
Abstract
Loss of the RNA-binding protein Bicaudal-C (Bicc1) provokes renal and pancreatic cysts as well as ectopic Wnt/β-catenin signaling during visceral left-right patterning. Renal cysts are linked to defective silencing of Bicc1 target mRNAs, including adenylate cyclase 6 (AC6). RNA binding of Bicc1 is mediated by N-terminal KH domains, whereas a C-terminal sterile alpha motif (SAM) self-polymerizes in vitro and localizes Bicc1 in cytoplasmic foci in vivo. To assess a role for multimerization in silencing, we conducted structure modeling and then mutated the SAM domain residues which in this model were predicted to polymerize Bicc1 in a left-handed helix. We show that a SAM-SAM interface concentrates Bicc1 in cytoplasmic clusters to specifically localize and silence bound mRNA. In addition, defective polymerization decreases Bicc1 stability and thus indirectly attenuates inhibition of Dishevelled 2 in the Wnt/β-catenin pathway. Importantly, aberrant C-terminal extension of the SAM domain in bpk mutant Bicc1 phenocopied these defects. We conclude that polymerization is a novel disease-relevant mechanism both to stabilize Bicc1 and to present associated mRNAs in specific silencing platforms.
INTRODUCTION
The asymmetric distribution and localized translation of mRNAs control the expression of many proteins in a wide range of cells and tissues. Well-known examples include maternal determinants of embryo patterning and asymmetric fates in Drosophila oocytes, such as bicoid, oskar (osk), gurken (grk), and nanos mRNAs. Multiple trans-acting factors associate with these mRNAs in dynamic ribonucleoprotein (RNP) complexes to orchestrate nuclear processing, translational silencing, transport, cytoskeletal anchoring, and derepression of translation at the final destination (Citation1). Defects at any of these steps can have dramatic consequences. In particular, the failure to activate the translation of osk mRNA in the posterior pole plasm prevents the formation of abdominal structures and germ cells, whereas precocious osk mRNA translation in anterior oocytes blocks head formation and can give rise to a bicaudal phenotype with posterior duplications (Citation2Citation–Citation6).
Posterior localization and translational regulation of osk mRNA are mediated by specific cis- and trans-acting elements and by multiple interacting factors and regulatory proteins (Citation7Citation–Citation9), including Bicaudal-C (Bic-C; reviewed in reference Citation10). Bicaudal-C comprises three KH domains with characteristic RNA-binding GXXG signatures, where G represents glycine and one or both residues X are basic (Citation11, Citation12). The three KH and two KH-like domains that are distinguished by the absence of GXXG signatures are linked to a C-terminal sterile alpha motif (SAM) by a long glycine/serine-rich intervening sequence (see Fig. S1A in the supplemental material). The loss of Bic-C in Drosophila oocytes leads to the premature derepression of osk translation (Citation13, Citation14) and ectopic anterior localization of grk mRNA (Citation15). A biochemical screen for direct targets revealed binding of Bic-C to stau mRNA and to several other transcripts (Citation16). In addition, Bic-C has been shown to recruit CCR4-NOT deadenylase to attenuate its own translation (Citation16).
Mutations in mouse and human Bic-C homologs, as well as knockdown of Xenopus Bic-C, revealed an essential role in renal tubule morphogenesis (Citation17Citation–Citation19). In jcpk mutant mice, Bicc1 is truncated before the first KH domain, whereas a GC insertion in bpk mutant mice changes the reading frame within the last exon so that 21 amino acids at the C terminus are replaced by 149 aberrant residues (Citation17) (see Fig. S1B in the supplemental material). Bicc1jcpk/jcpk mutants develop renal cysts along the entire nephron, combined with dilated pancreatic and liver bile ducts that are reminiscent of autosomal dominant polycystic kidney disease (ADPKD). Unlike homozygotes, which die soon after birth, heterozygotes develop glomerulocystic disease in 25% of the cases after ageing (Citation20). In contrast, bpk mutants are a noncongenital model of autosomal recessive polycystic kidney disease (ARPKD), with renal cysts arising first in proximal tubules and later in collecting ducts (Citation21). Compared to the time of cyst formation in jcpk mutants, cyst formation is delayed in bpk mutants, likely because bpk affects only one of two alternatively spliced transcripts (Citation17, Citation22). ADPKD is caused by mutations in the PKD1 or PKD2 gene, whereas ARPKD results from defects in PKHD1. The corresponding proteins, polycystin-1, polycystin-2, and fibrocystin, associate with each other and regulate calcium flux in response to mechanical stimulation by fluid flow (Citation23, Citation24). In embryonic mouse kidneys, Pkd1 enhances the expression of Bicc1, which in turn increases the levels of Pkd2 mRNA, indicating that Bicc1 acts both downstream and upstream of polycystins (Citation25, Citation26).
