
Abstract
The DNA damage response (DDR) is critical for genome stability and the suppression of a wide variety of human malignancies, including neurodevelopmental disorders, immunodeficiency, and cancer. In addition, the efficacy of many chemotherapeutic strategies is dictated by the status of the DDR. Ubiquitin-specific protease 28 (USP28) was reported to govern the stability of multiple factors that are critical for diverse aspects of the DDR. Here, we examined the effects of USP28 depletion on the DDR in cells and in vivo. We found that USP28 is recruited to double-strand breaks in a manner that requires the tandem BRCT domains of the DDR protein 53BP1. However, we observed only minor DDR defects in USP28-depleted cells, and mice lacking USP28 showed normal longevity, immunological development, and radiation responses. Our results thus indicate that USP28 is not a critical factor in double-strand break metabolism and is unlikely to be an attractive target for therapeutic intervention aimed at chemotherapy sensitization.
INTRODUCTION
In response to diverse DNA lesions, cells mount a DNA damage response (DDR) to prevent the accumulation of chromosomal instability, such as DNA breaks or chromosomal rearrangements (Citation1, Citation2). The DDR coordinates the regulation of cell cycle checkpoints, DNA repair, transcription, and cell fate pathways, such as apoptosis and senescence. Mutations in various DDR genes are involved in hereditary diseases characterized by increased cancer predisposition, premature aging, infertility, and developmental defects that particularly impair the immune system and central nervous system (CNS).
DNA double-strand breaks (DSBs) are recognized by the MRE11 complex, composed of MRE11, RAD50, and NBS1, which then rapidly activates the ATM kinase, the central transducer of the DDR to DSBs (Citation3, Citation4). Mutations in ATM, MRE11, NBS1, and RAD50 lead to similar, but clinically distinct, human diseases—ataxia-telangiectasia (AT), AT-like disease (ATLD), Nijmegen breakage syndrome (NBS), and NBS-like disease (NBSLD), respectively—characterized by chromosomal instability, hypersensitivity to DSB-inducing agents, and impaired DNA damage responses. Given the apical functions of ATM and the MRE11 complex in the DDR and their roles in preventing human disease, it is important to identify and characterize regulatory factors governing DDR events to gain a better understanding of the etiology of human pathologies associated with an impaired DDR.
Recently, it has become clear that ubiquitin ligases, deubiquitylating enzymes, and other mediators of ubiquitin signaling represent major components of the DDR and play significant roles in human disease pathologies and chemotherapeutic responses (Citation5). Indeed, there is growing evidence that the development of pharmacological agents against proteins mediating ubiquitylation and deubiquitylation may provide new opportunities for human disease management (Citation5, Citation6). ATM-dependent phosphorylation events are required for the recruitment of many DSB response factors, including the RING-E3 ubiquitin ligases, RNF8 and RNF168, which act sequentially to orchestrate the recruitment of critical DDR regulators, including 53BP1 and the tumor suppressor BRCA1, itself a ubiquitin RING-E3 ligase in conjunction with the BARD1 protein (Citation7, Citation8).
In addition to requiring ubiquitylation, the recruitment of 53BP1 to DSBs requires its oligomerization and recognition of histone H4 lysine 20 dimethylation (H4K20me2) through its Tudor domain (Citation9Citation–Citation11). Unlike BRCA1, 53BP1 does not exhibit any known enzymatic activities, but nonetheless it plays important roles in development and in response to DNA damage, in part by facilitating long-range end-joining events through the promotion of end synapsis and chromatin mobility (Citation12, Citation13). Indeed, mice lacking functional 53BP1 exhibit increased cell death in the developing thymus, severe deficiencies in immunoglobulin class switch recombination (CSR), chromosomal instability, elevated sensitivity to DSB-inducing agents, and predisposition to lymphoma (Citation14Citation–Citation17). A central role of 53BP1 appears to be the negative regulation of DNA-end resection pathways initiated by the Mre11 complex, CtIP, and BRCA1 (Citation18, Citation19). In this regard, it is notable that 53BP1 loss rescues the viability of BRCA1 mutant mice and restores the DNA repair proficiency to BRCA1-deficient cells by allowing DNA resection and repair through homology-directed repair pathways (Citation20, Citation21). Therefore, the proper regulation and balance between 53BP1 and BRCA1 recruitment can significantly influence tumor responses to chemotherapies.
Various deubiquitylating enzymes, including the ubiquitin-specific proteases (USPs) USP1, USP3, USP10, USP11, USP16, and USP28, have also been implicated in the DDR to DSBs, although in many cases their precise roles remain to be defined (Citation22). In particular, USP28 was first linked to the DDR through its physical association with the 53BP1 protein, and its depletion was reported to impair the stability of key DDR response proteins, including NBS1, CHK2, 53BP1, claspin, and TOPBP1, after a human cell line was treated with ionizing radiation (IR) (Citation23). Therefore, it was proposed that, by reversing DDR-induced ubiquitylation of such factors, USP28 protects them from proteasomal degradation. In addition, USP28 was shown to be an ATM substrate in response to DNA damage and was found to regulate CHK2-dependent apoptosis, in part through the induction of the proapoptotic PUMA protein. Subsequently, USP28 was implicated in the maintenance of cell cycle G2 arrest through its ability to stabilize the claspin protein, a key regulator of CHK1 activity (Citation24). Together, these data lead to a model in which USP28 is an ATM target and a crucial regulator of multiple DDR factors that control DNA repair, checkpoint responses, and apoptosis. Indeed, these findings suggested that USP28 deficiency would likely predispose to pathological outcomes associated with chromosomal instability and defective responses to DNA breaks, such as increased tumor predisposition and/or immunological deficiencies.
USP28 was independently identified in an shRNA screen for proteins required for apoptosis induced by the c-Myc oncogene and was shown subsequently to promote c-Myc protein stability in a posttranscriptional manner (Citation25). c-Myc is a potent inducer of replication stress, and its misregulated expression provokes a DDR involving ATM and the related ATR kinase that is critical for survival of cells overexpressing c-Myc (Citation26, Citation27). c-Myc levels are reduced following DNA damage, and it was proposed that this requires the dissociation of USP28 from the FBXW7 ubiquitin E3 ligase, which targets c-Myc for polyubiquitination and degradation via the proteosome (Citation28Citation–Citation30). This would then allow USP28 to associate with DDR proteins to promote their stabilization and enhance the DDR. These data therefore suggested that USP28 deficiency can impair tumorigenesis, particularly when driven by c-Myc oncogene overexpression.
Based on these previous studies, we hypothesized that USP28 could be an attractive therapeutic target, as its inhibition would be predicted to enhance DNA damage sensitivity through the destabilization of critical DDR mediators, such as 53BP1, NBS1, and claspin. Indeed, based on published evidence, USP28 deficiency would be predicted to influence tumor predisposition as well as tumor responsiveness to DNA damage-inducing cancer therapies and DDR-targeted drugs, as well as potentially selectively impairing the proliferation of tumor cells reliant on c-Myc activity. To address these possibilities, we examined the effects of USP28 depletion in both primary and transformed cell cultures, during mammalian development, and in response to radiation in vivo. We found that USP28 localized to DSBs in a manner that required the tandem BRCT domains of 53BP1. However, USP28 deficiency had no major effect on the stability of DDR proteins, cell cycle checkpoint responses, DNA repair, chromosome stability, or sensitivity to DNA damage in either primary or transformed cells. Consistent with USP28 being dispensable for effective DSB repair, we found that V(D)J recombination and class switch recombination (CSR) functioned essentially normally in USP28-deficient mice. In addition, USP28-deficient animals were fertile, and CHK2-dependent apoptosis occurred normally following IR exposure in cells and in tissues lacking USP28 expression. These data indicated that USP28 is not a critical factor for the DDR in vitro and in vivo, and its deficiency does not predispose to tumors in mice. Considering these findings, we suggest that USP28 is unlikely to be a potent tumor suppressor or represent a promising target for developing new cancer therapeutic strategies.
MATERIALS AND METHODS
Cell culture and transfections.
Human U2OS, HEK-293, and RPE1 cells and mouse embryonic fibroblasts (MEFs) were grown in Dulbecco's modified Eagle medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS; BioSera), 2 mM l-glutamine, 100 unit/ml penicillin, 100 μg/ml streptomycin, and amphotericin B (Fungizone; Sigma-Aldrich). Cell transfections with plasmid DNA or small interfering RNA (siRNA) duplexes () were performed by using FuGene HD (Roche) and Lipofectamine RNAiMax (Invitrogen), respectively, following the manufacturers' instructions. All siRNAs were purchased from Eurofins MWG Operon. Cells were analyzed 48 to 72 h after transfection.
