilation of analytical data on nine GSJ geochemical reference samples, “Sedimentary rock series” . Geostandards Newsl. , 20 : 165 – 216 .
- Jørgensen , N , Lausen , J , Viksna , A , Pind , N and Holm , PE . 2005 . Multi-elemental EDXRF mapping of polluted soil from former horticultural land . Environ. Int. , 31 : 43 – 52 .
- Kimura , K , Motoyoshi , H , Takeda , A and Yamasaki , S . 2008 . Trace elements in the arable soils of miyagi prefecture, Northeastern Japan . Jpn. J. Soil Sci. Plant Nutr. , 79 : 359 – 364 . (In Japanese with English summary.)
- Margui , E , Queralt , I , Carvaiho , ML and Hidalgo , M . 2005 . Comparison of EDXRF and ICP-OES after microwave digestion for element determination in plant specimens from an abandoned mining area . Anal. Chim. Acta , 549 : 197 – 204 .
- Matsunami , H , Matsuda , K and Miura , Y . 2009 . Determination of major and trace-element concentrations in farmyard manure using an energy dispersive x-ray fluorescence spectrometer (EDXRF) . Jpn. J. Soil Sci. Plant Nutr. , 80 : 219 – 225 . (In Japanese with English summary.)
- Mizushima , R , Yonezawa , M , Ejima , A , Koyama , H and Satoh , H . 1996 . Mincowave digestion using dual PTFE containers for analysis of trace elements in small amounts of biological samples . Tohoku J. Exp. Med. , 178 : 75 – 79 .
- Murphy , J and Riley , JP . 1962 . Modified single solution method for the determination of phosphate in natural water . Anal. Chim. Acta , 27 : 31 – 36 .
- Nakayama , K , Shibata , Y and Nakamura , T . 2007 . Glass beads/x-ray fluorescence analyses of 42 components in felsic rocks . X-Ray Spectrom. , 36 : 130 – 140 .
- Nanzyo , M , Yamasaki , S and Honna , T . 2002 . Changes in content of trace and ultratrace elements with an increase in noncrystalline materials in volcanic ash soils of Japan . Clay Sci. , 12 : 25 – 32 .
- National Bureau of Standards & Technology . 1993a . “ Certificate of AnalysisStandard reference material ” . 2709
- National Bureau of Standards & Technology . 1993b . “ Certificate of AnalysisStandard reference material ” . 2710
- National Bureau of Standards & Technology . 2000 . “ Report of InvestigationReference material ” . 8704
- National Bureau of Standards & Technology . 2002 . “ Certificate of AnalysisStandard reference material ” . 2711
- Nielson , KK , Mahoney , AW , Williams , LS and Rogers , VC . 1991 . X-ray fluorescence measurements of Mg, P, S, Cl, K, Ca, Mn, Fe, Cu, and Zn in fruits, vegetables, and grain products . J. Food Compost. Anal. , 4 : 39 – 51 .
- Orescanin , V , Mikelic , IL , Mikelic , L and Lulic , S . 2008 . Applicability of MiniPal 4 compact EDXRF spectrometer for soil and sediment analysis . X-Ray Spectrom. , 37 : 508 – 511 .
- Roubault , M , de la Roche , H and Govindaraju , K . 1970 . Present status (1970) of the cooperative studies on the geochemical standards of the “Centre de Recherches Pétrographique et Géochimiques” . Sci. Terre , 15 : 354 – 393 .
- Takeda , A , Kimura , K and Yamasaki , S . 2004 . Analysis of 57 elements in Japanese soils, with special reference to soil group and agricultural use . Geoderma , 119 : 291 – 307 .
- Terashima , S , Imai , N , Hirata , S and Taniguchi , M . 2000 . Preparation of a new GSJ geochemical reference material: JSO-2 soil . Bunseki Kagaku , 49 : 319 – 324 . (In Japanese with English summary.)
- Terashima , S , Imai , N , Taniguhci , M , Okai , T and Nishimura , A . 2002 . Preparation and preliminary characterisation of four new GSJ geochemical reference materials: soils, JSO-1 and JSO-2 soil; and marine sediments, JMS-1 and JMS-2 . Geostandards Newsl. , 26 : 85 – 94 .
- Terashima , S , Taniguchi , M , Mikoshiba , M and Imai , N . 1998 . Preparation of two new GSJ geochemical reference materials: basalt JB-1b and caol fly ash JCFA-1 . Geostandards Newsl. , 22 : 113 – 117 .
- Van Espen , P , Janssens , K and Nobels , J . 1986 . AXIL-PC, software for the analysis of complex X-ray spectra . Chemom. Intell. Lab. Syst. , 1 : 109 – 114 .
- Vekemans , B , Janssens , L , Vincze , L , Adams , F and Van Espen , P . 1994 . Analysis of X-Ray spectra by interactive least squares (AXIL): new developments . X-Ray spectrom. , 23 : 278 – 285 .
- Wada , K . 1986 . Ando Soils in Japan , 276 Fukuoka, , Japan : Kyushu University Press .
- Yamasaki , S . 1978 . An examination of the total analysis of major elements in soil samples . Soil Sci. Plant Nutr. , 24 : 305 – 308 .
- Yamasaki , S . 1996 . “ Inductively coupled plasma mass spectrometry ” . In Mass Spectrometry of Soils , Edited by: Boutton , T and Yamasaki , S . 459 – 491 . New York : Marcel Dekker .
- Yamasaki , S . 2000 . “ Inductively coupled plasma mass spectrometry in environmental analysis ” . In Encyclopedia of Analytical Chemistry: Instrumentation and Applications , Edited by: Meyers , RA . 2672 – 2692 . UK : John Wiley & Sons .
- Yamasaki , S , Katayama , M and Sasaki , T . 1980 . Total analysis of major constituents in soils by X-ray emission spectrometry with glass beads techniques . Soil Sci. Plant Nutr. , 26 : 25 – 36 .
- Yamasaki , S , Kimura , K , Motoyoshi , H , Takeda , A and Nanzyo , M . 2009 . Cadmium concentrations in soils of Japan . Jpn. J. Soil Sci. Plant Nutr. , 80 : 30 – 36 . (In Japanese with English summary.)
- Yamasaki , S , Takeda , A , Nanzyo , M , Taniyama , I and Nakai , M . 2001 . Background levels of trace and ultra-trace elements in soils of Japan . Soil Sci. Plant Nutr. , 47 : 755 – 765 .
- Yamasaki , S , Yoshida , T , Tsumura , A and Murase , G . 1990 . ICP-MS: one of the most promising techniques for comprehensive studies of heavy metals in soils . Trans. 14th Congress of Soil Sci , II : 96 – 101 .
- Yoshida , T , Yamasaki , S and Tsumura , A . 1992 . Determination of trace and ultra-trace elements in 32 international geostandards by ICP-MS . J. Min. Pet. Econ. Geol. , 87 : 107 – 122 .
- Yu , KN , Yeung , ZLL , Lee , LYL , Stokes , MJ and Kwok , RCW . 2002 . Determination of multi-element profiles of soil using energy dispersive X-ray fluorescence (EDXRF) . Appl. Radiat. Isot. , 57 : 279 – 284 .
- Present address: National Institute of Livestock and Grassland Science, National Agriculture and Food Research Organization, Tochigi 329-2793, Japan