Recent functional analysis identified adenylate cyclase 6 (AC6) and protein kinase inhibitor α (PKIα) mRNAs to be the first direct targets of mammalian Bicc1 (Citation27). PKIα inhibits protein kinase A (PKA), whereas AC6 stimulates it by synthesizing cyclic AMP (cAMP), suggesting a dynamic role for Bicc1 in regulating cAMP/PKA signaling. Importantly, cAMP and AC6 promote cystic growth in Pkd1 mutant mice and in ADPKD patients (Citation28Citation–Citation30). Bicc1 mutant mice also share other key features with human ADPKD, including impaired apical-basal sorting of the epidermal growth factor receptor; hyperactivation of its ligand, transforming growth factor α; and elevated mTOR signaling (Citation31Citation–Citation34). In addition, Bicc1 is needed during development for the alignment of motile node cilia by planar cell polarity (PCP) signals that govern visceral left-right patterning (Citation35, Citation36). Deregulation of PCP or canonical Wnt signaling can also trigger renal cysts (Citation23). Together, these observations highlight the relevance of Bicc1 mutants as a disease model and the importance of elucidating the molecular mechanisms that regulate Bicc1 activities at the crossroads of multiple signaling pathways.
Repression of AC6 and PKIα mRNAs by Bicc1 depends on specific regions in their proximal 3′ untranslated regions (UTRs) and on cognate microRNAs (miRNAs) (Citation27). While Bicc1 binds these RNAs independently of the SAM domain, deletion of the SAM domain blocks their loading into miRNA-induced silencing complexes (miRISCs) with Argonaute 2 (Ago-2) (Citation27). In addition, the SAM domain increases the potential of Bicc1 to inhibit the Wnt signaling component Dishevelled 2 (Dvl2) independently of KH domains in TOPflash reporter assays, possibly by localizing Bicc1 in a network of cytoplasmic foci that seem to interact with Dvl2 foci (Citation35). However, to our knowledge, a role for Bicc1 in localizing target mRNAs has not been evaluated.
Novel mechanistic insights into mRNA silencing may come from structure-function analysis of the Bicc1 SAM domain. SAM domains are found in numerous regulatory proteins and in some instances mediate the formation of homo- or heterooligomers (Citation37Citation–Citation39). The crystal structure of the SAM domain of the transcriptional repressor TEL established that self-association in a head-to-tail configuration via the so-called midloop (ML) and end helix (EH) surfaces can give rise to a helical polymer (Citation40, Citation41). Since a green fluorescent protein (GFP) fusion of the human BICC1 SAM domain can polymerize in vitro (Citation42), here we asked whether self-association controls the localization or function of full-length Bicc1. To address this question, we modeled the three-dimensional (3D) structure of the mouse Bicc1 SAM domain and designed point mutations to disrupt a potential SAM-SAM interface. We show that such mutations inhibit polymerization of full-length Bicc1 and its cytoplasmic clustering. Furthermore, we report that polymerization-competent Bicc1 recruits an associated AC6-3′ UTR reporter mRNA to cytoplasmic foci and that the SAM domain promotes mRNA silencing by mediating Bicc1 polymerization. bpk mutant Bicc1 (Bicc1bpk), which is associated with an ARPKD-like disease, phenocopied these defects, indicating that SAM polymerization is likely essential for Bicc1 functions in vivo.