TABLE 1 siRNA sequences
Immunostaining.
Human U2OS and RPE1 cells and MEFs were grown on coverslips or glass-bottom dishes for treatment with IR or laser microirradiation, respectively. Following the treatment, cells were either preextracted in CSK buffer [10 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (pH 7.0), 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2] containing 0.5% Triton X-100 for 5 min at room temperature and then fixed in 2% formaldehyde or fixed without preextraction. Samples were then blocked in 5% bovine serum albumin and stained with the appropriate primary antibody and secondary antibodies coupled to Alexa Fluor 488 or 594 (Molecular Probes). Confocal images were captured on a FluoView1000 Olympus microscope with a 40× or 60× oil objective lens.
Automated IRIF quantification using high-throughput microscopy.
RPE1 cells were seeded in 96 microwell plates (Cell Carrier; PerkinElmer) 48 h after siRNA transfections and treated the following day. Immortalized MEFs were seeded in the dishes described above 24 h prior to treatment. An Opera spinning-disk high-throughput microscopy platform (PerkinElmer) was used to image fixed immunostained cells, using a 20× water immersion objective lens (0.7 numerical aperture; Olympus). Six to 10 digital micrographs of two fluorescence channels (4′,6-diamidino-2-phenylindole [DAPI] and Alexa Fluor 488) were captured per microplate in a single optimal focal plane. For each experiment, 2 to 8 microplate wells per condition were imaged and automatically analyzed, leading to the quantification of >500 nuclei for each independent biological replicate. The images were analyzed using the spot detection script of the Acapella software (PerkinElmer). The DAPI channel was used to detect and segment the nuclei. Transfer of the nuclear area regions to the second fluorescence channel (γH2AX, 53BP1, or FK2) allowed the detection of IR-induced foci (IRIF) specifically within the nuclear areas of the detected cells.
Laser microirradiation and FRAP.
The experimental setup for fluorescence recovery after photobleaching (FRAP) was essentially as described previously (Citation31). In brief, cells were plated in glass-bottom dishes (Willco-Wells) and presensitized with 10 μM 5-bromo-2′-deoxyuridine (BrdU; Sigma-Aldrich) in phenol red-free medium (Invitrogen) for 24 h at 37°C. Laser microirradiation was performed by using a FluoView1000 confocal Olympus microscope equipped with a 37°C heating stage (Ibidi) and a 405-nm laser diode (6 mW) focused through a 60× UPlanSApo 1.35 numerical aperture lens. Localized DNA damage was generated by exposure of cells to a UV-A laser beam (Citation32).
For FRAP analyses, green fluorescent protein (GFP)-tagged proteins were allowed to accumulate and reach the steady-state level at the laser tracks. After a series of three prebleach images, a rectangular region placed over the laser-damaged line was subjected to a bleach pulse, followed by image acquisition in 6-s intervals for GFP-MDC1 and GFP-53BP1. Average fluorescent intensities in the bleached region were normalized against intensities in an undamaged nucleus in the same field after background subtraction to correct for overall bleaching of the GFP signal due to repetitive imaging. For mathematical modeling of GFP-tagged protein mobility, (It − I0)/Ipre values were plotted as a function of time, where I0 was the fluorescence intensity immediately after bleaching and Ipre was the average of the three prebleach measurements. Estimation of the mobile protein fraction (A) and residence time (τ) were performed using Prism 4 software and assuming the existence of one protein population, using the following equation: y(t) = A[1 − exp(−t/τ)].
Western blotting and immunoprecipitations.
Samples were lysed either in TNG-150 buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% Tween 20, 0.5% NP-40) with protease and phosphatase inhibitors or in SDS lysis buffer, then separated by SDS-PAGE and transferred to a polyvinylidene difluoride membrane (Millipore). Membranes were probed with antibodies for ATM and α-tubulin (both from Sigma), ATM pS1981 (Epitomics), ATR, H2AX, H3, H3 pS10, and β-actin (Abcam), 53BP1 (Novus Biological), USP28, CHK2 pT68, CHK1 pS345, and H2AX pS139 (Cell Signaling), USP28, CHK1, and RPA32 pS33 (Bethyl Laboratories), claspin, CHK1, and hemagglutinin (HA) (Santa Cruz Biotechnology), NBS1 (Novus Biological), CHK2 and H2AX pS139 (Millipore), anti-ubiquitin (FK2; Enzo Life Sciences), GFP (Roche). Primary antibodies were detected with appropriate secondary antibodies conjugated to horseradish peroxidase and visualized with an ECL-Plus apparatus (GE). HA- and GFP-tagged 53BP1 derivatives were immunoprecipitated using EZview red anti-HA affinity gel (Sigma-Aldrich) or GFP-trap agarose (ChromoTek), respectively.
Clonogenic survival assay.
Immortalized MEFs were seeded and treated with the indicated doses of IR or UV-C in an X-ray cabinet (Maxishot.200) or UV cross-linking oven (Stratalinker; Stratagene). U2OS cells were transfected with siRNA, and 48 h later they were seeded on 6-well dishes. Twenty-four hours later, cells were treated with a DNA-damaging agent, IR (CellRad Faxitron; Faxitron Bioptics), or camptothecin (Sigma). One or 2 weeks later, colonies were stained with 0.5% crystal violet–20% ethanol and counted. Results were normalized to plating efficiencies.
Checkpoint assays.
A G1/S checkpoint assay was performed essentially as described previously (Citation33) with modifications. Asynchronously growing RPE1 cells were pulsed for 30 min with 10 μM 5-ethynyl-2′-deoxyuridine (EdU), followed by mock or IR (2 Gy) treatment and incubation with 10 μM BrdU and 0.25 μg/ml nocodazole for 7 h. Cells were collected by trypsinization and fixed using Cytofix/Cytoperm reagent (BD Biosciences). EdU and BrdU were detected using Click-iT chemistry (Life Technologies) and anti-BrdU antibody (GE Healthcare), respectively. Genomic DNA was stained with DAPI, and samples were analyzed by flow cytometry. EdU-positive cells represent the S-phase population at the time of irradiation, while BrdU-positive cells represent the S-phase population following mock or IR treatment. Cells labeled with both EdU and BrdU represent the population of cells that continued DNA replication following the treatment.
The G2/M checkpoint was analyzed by assessing the mitotic index following irradiation with the indicated doses and recovery for 1, 6, or 24 h. Cells were fixed in 70% ethanol, washed, and stained with primary antibody for phospho-histone H3-S10 (Millipore), stained with secondary antibody conjugated to fluorescein isothiocyanate (FITC), and analyzed by flow cytometry.
Comet assay.
For the comet assay, cells were treated with 60 μg/ml phleomycin for 2 h at 37°C and allowed to recover for 2 h in culture medium at 37°C. Neutral comet assays were performed as specified in a comet assay kit (Trevigen) using GelBond films (Lonza) to support agarose gels. Samples stained with SYBR green I were imaged on an epifluorescence microscope (Olympus IX71) with a UPlanFLN 10× objective. Images were analyzed with CometScore software (TriTek). A total of 50 to 100 cells were scored in each sample.
Homologous recombination assay.
Homologous recombination (HR) efficiency was evaluated using the Traffic Light Reporter (TLR) system, the four-color system to track nuclease and donor template delivery simultaneously (described in reference Citation34). We performed assays using U2OS cells with a stably integrated TLR construct, which if cut with ISceI and repaired accurately using the provided donor sequence results in the restoration of an intact GFP sequence. At 6 h after siRNA transfections, the cells were cotransfected with DNA plasmids containing an HR donor and ISceI enzyme (plasmids contained blue fluorescent protein [BFP] and infrared fluorescent protein [IFP] expression markers, respectively). At 72 h after the first transfection, cells were harvested and analyzed by flow cytometry. GFP-positive cells were accounted for only in the population of dually transfected cells positive for BFP and IFP signals.
Generation of mice and mouse embryo fibroblasts.