MATERIALS AND METHODS
Plasmids, cloning, and mutagenesis.
The luciferase reporter plasmids pCS+::AC6-3′UTRprox and pCS+::PKIα-3′UTRprox have been described previously (Citation27). To construct the AC6-27×MS2 reporter plasmid, 24 repeats of a motif that binds the bacteriophage MS2 coat protein were amplified by PCR from pCR4-24MS2SL-stable (catalog number 31865; Addgene) and added to 3 existing MS2 sites in plasmid pCS+::AC6-3′UTRprox by insertion into a unique PstI site. To subclone the mutant Bicc1 cDNAs encoding the mutation in mutant A (mutA) to mutF and bpk, the plasmid pCMV-SPORT6::HA-Bicc1 (Citation35) was modified by introducing a silent BglII site upstream of the SAM-coding sequence. Mutant cDNA fragments were then cloned between this engineered BglII site and a unique XbaI site. Mutant cDNA fragments encoding the mutations in mutA to mutF were generated by overlap extension PCR. The plasmids carrying Bicc1bpk and the splice variant B of wild-type (WT) Bicc1 were obtained by introducing a synthetic cDNA (Proteogenix) into the pCMV-SPORT6::HA-Bicc1 vector. The sequences of all mutated expression vectors were verified by Sanger sequencing. The plasmids pTOPFLASH (Citation43), pCDNA3.1::mDvl2 (Citation44), pDCP1a-GFP (Citation45), and pGEX-1λT::SAM (Citation27) were previously described.
Recombinant GST-SAM and custom anti-Bicc1 antibody.
The pGEX-1λT::SAM plasmid was used to produce the recombinant glutathione S-transferase (GST)–SAM protein in the Escherichia coli BL21 strain (Novagen) as described previously (Citation27). The GST-SAM protein was purified from cell extracts under native conditions, using glutathione-Sepharose 4B, as recommended by the manufacturer (GE Healthcare). Purification was carried out in a buffer consisting of 50 mM Tris-HCl, pH 8, 200 mM NaCl, 1 mM dithiothreitol (DTT). When necessary, 20 mM glutathione was added for elution. A custom polyclonal Bicaudal-C antibody against GST-SAM was raised by Proteogenix (Oberhausbergen, France) in rabbit by development of an antibody against the mouse Bicc1 SAM domain (residues 870 to 984) fused to an N-terminal His tag. The antibody was affinity purified and tested for its activity against the antigen by enzyme-linked immunosorbent assay.
Bicc1 mutant mice.
Mice heterozygous for a targeted null allele of Bicc1 were maintained on a C57BL/6 mouse genetic background in individually ventilated cages at the Ecole Polytechnique Fédérale de Lausanne (EPFL) animal facility as described previously (Citation35). Mice homozygous for the hypomorphic bpk allele of Bicc1 have been described previously (Citation17, Citation21, Citation34). All animal experiments were approved by the Veterinary Service of the Swiss canton of Vaud or by the Institutional Animal Care and Use Committee and adhered to the guidelines in the Guide for the Care and Use of Laboratory Animals (Citation46).
Cell culture and transfections.
HEK293T and COS-1 cells were cultured in Dulbecco modified Eagle medium (Sigma) supplemented with 10% fetal bovine serum (FBS; Sigma), glutamine (1%; Invitrogen), and gentamicin (1%; Invitrogen). Plasmids were transfected using the jetPEI transfection reagent (Polyplus Transfection) according to the manufacturer's instructions. Scrambled and Lsm1 small interfering RNAs were transfected 24 h prior to plasmid transfection using the INTERFERin transfection reagent (Polyplus Transfection).
Indirect immunofluorescence analysis and histological stainings.