The EUCE0037g10 embryonic stem cell line was obtained from the EUCOMM repository and injected into 3.5-day-old blastocysts derived from C57B6/J mice. Blast cells were reimplanted into 2.5-day-old pseudopregnant foster mice. Chimeras were scored for coat color and mated with C57B6/J wild-type mice. Agouti offspring were screened for the presence of the mutation by using PCR primers specific for the gene trap (for details on these methods, see the supplemental material). For the generation of primary MEFs, embryos were extracted at embryonic day 14.5 and trypsinized overnight. Disaggregated embryos were plated and cultured in MEF medium (15% FBS [HyClone], 1× Glutamax [Gibco], and 1× penicillin-strepdavidin [Gibco]). Primary MEFs were transformed by the transfection (MEF2 reagent Amaxa Nucleofector; Lonza) of a linearized, origin-less plasmid containing the simian virus 40 genome (p129).
Real-time quantitative PCR.
For real-time PCR of murine Usp28, total RNA was isolated using Tri reagent (Sigma) and reverse transcribed using a high-capacity RNA-to-cDNA kit (Applied Biosystems). A TaqMan (Applied Biosystems) master mix was used to amplify the cDNA of either Usp28 or GAPDH as an endogenous control on a 7900HT machine (Applied Biosystems). Data were plotted using SDS2.3 software and the 2−ΔΔCT method and analyzed with the RQ manager.
Metaphase spread preparations.
Early-passage (p2) MEFs were untreated or treated with aphidicolin (0.3 ng/ml) for 24 h, followed by incubation with colcemid (0.1 μg/ml) for 1 to 3 h. MEFs were swelled in 0.075 M KCl for 15 min at 37°C and then fixed in ice-cold fixative (75% methanol and 25% acetic acid) and washed several times in fixative. Metaphase preparations were spread on glass slides, steam treated using an 80°C water bath for 3 to 5 s, dried, and stained with 5% Giemsa solution.
Analysis of thymocytes.
Thymi were dissected from 7-to 9-week-old animals, and 1 × 106 cells were mock treated or irradiated using an X-ray cabinet (1.5 Gy/min; Maxishot 200; Yxlon International). Viable (double-negative [DN]) cells were identified by flow cytometry after propidium iodide and anti-annexin V–FITC antibody (BD Biosciences) staining. A viability ratio (the percentage of treated DN cells/percentage of mock-treated DN cells) or graphs of viable and apoptotic cells were plotted. To identify T-cell receptor (TCR) rearrangements, genomic DNA was prepared using the PureLink genomic DNA minikit (Invitrogen) following the manufacturer's instructions. To assay for recombination within and between TCRγ and TCRβ loci, a nested PCR was performed as described previously, using 100 ng of genomic DNA as the template (Citation35). The primers used were as follows: for TCRγ intralocus, TCRGV3S1a (5′-ACCATACACTGGTACCGGCA) and TCRGJ1/2a (5′-TCATCACTGGAATAAAGCAG); for TCRβ intralocus, TCRBV5S1a (5′-TGGTATCAACAGACTCAGGGG) and TCRBJ2a (5′-TCTACTTCCAAACTACTCCAG); for the TCRγ and TCRβ interlocus, TCRGV3S1a and TCRBJ2a. To assess thymic subpopulations during development, isolated thymocytes were stained with fluorescence-conjugated antibodies for CD3e, CD4, and CD8 (Becton Dickinson). Cells were analyzed by flow cytometry, and CD3e-positive cells were gated and analyzed using the FlowJo program.
Analysis of B lymphocytes and class switch recombination.
Eight- to 12-week-old mice were sacrificed, and B cells were isolated from the spleen by using a magnetically activated cell sorting B-cell isolation kit (Miltenyi Biotec). Isolated B cells were stained with carboxyfluorescein succinimidyl ester (CFSE; Sigma) and stimulated in RPMI 1640 medium (Sigma) supplemented with 10% FBS (HyClone), 1% penicillin-strepdavidin (Gibco), 1% glutamine (Gibco), 1 mM sodium pyruvate (Gibco), 53 μM β-mercaptoethanol (Gibco), 10 mM HEPES (Gibco), and 1× nonessential amino acids (Gibco) with 5 ng/ml interleukin-4 (IL-4; Sigma) and 25 μg/ml lipopolysaccharide (LPS; Sigma) for 1 to 4 days. Stimulated B cells were either processed for fluorescence-activated cell sorting analysis or Western blotting as described above. For analysis by flow cytometry, B cells were incubated in FcR blocking reagent and stained with biotin-labeled primary antibodies (IgG1 and IgM; Miltenyi Biotec) for 10 min at 4°C and then washed and stained with anti-biotin-allophycocyanin for 10 min at 4°C. Finally, DAPI was added at a concentration of 1 μg/ml to exclude dead cells. Data were collected by flow cytometry and analyzed using the FlowJo software. For analysis of CSR in vivo, 2-month-old mice were immunized intraperitoneally with 100 μg trinitrophenyl-keyhole limpet hemocyanin (TNP-KLH; Biosearch Technologies) in 200 μl Imject alum (Pierce Chemical Co.). Serum was collected on day 0 before immunization and on days 7, 14, 21, and 40 after immunization. A second immunization was performed on day 41, and serum was collected after 8 days. All samples were processed for analysis of immunoglobulin subtypes (IgG1 and IgG2b) in an enzyme-linked immunosorbent assay (ELISA).
Immunohistochemistry.
Seven- to 9-month-old mice were irradiated with 12.5 Gy by using a Faxitron X-ray cabinet and sacrificed 4 h later. The spleen, thymus, intestine were isolated and fixed in 10% (vol/vol) neutral buffered formalin, paraffinized, and sectioned using standard methods. Serial sections were stained with antibodies against cleaved caspase-3 (Cell Signaling), Ki67 (Novocastra Leica Biosystems), or yH2AX (Millipore).
RESULTS
USP28 is recruited to DNA breaks via 53BP1 tandem BRCT domains.
Previous work identified USP28 through an interaction with 53BP1, but whether this interaction required specific domains of 53BP1 or affected USP28 localization remained unclear (Citation23). To address this, we expressed HA epitope-tagged 53BP1 derivatives (see Fig. S1 in the supplemental material) in human HEK293 cells and analyzed the ability of endogenous USP28 to coimmunoprecipitate with these by using HA-agarose beads. This experiment revealed that the tandem BRCT domains were robustly associated with USP28 (). By contrast, we could not detect USP28 in immunoprecipitates of the C-terminal portion of 53BP1 from which the BRCT domains were deleted (CΔBRCT) (). We then generated U2OS cells that stably expressed GFP-tagged full-length 53BP1 or its derivative lacking the BRCT domains (ΔBRCT). In these cell lines, the endogenous 53BP1 protein can be efficiently depleted by siRNA against the 3′ untranslated region (UTR) of the 53BP1 mRNA (), leaving only expression of the exogenous GFP-tagged protein. Following siRNA-mediated depletion of the endogenous 53BP1, we performed GFP pulldown assays of the full-length and ΔBRCT proteins and found that the BRCT domains of 53BP1 were required for coimmunoprecipitation with USP28 (). In agreement with previously published work (Citation23), IR had little or no effect on the ability of USP28 to coimmunoprecipitate with 53BP1 ( and , respectively).
FIG 1 Interaction of USP28 with 53BP1 and its recruitment to DSBs is dependent on the tandem BRCT domains of 53BP1. (A) HA tag alone or HA-tagged derivatives of 53BP1 (BRCT and CΔBRCT) were expressed in HEK293 cells and immunoprecipitated using HA-agarose. Western blots were probed for the endogenous USP28. (B) siRNA against the 3′ UTR of 53BP1 leads to efficient depletion of the endogenous protein. (C) U2OS cells stably expressing GFP-tagged 53BP1, either the WT or the protein lacking the tandem BRCTs (ΔBRCT), were depleted of the endogenous protein by using siRNA directed against the 3′ UTR. Proteins precipitated using GFP-agarose were assayed for presence of endogenous USP28. (D) Recruitment of endogenous USP28 in U2OS cells stably expressing GFP-53BP1, WT and ΔBRCT, and depleted for the endogenous 53BP1 by using the 3′ UTR siRNA.
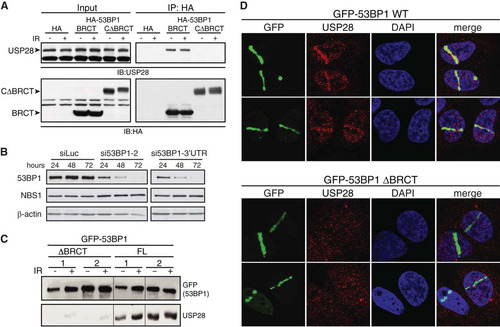
53BP1 localizes rapidly to sites of DNA damage in a manner that is independent of its BRCT domains (Citation16, Citation36, Citation37). We next asked whether USP28 also localized to DNA damage sites and, if so, whether interaction with 53BP1 was required for this. To address this question, cells stably expressing GFP-53BP1 full length or ΔBRCT were depleted for endogenous 53BP1 () and then subjected to laser microirradiation to induced DNA damage tracks (Citation38). Ensuing imaging revealed that endogenous USP28 was effectively recruited to sites of DNA damage in cells complemented with wild-type (WT) 53BP1 but not in cells expressing the ΔBRCT 53BP1 mutant (, upper and lower panels, respectively). Thus, as reported for EXPAND1/MUM1 (Citation39), USP28 recruitment to DSB sites depends on the tandem BRCT domains of 53BP1.
USP28 has a minor influence on DDR focus composition and dynamics.
As USP28 has been implicated in the stability of many key DDR mediator proteins, we examined the influence of USP28 on the recruitment and retention of DDR proteins to IRIF. As a first approach to address this issue, we used FRAP analysis in U2OS cells stably expressing GFP-MDC1 or GFP-53BP1. This experiment revealed that siRNA-mediated USP28 depletion had no significant effect on the association kinetics of MDC1 or 53BP1 with sites of DNA damage following IR treatment ( and ).
FIG 2 USP28 depletion has minor effects on the dynamics of DDR factors at IRIF. (A) FRAP analysis of GFP-MDC1 in U2OS cells stably expressing the fusion protein transfected with either siLuc (n = 13) or siUSP28 (n = 21). (B) FRAP analysis of GFP-53BP1 in U2OS cells stably expressing GFP-53BP1 transfected with either siLuc (n = 16) or siUSP28 (n = 15). Error bars represent standard deviations. (C to H) Quantification of IRIF numbers via use of antibodies against 53BP1, γH2AX, or conjugated ubiquitin adducts (FK2) following treatment of RPE-1 cells (C, D, and E) or MEFs (F, G, and H) of the indicated genotypes with 2 Gy of IR, viewed with an Opera microscope. MEF data represent the averages of 2 experiments, each using 2 littermate pairs of WT and Usp28-deficient MEFs.
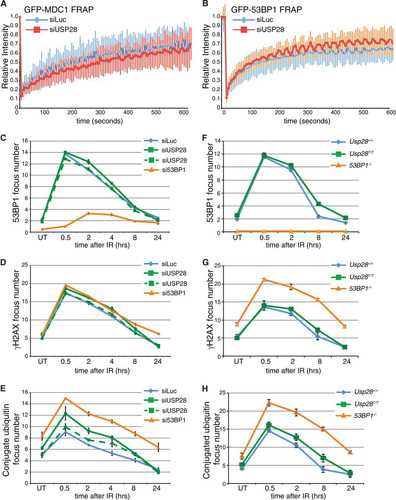
We next conducted a quantitative analysis of IRIF formation of established DDR markers in IR treated human RPE cells transfected with siRNAs targeting a control gene (luciferase; siLuc), 53BP1, or USP28. While the depletion of 53BP1 abolished 53BP1 foci as expected, depletion of USP28 with multiple siRNAs had no discernible effect on the localization of 53BP1 to IRIF over the 24-hour monitoring period (; see also Fig. S2 in the supplemental material). In addition, γH2AX focus numbers were also unaffected by USP28 depletion (), suggesting that lack of USP28 does not lead to DSB persistence. By contrast, the amounts of polyubiquitin chains, as detected with the FK2 antibody, were slightly but reproducibly increased in cells depleted of USP28, albeit to a lesser extent than in cells depleted of 53BP1 (; see also Fig. S2). This suggested that USP28 deficiency leads to a minor effect in the ubiquitin composition of IRIF.
To ensure that the lack of effects in the above studies was not due to the function of residual USP28 protein, we corroborated our analyses by using MEFs lacking USP28 expression due to a gene trap insertion between the first and second exons of the gene (Usp28T) (see Fig. S3 in the supplemental material). In homozygosity, the Usp28T allele led to undetectable Usp28 mRNA production (see Fig. S3). As observed in siRNA-treated RPE1 cells, the kinetics of 53BP1 IRIF formation in Usp28T/T MEFs was similar to that of littermate controls (). However, we did observe a slight increase in 53BP1 and conjugated ubiquitin (FK2) foci in cells lacking USP28 at later time points, consistent with the data for RPE1 cells ( and ). Collectively, these data showed that USP28 had a minor influence on the levels of polyubiquitinylated proteins in IRIF but did not detectably affect the dynamics of IRIF formation by the major DDR regulators studied.
USP28 deficiency does not strongly affect the DDR.
The composition and dynamics of IRIF do not easily translate into functional outcomes of the DDR, such as checkpoint activation or DNA repair efficiency. To determine if USP28 regulates DDR signaling, we performed Western immunoblotting using antibodies directed against key DDR proteins and phosphorylation events. This experiment established that siRNA-mediated USP28 depletion does not perceptibly affect the kinetics of ATM, CHK2, CHK1, RPA, or histone H2AX phosphorylation following treatment with IR or the topoisomerase I inhibitor camptothecin (CPT); furthermore, we did not observe marked differences in the levels of any of the DDR proteins examined ( and ). Consistent with these findings, USP28 loss did not affect the levels of ATM, ATR, 53BP1, claspin, NBS1, or CHK1 in primary MEFs treated with IR or UV-C light ( and ).
FIG 3 Normal DNA damage-signaling and checkpoint responses in USP28-depleted cells. Western blotting of 2 or 10 Gy IR-treated (A) or camptothecin-treated (CPT at 1 μM for 1 h) (B) U2OS cells at the indicated time points. Cells were transfected with siRNAs for luciferase (siLuc) or USP28. Western blotting results for MEFs treated with 5 Gy IR (C) or 10 J/m2 UV-C (D) at the indicated time points are shown. (E) The G1 checkpoint was intact in RPE-1 cells depleted of siRNA for USP28 or 53BP1 but not ATM. (F) G2/M checkpoint responses were normal in MEFs lacking USP28.
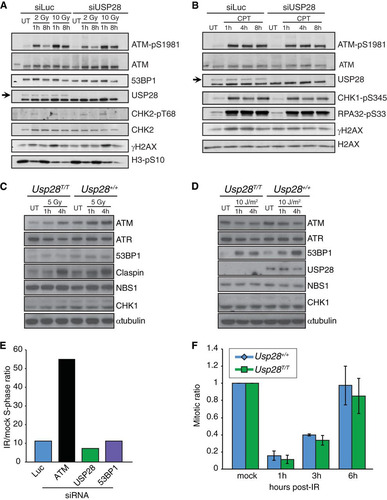
To determine if USP28 depletion affected processes regulated by the DDR, such as cell cycle checkpoint arrest or DNA repair, we examined the G1/S and G2/M checkpoints in human and murine cells, assessed DSB repair proficiency in multiple assays, and examined metaphase chromosome preparations in cells lacking USP28. This work established that human RPE1 cells depleted of USP28 or 53BP1 are able to efficiently induce G1 checkpoint arrest, while as expected, cells transfected with siRNA against ATM showed a strong G1/S checkpoint defect (). Furthermore, MEFs lacking USP28 expression showed normal G2/M checkpoint arrest and recovery following IR treatment (). This was consistent with the similar levels of the mitotic mark histone H3 phospho-serine 10 that were observed in U2OS cells transfected with either siLuc or siUSP28 (). We concluded that USP28 is not required for the activation of major IR-induced checkpoint responses.