For immunostaining, COS-1 cells were transfected with hemagglutinin (HA)-tagged Bicc1 (HA-Bicc1) and GFP-Dcp1a (1 μg DNA each) in 6-well plates. After 24 h, the cells were split and grown on sterile coverslips in 24-well plates. At 48 h after transfection, the cells were fixed for 10 min at −20°C in methanol and washed with phosphate-buffered saline (PBS). The coverslips were incubated for 1 h at room temperature for blocking in PBS containing 1% bovine serum albumin (BSA) and then for 2 h at room temperature in blocking buffer containing the anti-HA primary antibody (1/500, rabbit; Sigma). After washes in PBS, the Alexa Fluor 568-conjugated anti-rabbit secondary antibody was added, and the mixture was incubated in blocking buffer for 1 h at room temperature in the presence of DAPI (4′,6-diamidino-2-phenylindole; 1/10,000). Pictures were acquired on a Zeiss LSM700 confocal microscope.
For cryosectioning, organs were collected from Bicc1 mutants and control littermates at postnatal day 2, fixed for 4 h in 4% paraformaldehyde, soaked in 15% sucrose overnight at 4°C, and embedded in optimum-cutting-temperature (OCT) compound on isopentane. Sections (8 μm) were permeabilized for 10 min at room temperature with PBS, 0.2% Triton X-100 and then incubated for 2 h at room temperature for blocking in PBS containing 1% BSA. An additional blocking step using a streptavidin-biotin blocking kit (catalog number SP-2002; Vector Laboratories) was added specifically for biotinylated Lotus tetragonolobus lectin (LTL) detection. Primary antibodies anti-Bicc1 SAM (1/300, rabbit) (27), anti-Dcp1a (1/100, mouse; Abnova), anti-CK19 (1/50, human; Santa Cruz Biotechnology), and biotinylated LTL (1/200; Vector Laboratories) were added and incubated overnight at 4°C. Bicc1 antibody was preabsorbed with embryo powder (1/100) prior to incubation. After washing in PBS–0.1% Triton X-100, secondary antibodies (anti-rabbit antibody–Alexa Fluor 488, anti-mouse antibody–Alexa Fluor 647, anti-human antibody–Alexa Fluor 568, or streptavidin-Alexa Fluor 568) were incubated in PBS–0.1% Triton X-100 for 1 h at room temperature. Nuclei were stained with DAPI (1/10,000) during the final washes. Stained sections were mounted in DABCO (1,4-diazabicyclo[2.2.2]octane). Images were acquired on a Zeiss LSM700 confocal microscope.
Molecular modeling and MD simulation.
The X-ray structure of the diacylglycerol kinase δ1 (DGKδ1) SAM dimer (PDB accession number 3BQ7) (Citation47) was identified with tools from the SWISS-MODEL work space (Citation48). The software MODELLER (v9.5) (Citation49) was used to align the Bicc1 and DGKδ1 sequences and to construct 100 models of the Bicc1 SAM domain dimer. The best model, according to the discrete optimized protein energy (DOPE) (Citation50) score, was set up for molecular dynamics (MD) simulation with a GROMACS (v4.5) simulation tool kit (Citation51). The protein was simulated in water and subjected to energy minimization with the steepest-descent method (Citation52) and state-of-the-art equilibration. The MD simulation was then carried out using GROMACS (v4.5) software and the CHARMM27 force field (Citation53). The system was simulated for 10 ns in the isobaric-isothermal ensemble, with the pressure being maintained at 1 atm and the temperature being maintained at 300 K. Final models were rendered in the PyMOL program (Schrödinger).
Electron microscopy.
Twenty microliters of protein sample at 0.5 μg/μl was deposited on a carbon-Formvar grid (Ted Pella Inc.), incubated for 2 min, and blotted dry. The grid was then covered with 20 μl of 2% uranyl acetate, incubated for 2 min, and blotted dry. Samples were visualized on a JEOL JEM-2200FS transmission electron microscope.
Immunoprecipitation and RT-PCR analyses.