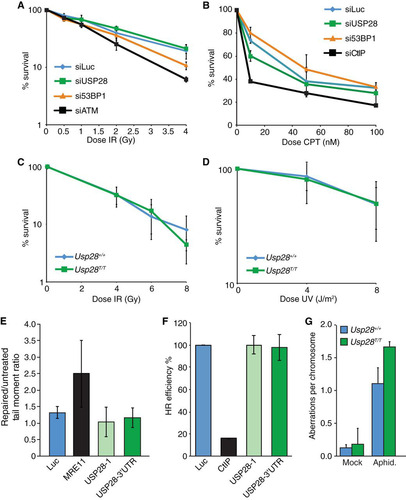
We next examined the effects of USP28 depletion on the sensitivity of cells to multiple genotoxic agents, including IR, CPT, and UV-C. While siRNA-mediated depletion of the key DDR factors ATM and CtIP (Citation40) led to IR and camptothecin hypersensitivity in human U2OS cells, respectively, as expected, no significant effects on clonogenic cell survival were observed upon USP28 depletion ( and ). Similarly, clonogenic survival curves in response to IR or UV-C were essentially identical in MEFs containing or lacking USP28 ( and ). To more directly assess DNA repair proficiency in control cells and in those depleted of USP28, we performed a neutral comet assay for DSB quantification and traffic light reporter system to test DSB repair by homologous recombination (Citation34). In each case, depletion of USP28 did not have a discernible effect on repair, while depletion of the critical DNA repair factor MRE11 led to an increase in tail moment in the comet assay following treatment with phleomycin, and depletion of CtIP led to a defect in homology-directed repair ( and ). Supporting the integrity of DNA repair pathways in cells lacking USP28, we did not observe increased levels of chromosome aberrations in primary Usp28T/T MEFs compared to controls, although USP28 loss did lead to slightly elevated levels of chromosomal aberrations when cells were subjected to acute aphidicolin exposure to impair DNA replication (). Collectively, these data indicated that, while USP28 may have a minor influence on chromosomal stability in response to replication inhibition, USP28 depletion has no marked effect on DNA damage signaling, DDR factor stability, DNA damage sensitivity, or DNA repair in multiple cell lines.
FIG 4 Normal DNA damage sensitivity, DNA repair kinetics, and genome stability in cells depleted for USP28. (A to D) Colony formation assays using U2OS cells transfected with indicated siRNAs (A and B) or MEFs of the indicated genotypes (C and D) were conducted, with cells treated with the indicated doses of IR, camptothecin (CPT), or UV-C. (E) Comet assay results in U2OS cells transfected with siLuc, siMRE11, or two different siRNAs to USP28; the tail moment ratios between repaired and untreated cells are indicated. (F) Efficiency of homologous recombination in a U2OS-based TLR cell line transfected with siLuc, siCtIP, or two different siRNAs to USP28. Values were normalized to the proportion of S/G2 phase cells. (G) Analysis of metaphase aberrations from early-passage MEFs that were mock treated or treated with aphidicolin (300 nM) for 24 h.
Physiological end-joining events are intact in USP28-deficient mice.
In cultured cells, numerous DNA end-joining pathways are available that can sometimes obscure more subtle phenotypes. For example, 53BP1-depleted U2OS cells are not strongly hypersensitive to DNA damage induced by IR, but mice lacking 53BP1 nevertheless have a severe immunoglobulin gene CSR defect and are predisposed to lymphomas (Citation14, Citation15, Citation17). To determine if USP28 plays important roles in physiological DNA repair events, such as those in the immune system, we examined both V(D)J recombination-dependent development of T lymphocytes and CSR in B lymphocytes. In contrast to mice lacking either ATM or 53BP1, USP28-deficient animals had similar total numbers of cells in the thymus () and a normal profile of CD4 and CD8 single-positive and double-positive T lymphocytes (CD3+) compared to littermate controls ( and ). Consistent with these findings, normal TCR joining events were readily detectable in these animals, and trans-rearrangements, characteristic of ATM deficiency (Citation41), were not detectable ().
FIG 5 Physiological end-joining processes dependent on ATM and 53BP1 are unaffected by USP28 deficiency. (A) The number of thymocytes in dissected thymi from mice of the indicated genotypes. Results were pooled from multiple WT (n = 6) and Usp28T/T mice (n = 7). (B) Distribution of thymocyte populations from 2-month-old mice of the indicated genotypes (n = 2 for each genotype). CD3-positive cells were stained for CD4 and CD8, and percentages were determined by flow cytometry. DN or DP indicates cells double negative or double positive for CD4 and CD8. (C) Examples of the primary flow cytometry data plotted in panel B. (D) Nested PCR analysis of expected TCRγ and TCRβ loci junction products or transrearrangements between loci in WT (lanes 1) or Usp28T/T (lanes 2) thymocytes. Results for a water control are shown in lanes 3. (E and F) The splenic weight (E) and cell number (F) in WT (n = 6) and Usp28T/T-deficient (n = 7) mice. (G) Class switching to IgG1 in vitro following LPS or IL-4 stimulation (n = 3 for each genotype). (H) Examples of the primary data plotted in panel G, showing CFSE staining to monitor proliferation. (I to K) ELISA measurements of IgG1 (I), IgG2b (J), and IgE (K) levels from animals of the indicated genotype injected with TNP-KLH at days 0 and 41. Each data point is the average for 3 animals, with the standard deviation plotted.
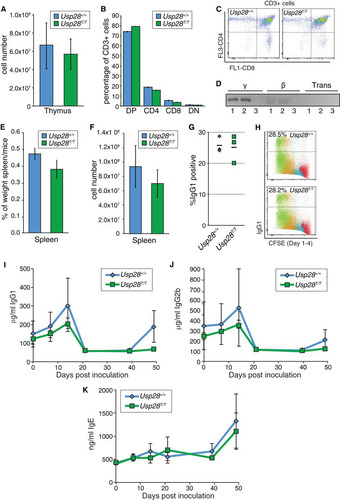
In contrast to the situation regarding the thymus, Usp28T/T animals had slightly smaller spleens with reduced cellularity ( and ). However, B-cell proliferation and CSR to IgG1 were indistinguishable in Usp28T/T and wild-type littermates following treatment with LPS or IL-4 ( and ). To more rigorously determine if USP28 impacts CSR, we inoculated a cohort of mice with TNP-KLH and examined IgG1, IgG2b, and IgE levels in serum at different times following injection. This revealed that serum levels of IgG1, IgG2B, and IgE were similar in WT and Usp28T/T animals in vivo following inoculation with TNP-KLH (, , and ). These data indicated that USP28 is not required for physiological end-joining events that take place during V(D)J and CSR and are required for immune system development.
DSB induced apoptosis is normal in USP28-deficient mice.
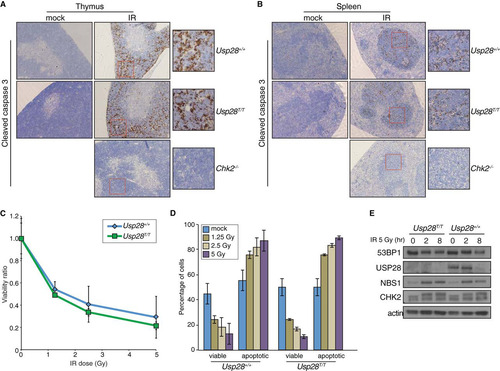
USP28 has been proposed to be required for CHK2 stability following DNA damage and to affect CHK2 function in apoptosis following IR treatment (Citation23). Following whole-body irradiation of wild-type mice, or those deficient for either Usp28 or Chk2, we examined apoptosis in the thymus and spleen by staining for cleaved caspase-3. While, as expected, little apoptosis was observed in mice lacking Chk2 following whole-body irradiation (Citation42), comparable levels were seen in wild-type mice and those lacking Usp28 expression ( and ). The IR-induced apoptosis of thymocytes is dependent on both CHK2 and ATM, which together fully activate p53-dependent responses, while intermediate defects can be detected more readily by analyzing apoptosis in thymocytes ex vivo (Citation43). Importantly, we found that cells lacking USP28 exhibited normal levels of apoptosis in response to IR over a range of doses, indicating that both the CHK2 and ATM branches of p53 signaling were functional in such cells ( and ) (Citation4). Additionally, CHK2 hyperphosphorylation, as well as CHK2 and NBS1 protein levels, were normal in B lymphocytes lacking USP28 (). Collectively, our data thereby indicated that USP28 does not strongly influence well-characterized apoptotic pathways that are dependent upon NBS1, CHK2, ATM, and p53 in the radiosensitive tissues of the immune system.
FIG 6 Apoptosis in response to IR is not affected by loss of USP28. (A and B) IHC staining of the thymi (A) or spleens (B) of mice of the indicated genotypes with an antibody for cleaved caspase 3 after mock or IR treatment. (C and D). Ex vivo analysis of thymocyte viability after exposure to a range of IR doses. The ratio of viable (double negative for PI and annexin V) cells relative to the mock treated group are plotted on the curve (C), and the absolute percentages of viable or apoptotic cells are graphed for the indicated genotypes (D). (E) Western blot results for 53BP1, USP28, NBS1, and CHK2 following IR treatment of B lymphocytes. Actin served as a loading control.