To monitor protein-RNA interactions, immunoprecipitations were conducted with HEK293T cells transfected in 10-cm dishes (for Bicc1 WT, 1 μg DNA was used; for Bicc1 mutD, mutant Bicc1 lacking SAM [Bicc1ΔSAM], and the Bicc1 bpk mutant, 2 μg DNA was used). At 36 h after transfection, cells were washed with PBS and resuspended in extraction buffer, consisting of 20 mM Tris-HCl, pH 7.4, 2.5 mM MgCl2, 100 mM NaCl, 5% glycerol, 1 mM DTT, 0.05% NP-40, RNasin (Promega), and protease inhibitors (Roche). After a brief sonication and centrifugation at 10,000 × g for 10 min, the supernatants were incubated for 2 h at 4°C with anti-HA beads (Sigma) on a wheel. After five washes of 5 min each in washing buffer, consisting of 20 mM Tris-HCl, pH 7.4, 2 mM MgCl2, 200 mM NaCl, 1 mM DTT, and 0.1% NP-40, RNAs were isolated by phenol-chloroform extraction, followed by ethanol precipitation and RQ1 DNase (Promega) treatment. Reverse transcription-PCR (RT-PCR) analyses were carried out using SuperScript II reverse transcriptase (Life Technologies) according to the manufacturer's recommendations. The reverse transcription was performed using the following primer: CAGTGCAGGGTCCGAGGTATTCGGCCTCTGCGCTTTCTC. The PCR was carried out using the forward primer GAAGATCGGGTTGAACATGGGTCC and the reverse primer CAGTGCAGGGTCCGAGGTATTC.
GST pulldown assays.
To monitor interactions between GST-SAM and HA-Bicc1, HEK293T cells were transfected in 6-well plate dishes (for the Bicc1 WT, mutA, mutB, and mutF, 1 μg DNA was used; for Bicc1 mutC, mutD, and mutE and the ΔSAM mutants, 2 μg DNA was used) or 10-cm dishes (for Bicc1bpk, 8 μg DNA was used). Cell extracts were prepared as mentioned above for the immunoprecipitation assays. Supernatants were incubated for 2 h at 4°C with glutathione-Sepharose 4B beads saturated with GST-SAM or GST for the negative control. After washing as described above, the beads were resuspended in Laemmli buffer, fractionated on SDS-polyacrylamide gels, and analyzed by Western blotting. Anti-HA antibodies (1/1,000, rabbit; Sigma) were used for the detection of HA-Bicc1. The retention of the GST-SAM bait was monitored directly by Coomassie staining.
Sucrose gradient fractionation assays.
HEK293T cells were transfected in 10-cm dishes, using 3 plates per condition (2 μg DNA/plate). Cell extracts were prepared as mentioned above for the immunoprecipitation assays. Continuous 15 to 60% sucrose gradients were prepared manually by layering and passive diffusion of sucrose solutions prepared in buffer consisting of 20 mM Tris-HCl, pH 7.4, 100 mM NaCl, 0.05% NP-40. Identical volumes of cell extracts were fractionated at 4°C by centrifugation at 100,000 × g for 3 h. Fractions were recovered manually from the top, fractionated on SDS-polyacrylamide gels, and analyzed by Western blotting. Anti-HA antibodies (1/1,000, rabbit; Sigma) were used for the detection of HA-Bicc1. Anti-γ-tubulin (1/2,000, mouse; Sigma) and anti-ribosomal protein S6 (anti-RPS6; 1/1,000, mouse; Cell Signaling) antibodies were used to detect, respectively, γ-tubulin and RPS6 proteins, which were used as internal controls.
Protein half-life measurements.
HEK293T cells were transfected in 6-well plate dishes (1 μg DNA). At 24 h after transfection, cycloheximide (CHX; Sigma) was added at a final concentration of 100 μg/ml. Cells were collected before treatment (0 h) and 8, 24, 32, 48, and 56 h after cycloheximide addition. Cell extracts were prepared as mentioned above for immunoprecipitation assays. Anti-HA antibodies (1/1,000, rabbit; Sigma) were used for the detection of HA-Bicc1, and anti-γ-tubulin antibodies (1/2,000, mouse; Sigma) were used for normalization.
Luciferase assays.
HEK293T cells were plated in 24-well plates. After 12 h, quadruplicate samples were transfected with the plasmids indicated below (a 1× dose was 0.1 μg/well) and with a lacZ expression vector (0.05 μg/well) using jetPEI (Polyplus Transfection). At 36 h after transfection, cells extracts were prepared in buffer consisting of 25 mM Tris-phosphate, pH 7.8, 2 mM DTT, 2 mM CDTA (1,2-diaminocyclohexanetetraacetic acid), 10% glycerol, 0.5% Triton X-100. The measurements of luciferase expression levels were carried out using 20-fold-diluted extracts, and luminescent counts were normalized to β-galactosidase activity. Results represent mean values from at least 3 independent experiments performed in quadruplicate, and error bars show the standard errors of the means (SEMs). Student's t test was used to calculate P values.
RESULTS
Cytoplasmic clustering of Bicc1 in mouse kidney and liver epithelial cells.
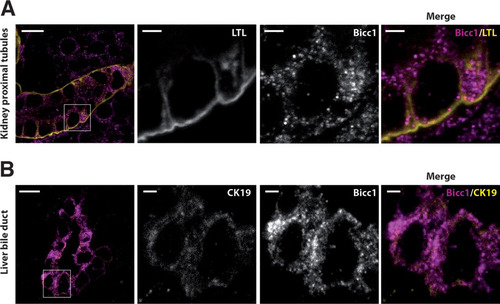
Bicc1-deficient mice are born with cysts in their kidneys and pancreas and dilated liver bile ducts (Citation25, Citation27, Citation54). Concordant with a function in renal morphogenesis, the Bicc1 protein is expressed in the newborn mouse kidney cortex (Citation25, Citation27). However, the distribution of Bicc1 in this or other tissues has not been resolved at a subcellular level. To address this, we stained frozen sections of postnatal kidneys and livers using a novel custom anti-Bicc1 antibody that specifically reacts with the tissues of wild-type but not Bicc1−/− mice (see Fig. S2 in the supplemental material). Lotus tetragonolobus lectin (LTL), which is expressed in proximal tubules, was used to mark cortical renal structures, whereas the cholangiocytes of liver bile ducts were marked by cytokeratin-19 (CK19). High-resolution imaging revealed a nonhomogeneous distribution of endogenous Bicc1 in clusters of cytoplasmic puncta both in renal proximal tubule cells and in cholangiocytes (). The volumes of these puncta varied from 0.05 to 0.6 μm3, similar to those of cytoplasmic foci detected by anti-HA staining of tagged Bicc1 in transfected HEK293T cells, although the latter were larger, on average, possibly due to overexpression rather than alternative fixation or the use of anti-HA antibody instead of anti-SAM antibody (see Fig. S3 in the supplemental material). These data show that Bicc1 clusters in cytoplasmic puncta and that this pattern is not restricted to one tissue but could be a general feature of Bicc1.
FIG 1 Bicc1 protein forms cytoplasmic clusters in mouse kidney cells and bile duct cholangiocytes. (A) Frozen sections of WT mouse kidney labeled with anti-Bicc1 antibodies and with the proximal tubule marker LTL on postnatal day 4. (B) Frozen sections of liver of a WT mouse obtained postnatally labeled with anti-Bicc1 and anti-CK19 antibodies. CK19 is an intermediate filament protein of epithelial tissues. The boxed areas in the left panels are magnified in the three panels to the right. Bars, 10 μm (large views) and 2 μm (magnified views).
Cytoplasmic Bicc1 foci contain target mRNA.