DISCUSSION
Proper regulation of the DDR is critical for the maintenance of genome stability. Recent work from various groups has demonstrated that the ubiquitin-proteasome pathway plays important roles in the DDR, including regulating repair pathway choice and many aspects of mammalian development, particularly those involving programmed DNA breakage and rearrangement in the immune system and germ line cells (Citation22, Citation44Citation–Citation50). The ubiquitin-specific protease USP28 has been reported to play a role in the DDR through the stabilization of key proteins, such as NBS1, CHK2, claspin, and 53BP1 (Citation23, Citation24). Furthermore, USP28 has been implicated in regulating the levels of the c-Myc oncoprotein and has been proposed to control the balance between c-Myc expression and the DDR, making it a particularly interesting enzyme, and potentially a drug target, with regard to cancer therapeutics (Citation25, Citation28).
While the BRCT domains of 53BP1 are dispensable for its known functions in the regulation of DNA repair pathway choice, telomere protection, and CSR (Citation10, Citation45, Citation51, Citation52), we have found that they mediate binding to USP28 and are critical mediators of USP28 localization to sites of DNA damage induced by laser microirradiation. Notably, previous work established that 53BP1 BRCT domains also mediate binding to p53 (Citation53Citation–Citation55) and MUM1/EXPAND1 (Citation39). It will therefore be interesting in future studies to assess whether 53BP1 binding to p53 or MUM1 is affected by USP28 status or vice versa. Importantly, while p53 plays key roles in damage-induced apoptosis, G1/S checkpoint arrest, and senescence (Citation53Citation–Citation55), we found that both the G1/S checkpoint and apoptosis were apparently fully intact in cells depleted of USP28, suggesting that p53-dependent responses were unaffected by USP28 status. Furthermore, although we did not examine cell senescence per se, it seems likely that p53-mediated senescence is also not affected by USP28 status, because we did not observe any reduced or enhanced growth in multiple preparations of primary MEF cultures under atmospheric (∼20%) oxygen conditions, which are known to promote senescence in certain DDR-deficient backgrounds (see Fig. S4 in the supplemental material) (Citation56).
Depletion of USP28 led to a mild increase in conjugated ubiquitin, as detected by the FK2 antibody which recognizes Lys-29-, Lys-K48-, and Lys-63-mediated ubiquitin chains, at IRIF (), but the targets of these modifications remain unclear and they appear to have little bearing on the DDR parameters that we have examined in vitro or in vivo. Tandem BRCT domains can function as phospho-recognition domains (Citation57, Citation58). While we cannot rule out that phosphorylation of USP28 contributes to its interaction with 53BP1, in line with a previous report (Citation23), we found it to occur independently of DNA damage (), suggesting that it does not require DNA damage-induced phosphorylation events.
The E3 ubiquitin ligase FBXW7, a well-studied oncogene and regulator of c-Myc, has been shown to interact with USP28 (Citation28, Citation59, Citation60). However, it remains unclear whether FBXW7 plays a role in regulating the stability of DDR factors. Furthermore, recent work identified the PIRH2 E3 ligase as a regulator of CHK2 stability and showed that USP28 can interact with PIRH2 and CHK2 in the context of USP28 overexpression (Citation61). Interestingly, PIRH2 was also implicated in regulating c-Myc stability and the DDR (Citation61Citation–Citation63). While again consistent with previous data suggesting that USP28 regulates CHK2, the functional relevance of this interaction for the DDR remains unclear, as we have found that Usp28 knockout animals do not recapitulate any of the predicted phenotypes, such as destabilized CHK2 or impaired IR-induced apoptosis. Finally, in this regard we note that recent work has indicated that USP28 interacts with and stabilizes the lysine-specific demethylase 1 (LSD1) that localizes to sites of DNA damage in an RNF168-dependent manner and reverses the histone mark H3K4me2 to suppress transcription (Citation64). The effects of USP28 on LSD1 have been proposed to favor the expression of genes controlling cancer stem cell (CSC) traits and thereby contribute to enhanced tumorigenesis (Citation65). Thus, the loss of USP28 would be predicted to impair transcriptional suppression at DNA damage sites and suppress CSC traits. We suggest that the requirement of USP28 for LSD1 stabilization is likely to be context specific, as mice lacking LSD1 have severe phenotypes in early embryogenesis that are not recapitulated in Usp28-deficient animals (Citation66) (see Fig. S5A and B in the supplemental material).
Usp28-deficient cells and animals also do not appear to exhibit any of the major phenotypes observed in mice lacking NBS1, ATM, CHK2, or 53BP1, including tumor predisposition, DNA repair defects, impaired apoptosis, chromosomal instability, immunodeficiency, or infertility ( to ; see also Fig. S5 in the supplemental material) (Citation10, Citation15, Citation17, Citation67, Citation68). These data suggest that the loss of USP28 alone does not have a major influence on pathological outcomes associated with impaired DSB metabolism. It is possible that robust compensation exists through the activities of other deubiquitylating enzymes or that USP28's primary functions reside outside of cellular responses to DSBs. With regard to this, we have not formally addressed the effects of USP28 deficiency on c-Myc stability. The most robust phenotype we observed in vivo was a modest reduction of splenic cellularity, and we speculate that this reflects deregulation of c-Myc levels that are critical for fine-tuning germinal center B-cell populations, but we have not further investigated this possibility (Citation69).
In conclusion, we have characterized the effects of USP28 in vitro and in vivo on DSB-induced DDR processes in primary and transformed cells, as well as in developing and adult tissues. As cells and animals lacking USP28 did not exhibit major DDR signaling defects or developmental defects consistent with impaired DSB metabolism, we conclude that USP28 is not a critical player in this response and is unlikely to represent a promising therapeutic target for cancer, based on opportunities relating to sensitization to chemotherapies, radiotherapy, or DDR enzyme-targeting drugs. However, we cannot exclude the possibility that it can significantly influence pathological outcomes by modifying the effects of other mutations in DDR or c-Myc-dependent pathways during tumor evolution, play tissue-specific roles that we did not uncover in our study, or exert additional functions under particular environmental conditions, such as hypoxia (Citation70). In addition, we note that pharmacological inhibition of USP28 could have consequences distinct from those of USP28 loss or depletion. Further work will be required to understand USP28's primary physiological roles and relevance to human health and disease.
tmcb_a_12275234_sm0001.pdf
Download PDF (3.7 MB)ACKNOWLEDGMENTS
We are grateful to the IRB mutant mouse facility for the generation of Usp28-deficient animals and technical assistance, Lluis Palenzuela for assistance with mouse colony maintenance, Jeremy Daniels and Michela Di Virgilio for advice regarding B-cell culture and CSR, Josep Forment for help with TLR assays, Maria Angeles Tapia-Laliena for technical assistance, Rafael Carazo Salas for sharing the Opera system with us, Ross Chapman for 53BP1 knockout MEFs, and Martin Eilers and Axel Behrens for sharing unpublished data.
Research in the S.P.J. laboratory is funded by Cancer Research UK program grant C6/A11224, the European Research Council, and the European Community Seventh Framework Program grant agreement no. HEALTH-F2-2010-259893 (DDResponse). Core infrastructure funding was provided by Cancer Research UK grant C6946/A14492 and Wellcome Trust grant WT092096. T.H.S. is a Ramon y Cajal fellow and has been supported by the Ministerio de Ciencia e Innovación (SAF2009-10023) and the Ministerio de Economía y Competitividad (BFU2012-39521), C.K.S. was supported by a FEBS Return-to-Europe grant, and P.A.K. was supported by an Early Postdoc Mobility fellowship from the Swiss National Science Foundation.
SUPPLEMENTAL MATERIAL
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00197-14.
REFERENCES
- Jackson SP, Bartek J. 2009. The DNA-damage response in human biology and disease. Nature 461:1071–1078. http://dx.doi.org/10.1038/nature08467.
- Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol. Cell 40:179–204. http://dx.doi.org/10.1016/j.molcel.2010.09.019.
- Stracker TH, Petrini JH. 2011. The MRE11 complex: starting from the ends. Nat. Rev. Mol. Cell Biol. 12:90–103. http://dx.doi.org/10.1038/nrm3047.
- Stracker TH, Roig I, Knobel PA, Marjanovic M. 2013. The ATM signaling network in development and disease. Front Genet. 4:37. http://dx.doi.org/10.3389/fgene.2013.00037.
- Jackson SP, Durocher D. 2013. Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 49:795–807. http://dx.doi.org/10.1016/j.molcel.2013.01.017.
- Cohen P, Tcherpakov M. 2010. Will the ubiquitin system furnish as many drug targets as protein kinases? Cell 143:686–693. http://dx.doi.org/10.1016/j.cell.2010.11.016.