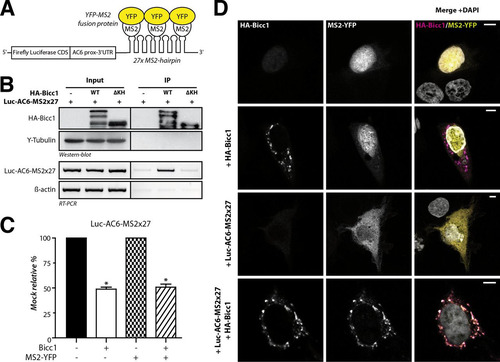
Bicc1 clustering may influence the localization of associated target mRNAs or affect RNA binding, or both. To distinguish between these possibilities, we monitored the localization of a reporter mRNA containing the Bicc1 binding region of the AC6 3′ UTR (Citation27) and 27 repeats of a motif that binds the bacteriophage MS2 coat protein (the MS2×27 motif) (Citation55). Coimmunoprecipitation and luciferase assays with HA-Bicc1 from extracts of transfected HEK293T cells showed that Bicc1 specifically binds this reporter mRNA and inhibits its expression, as expected ( to ). To assess whether Bicc1 influences mRNA localization, we cotransfected a fusion protein of MS2 with nuclear yellow fluorescent protein (YFP) into COS-1 cells. Like HEK293T cells, COS-1 cells show no endogenous Bicc1 expression but are more adhesive, have a larger cytoplasm, and, thus, are more amenable for imaging. MS2-YFP without Luc-AC6-MS2×27 reporter mRNA localized to nuclei both in the presence and in the absence of HA-Bicc1 (, rows 1 and 2). However, when coexpressed with Luc-AC6-MS2×27 and without Bicc1, MS2-YFP accumulated diffusely throughout the cytoplasm, confirming that MS2-YFP was exported from the nucleus together with the reporter mRNA (, row 3). Interestingly, the combined expression of the Luc-AC6-MS2×27 reporter with HA-Bicc1 led to the concentration of MS2-YFP in cytoplasmic Bicc1 puncta, with 89% ± 8% of the cytoplasmic YFP signal colocalizing with Bicc1 foci (, row 4). We therefore asked whether the Luc-AC6-MS2×27 reporter also enters cytoplasmic foci in the mIMCD3 mouse inner medullary collecting duct cell line that expresses endogenous Bicc1 (Citation56). Transfection of the reporter RNA was sufficient to translocate the MS2-YFP fusion protein from the nucleus to cytoplasmic Bicc1-stained foci independently of exogenous Bicc1 (see Fig. S4A in the supplemental material). Even though we could not sufficiently deplete endogenous Bicc1 foci by RNA interference to more conclusively validate their function (not shown), their colocalization with reporter RNA strongly corroborates our conclusion that target mRNAs are recruited to endogenous Bicc1 in cytoplasmic clusters.
FIG 2 Bicc1 concentrates an associated reporter mRNA in cytoplasmic foci. (A) Principle of the MS2-YFP colocalization assay. The 3′ extremity of the Luc-AC6 reporter mRNA was fused to 27 MS2 hairpins, which constitute multiple binding sites for the MS2 protein fused to YFP. CDS, coding sequence; prox, proximal. (B) RNA coimmunoprecipitation. The Luc-AC6-MS2×27 reporter mRNA was expressed in HEK293T cells together with HA-Bicc1 or empty vector. After HA immunoprecipitation (IP), the various fractions were analyzed by Western blotting and by RT-PCR. Five percent of the total extract was used as the input. The β-actin mRNA was used as a negative control for the RT-PCR. HA-Bicc1 lacking all KH and KH-like domains (ΔKH) was used as an additional negative control for RNA-binding specificity. (C) Cotransfection of the fluorescent YFP-MS2 fusion protein does not affect the silencing of the Luc-AC6-MS2×27 reporter by HA-Bicc1. β-Galactosidase was used as a control for normalization, and the data represent the percent expression relative to that of a mock-treated control. Error bars show SEMs. *, P < 0.005. (D) Localization by indirect immunofluorescence staining of the Luc-AC6-MS2×27 reporter mRNA and HA-Bicc1 in COS-1 cells. The MS2-tagged mRNA was detected by the relocalization of fluorescent MS2-YFP fusion protein, which binds the MS2 RNA hairpins. Bars, 5 μm.
3D model of Bicc1 SAM-SAM interface.