- Ruffner H, Joazeiro CA, Hemmati D, Hunter T, Verma IM. 2001. Cancer-predisposing mutations within the RING domain of BRCA1: loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proc. Natl. Acad. Sci. U. S. A. 98:5134–5139. http://dx.doi.org/10.1073/pnas.081068398.
- Xia Y, Pao GM, Chen HW, Verma IM, Hunter T. 2003. Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J. Biol. Chem. 278:5255–5263. http://dx.doi.org/10.1074/jbc.M204591200.
- Zgheib O, Pataky K, Brugger J, Halazonetis TD. 2009. An oligomerized 53BP1 tudor domain suffices for recognition of DNA double-strand breaks. Mol. Cell. Biol. 29:1050–1058. http://dx.doi.org/10.1128/MCB.01011-08.
- Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D, Callen E, Nussenzweig A, Nussenzweig MC. 2011. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol. Cell 42:319–329. http://dx.doi.org/10.1016/j.molcel.2011.03.019.
- Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. 2006. Structural basis for the methylation state-specific recognition of histone H4–K20 by 53BP1 and Crb2 in DNA repair. Cell 127:1361–1373. http://dx.doi.org/10.1016/j.cell.2006.10.043.
- Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, Kruhlak MJ, Callen E, Livak F, Nussenzweig MC, Sleckman BP, Nussenzweig A. 2008. 53BP1 facilitates long-range DNA end-joining during V(D)J. recombination. Nature 456:529–533. http://dx.doi.org/10.1038/nature07476.
- Dimitrova N, Chen YC, Spector DL, de Lange T. 2008. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 456:524–528. http://dx.doi.org/10.1038/nature07433.
- Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 2004. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat. Immunol. 5:481–487. http://dx.doi.org/10.1038/ni1067.
- Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, Cascalho M, Chen L, Nussenzweig A, Livak F, Nussenzweig MC, Chen J. 2004. 53BP1 is required for class switch recombination. J. Cell Biol. 165:459–464. http://dx.doi.org/10.1083/jcb.200403021.
- Morales JC, Xia Z, Lu T, Aldrich MB, Wang B, Rosales C, Kellems RE, Hittelman WN, Elledge SJ, Carpenter PB. 2003. Role for the BRCA1 C-terminal repeats (BRCT) protein 53BP1 in maintaining genomic stability. J. Biol. Chem. 278:14971–14977. http://dx.doi.org/10.1074/jbc.M212484200.
- Ward IM, Minn K, van Deursen J, Chen J. 2003. p53 binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell. Biol. 23:2556–2563. http://dx.doi.org/10.1128/MCB.23.7.2556-2563.2003.
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, Xu X, Deng CX, Finkel T, Nussenzweig M, Stark JM, Nussenzweig A. 2010. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141:243–254. http://dx.doi.org/10.1016/j.cell.2010.03.012.
- Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A, Deng CX, Finkel T. 2009. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol. Cell 35:534–541. http://dx.doi.org/10.1016/j.molcel.2009.06.037.
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, Haffty BG, Tommiska J, Blomqvist C, Drapkin R, Adams DJ, Nevanlinna H, Bartek J, Tarsounas M, Ganesan S, Jonkers J. 2010. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 17:688–695. http://dx.doi.org/10.1038/nsmb.1831.
- Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, Drost R, Wientjens E, Ji J, Aly A, Doroshow JH, Cranston A, Martin NM, Lau A, O'Connor MJ, Ganesan S, Borst P, Jonkers J, Rottenberg S. 2012. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 3:68–81. http://dx.doi.org/10.1158/2159-8290.CD-12-0049.
- Jacq X, Kemp M, Martin NM, Jackson SP. 2013. Deubiquitylating enzymes and DNA damage response pathways. Cell Biochem. Biophys. 67:25–43. http://dx.doi.org/10.1007/s12013-013-9635-3.
- Zhang D, Zaugg K, Mak TW, Elledge SJ. 2006. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell 126:529–542. http://dx.doi.org/10.1016/j.cell.2006.06.039.
- Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M. 2008. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell 134:256–267. http://dx.doi.org/10.1016/j.cell.2008.05.043.
- Popov N, Wanzel M, Madiredjo M, Zhang D, Beijersbergen R, Bernards R, Moll R, Elledge SJ, Eilers M. 2007. The ubiquitin-specific protease USP28 is required for MYC stability. Nat. Cell Biol. 9:765–774. http://dx.doi.org/10.1038/ncb1601.
- Schoppy DW, Ragland RL, Gilad O, Shastri N, Peters AA, Murga M, Fernandez-Capetillo O, Diehl JA, Brown EJ. 2012. Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J. Clin. Invest. 122:241–252. http://dx.doi.org/10.1172/JCI58928.
- Campaner S, Amati B. 2012. Two sides of the Myc-induced DNA damage response: from tumor suppression to tumor maintenance. Cell Div. 7:6. http://dx.doi.org/10.1186/1747-1028-7-6.
- Popov N, Herold S, Llamazares M, Schulein C, Eilers M. 2007. Fbw7 and Usp28 regulate myc protein stability in response to DNA damage. Cell Cycle 6:2327–2331. http://dx.doi.org/10.4161/cc.6.19.4804.
- Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, Clurman BE. 2004. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. U. S. A. 101:9085–9090. http://dx.doi.org/10.1073/pnas.0402770101.
- Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. 2004. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 23:2116–2125. http://dx.doi.org/10.1038/sj.emboj.7600217.
- Galanty Y, Belotserkovskaya R, Coates J, Polo S, Miller KM, Jackson SP. 2009. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature 462:935–939. http://dx.doi.org/10.1038/nature08657.
- Limoli CL, Ward JF. 1993. A new method for introducing double-strand breaks into cellular DNA. Radiat Res. 134:160–169.
- Cescutti R, Negrini S, Kohzaki M, Halazonetis TD. 2010. TopBP1 functions with 53BP1 in the G1 DNA damage checkpoint. EMBO J. 39:3723–3732. http://dx.doi.org/10.1038/emboj.2010.238.
- Certo MT, Ryu BY, Annis JE, Garibov M, Jarjour J, Rawlings DJ, Scharenberg AM. 2011. Tracking genome engineering outcome at individual DNA breakpoints. Nat. Methods 8:671–676. http://dx.doi.org/10.1038/nmeth.1648.
- Lista F, Bertness V, Guidos CJ, Danska JS, Kirsch IR. 1997. The absolute number of trans-rearrangements between the TCRG and TCRB loci is predictive of lymphoma risk: a severe combined immune deficiency (SCID) murine model. Cancer Res. 57:4408–4413.
- Iwabuchi K, Basu BP, Kysela B, Kurihara T, Shibata M, Guan D, Cao Y, Hamada T, Imamura K, Jeggo PA, Date T, Doherty AJ. 2003. Potential role for 53BP1 in DNA end-joining repair through direct interaction with DNA. J. Biol. Chem. 278:36487–36495. http://dx.doi.org/10.1074/jbc.M304066200.
- Ward IM, Minn K, Jorda KG, Chen J. 2003. Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J. Biol. Chem. 278:19579–19582. http://dx.doi.org/10.1074/jbc.C300117200.
- Polo SE, Jackson SP. 2011. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 25:409–433. http://dx.doi.org/10.1101/gad.2021311.
- Huen MS, Huang J, Leung JW, Sy SM, Leung KM, Ching YP, Tsao SW, Chen J. 2010. Regulation of chromatin architecture by the PWWP domain-containing DNA damage-responsive factor EXPAND1/MUM1. Mol. Cell 37:854–864. http://dx.doi.org/10.1016/j.molcel.2009.12.040.
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. 2007. Human CtIP promotes DNA end resection. Nature 450:509–514. http://dx.doi.org/10.1038/nature06337.
- Kobayashi Y, Tycko B, Soreng AL, Sklar J. 1991. Transrearrangements between antigen receptor genes in normal human lymphoid tissues and in ataxia telangiectasia. J. Immunol. 147:3201–3209.
- Takai H, Naka K, Okada Y, Watanabe M, Harada N, Saito S, Anderson CW, Appella E, Nakanishi M, Suzuki H, Nagashima K, Sawa H, Ikeda K, Motoyama N. 2002. Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. EMBO J. 21:5195–5205. http://dx.doi.org/10.1093/emboj/cdf506.
- Stracker TH, Couto SS, Cordon-Cardo C, Matos T, Petrini JH. 2008. Chk2 suppresses the oncogenic potential of DNA replication-associated DNA damage. Mol. Cell 31:21–32. http://dx.doi.org/10.1016/j.molcel.2008.04.028.
- Bohgaki T, Bohgaki M, Cardoso R, Panier S, Zeegers D, Li L, Stewart GS, Sanchez O, Hande MP, Durocher D, Hakem A, Hakem R. 2011. Genomic instability, defective spermatogenesis, immunodeficiency, and cancer in a mouse model of the RIDDLE syndrome. PLoS Genet. 7(4):e1001381. http://dx.doi.org/10.1371/journal.pgen.1001381.
- Chapman JR, Taylor MR, Boulton SJ. 2012. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 47:497–510. http://dx.doi.org/10.1016/j.molcel.2012.07.029.
- Ramachandran S, Chahwan R, Nepal RM, Frieder D, Panier S, Roa S, Zaheen A, Durocher D, Scharff MD, Martin A. 2010. The RNF8/RNF168 ubiquitin ligase cascade facilitates class switch recombination. Proc. Natl. Acad. Sci. U. S. A. 107:809–814. http://dx.doi.org/10.1073/pnas.0913790107.
- Santos MA, Huen MS, Jankovic M, Chen HT, Lopez-Contreras AJ, Klein IA, Wong N, Barbancho JL, Fernandez-Capetillo O, Nussenzweig MC, Chen J, Nussenzweig A. 2010. Class switching and meiotic defects in mice lacking the E3 ubiquitin ligase RNF8. J. Exp. Med. 207:973–981. http://dx.doi.org/10.1084/jem.20092308.
- Fradet-Turcotte A, Canny MD, Escribano-Diaz C, Orthwein A, Leung CC, Huang H, Landry MC, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, Durocher D. 2013. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499:50–54. http://dx.doi.org/10.1038/nature12318.
- Hao Z, Duncan GS, Su YW, Li WY, Silvester J, Hong C, You H, Brenner D, Gorrini C, Haight J, Wakeham A, You-Ten A, McCracken S, Elia A, Li Q, Detmar J, Jurisicova A, Hobeika E, Reth M, Sheng Y, Lang PA, Ohashi PS, Zhong Q, Wang X, Mak TW. 2012. The E3 ubiquitin ligase Mule acts through the ATM-p53 axis to maintain B lymphocyte homeostasis. J. Exp. Med. 209:173–186. http://dx.doi.org/10.1084/jem.20111363.
- Li L, Halaby MJ, Hakem A, Cardoso R, El Ghamrasni S, Harding S, Chan N, Bristow R, Sanchez O, Durocher D, Hakem R. 2010. Rnf8 deficiency impairs class switch recombination, spermatogenesis, and genomic integrity and predisposes for cancer. J. Exp. Med. 207:983–997. http://dx.doi.org/10.1084/jem.20092437.
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 2013. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 339:700–704. http://dx.doi.org/10.1126/science.1231573.
- Lottersberger F, Bothmer A, Robbiani DF, Nussenzweig MC, de Lange T. 2013. Role of 53BP1 oligomerization in regulating double-strand break repair. Proc. Natl. Acad. Sci. U. S. A. 110:2146–2151. http://dx.doi.org/10.1073/pnas.1222617110.
- Derbyshire DJ, Basu BP, Serpell LC, Joo WS, Date T, Iwabuchi K, Doherty AJ. 2002. Crystal structure of human 53BP1 BRCT domains bound to p53 tumour suppressor. EMBO J. 21:3863–3872. http://dx.doi.org/10.1093/emboj/cdf383.
- Joo WS, Jeffrey PD, Cantor SB, Finnin MS, Livingston DM, Pavletich NP. 2002. Structure of the 53BP1 BRCT region bound to p53 and its comparison to the Brca1 BRCT structure. Genes Dev. 16:583–593. http://dx.doi.org/10.1101/gad.959202.
- Iwabuchi K, Bartel PL, Li B, Marraccino R, Fields S. 1994. Two cellular proteins that bind to wild-type but not mutant p53. Proc. Natl. Acad. Sci. U. S. A. 91:6098–6102.
- Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. 2003. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat. Cell Biol. 5:741–747. http://dx.doi.org/10.1038/ncb1024.
- Yu X, Chini CC, He M, Mer G, Chen J. 2003. The BRCT domain is a phospho-protein binding domain. Science 302:639–642. http://dx.doi.org/10.1126/science.1088753.
- Manke IA, Lowery DM, Nguyen A, Yaffe MB. 2003. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 302:636–639. http://dx.doi.org/10.1126/science.1088877.
- Wang Z, Inuzuka H, Zhong J, Wan L, Fukushima H, Sarkar FH, Wei W. 2012. Tumor suppressor functions of FBW7 in cancer development and progression. FEBS Lett. 586:1409–1418. http://dx.doi.org/10.1016/j.febslet.2012.03.017.
- Welcker M, Clurman BE. 2008. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer. 8:83–93. http://dx.doi.org/10.1038/nrc2290.
- Bohgaki M, Hakem A, Halaby MJ, Bohgaki T, Li Q, Bissey PA, Shloush J, Kislinger T, Sanchez O, Sheng Y, Hakem R. 2013. The E3 ligase PIRH2 polyubiquitylates CHK2 and regulates its turnover. Cell Death Differ 20:812–822. http://dx.doi.org/10.1038/cdd.2013.7.
- Hakem A, Bohgaki M, Lemmers B, Tai E, Salmena L, Matysiak-Zablocki E, Jung YS, Karaskova J, Kaustov L, Duan S, Madore J, Boutros P, Sheng Y, Chesi M, Bergsagel PL, Perez-Ordonez B, Mes-Masson AM, Penn L, Squire J, Chen X, Jurisica I, Arrowsmith C, Sanchez O, Benchimol S, Hakem R. 2011. Role of Pirh2 in mediating the regulation of p53 and c-Myc. PLoS Genet. 7(11):e1002360. http://dx.doi.org/10.1371/journal.pgen.1002360.
- Halaby MJ, Hakem R, Hakem A. 2013. Pirh2: an E3 ligase with central roles in the regulation of cell cycle, DNA damage response, and differentiation. Cell Cycle 12:2733–2737. http://dx.doi.org/10.4161/cc.25785.
- Mosammaparast N, Kim H, Laurent B, Zhao Y, Lim HJ, Majid MC, Dango S, Luo Y, Hempel K, Sowa ME, Gygi SP, Steen H, Harper JW, Yankner B, Shi Y. 2013. The histone demethylase LSD1/KDM1A promotes the DNA damage response. J. Cell Biol. 203:457–470. http://dx.doi.org/10.1083/jcb.201302092.
- Wu Y, Wang Y, Yang XH, Kang T, Zhao Y, Wang C, Evers BM, Zhou BP. 2013. The deubiquitinase USP28 stabilizes LSD1 and confers stem-cell-like traits to breast cancer cells. Cell Rep. 5:224–236. http://dx.doi.org/10.1016/j.celrep.2013.08.030.
- Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, Krones A, Ohgi KA, Zhu P, Garcia-Bassets I, Liu F, Taylor H, Lozach J, Jayes FL, Korach KS, Glass CK, Fu XD, Rosenfeld MG. 2007. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 446:882–887. http://dx.doi.org/10.1038/nature05671.
- Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, Naka K, Xia Z, Camerini-Otero RD, Motoyama N, Carpenter PB, Bonner WM, Chen J, Nussenzweig A. 2002. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 4:993–997. http://dx.doi.org/10.1038/ncb884.
- Jankovic M, Feldhahn N, Oliveira TY, Silva IT, Kieffer-Kwon KR, Yamane A, Resch W, Klein I, Robbiani DF, Casellas R, Nussenzweig MC. 2013. 53BP1 Alters the Landscape of DNA rearrangements and suppresses AID-induced B cell lymphoma. Mol. Cell 49:623–631. http://dx.doi.org/10.1016/j.molcel.2012.11.029.
- Dominguez-Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, Dalla-Favera R. 2012. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat. Immunol. 13:1083–1091. http://dx.doi.org/10.1038/ni.2428.
- Flugel D, Gorlach A, Kietzmann T. 2012. GSK-3β regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1α. Blood 119:1292–1301. http://dx.doi.org/10.1182/blood-2011-08-375014.
- Biton S, Dar I, Mittelman L, Pereg Y, Barzilai A, Shiloh Y. 2006. Nuclear ataxia-telangiectasia mutated (ATM) mediates the cellular response to DNA double strand breaks in human neuron-like cells. J. Biol. Chem. 281:17482–17491. http://dx.doi.org/10.1074/jbc.M601895200.