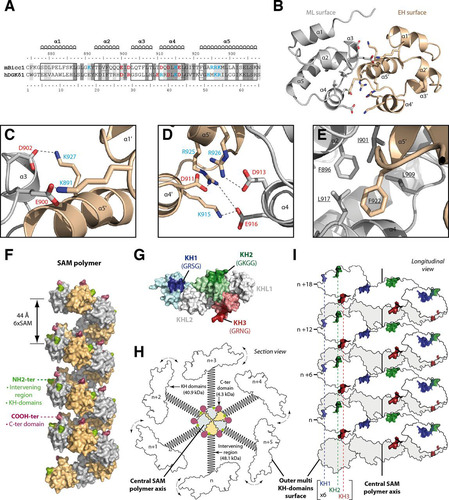
Bicc1 is localized in cytoplasmic foci by its SAM domain independently of the RNA-binding KH domains (Citation35). Certain SAM domains can form dimers or polymeric structures (Citation57), and a polymer of the human Bicc1 SAM domain fused to GFP has been observed in vitro by electron microscopy (EM) (Citation42). To test whether SAM polymerization is responsible for Bicc1 clustering, we searched for mutations that specifically disrupt polymerization. Since structure data for Bicc1 or its SAM domain are currently unavailable, mutations were designed on the basis of homology modeling, where the known structure of a related protein serves as a template. Among the available templates, we selected the SAM domain dimer of the diacylglycerol kinase δ1 (DGKδ1) E35G (PDB accession number 3BQ7) because it shares the highest sequence similarity (54%) and identity (31%) with the Bicc1 SAM domain () and can form head-to-tail polymers (Citation47). A model of dimeric Bicc1 SAM obtained after energy minimization revealed a common globular fold of five α helices, with two SAM subunits being docked to one another at characteristic ML and EH surfaces () (Citation40). Residues involved in the dimerization of the DGKδ1 SAM are conserved or replaced by similar amino acids in the Bicc1 SAM domain (highlighted in ). At the Bicc1 SAM-SAM interface, 4 negatively charged amino acids on the ML surface (Glu900, Asp902, Asp913, Glu916) and 5 positively charged amino acids from the EH surface (Lys891, Lys915, Arg925, Arg926, Lys927) form strongly polarized electrostatic networks in two independent regions of contact ( and ; see also Fig. S5 in the supplemental material). In addition, residue Phe922 from the EH surface reaches into a hydrophobic pocket of the ML surface comprising Phe896, Ile901, Leu909, and Leu917 ().
FIG 3 Molecular modeling of a Bicc1 SAM polymer. (A) ClustalW alignment of mouse (Mus musculus) Bicc1 (mBicc1) and human (Homo sapiens) DGKδ1 (hDGKδ1) SAM domains. The X-ray structure of dimeric DGKδ1 SAM (PDB accession number 3BQ7) served as the template to model the Bicc1 SAM dimer. The two SAM domains share 31% identity and 54% similarity. Predicted α helices are framed. Residues of the ML surface (red), residues from the EH surface (blue), and hydrophobic residues (underlined) are highlighted. Dark and light gray shading corresponds to identical and similar amino acids, respectively, between Bicc1 and the DGKδ1 template. (B) Dimeric Bicc1 SAM model obtained using MODELLER (v9.5) software. The α helix numbers and the side chains of the residues involved in the interface are displayed. (C to E) Magnified views of the main interacting patches in the predicted Bicc1 SAM dimer interface. Acidic and basic residues are displayed in red and blue, respectively. (F) Model of a Bicc1 SAM polymer of 24 units in surface representation. The NH2 terminus and the COOH terminus of each SAM domain are displayed in green and purple, respectively. (G) Model of the Bicc1 KH domain region in surface representation. Models for individual KH domains were obtained by homology modeling using the SWISS-MODEL work space (Citation48) and templates consisting of the structures with PDB accession numbers 1VIG (KH1), 2CTM (KH2), 1WVN (KH3), and 3N89 (KHL1 and -2). Individual KH domains were then superimposed with their homologous domain in the X-ray structure of ceGLD-3 KH domains (PDB accession number 3N89) (Citation58). The KH domains harboring the GXXG signatures for RNA binding are highlighted in color. Their putative RNA-binding surfaces are darkened, and the identity of their GXXG signature sequence is given in parentheses. The KH-like domains (KHL1 and -2) are displayed in gray. (H) Diagram of a transversal section through a polymer of full-length Bicc1. The SAM polymer is located at the center and displays other Bicc1 domains at its periphery. A schematic representation was used for the other domains. C-ter domain, C-terminal domain. (I) Diagram in longitudinal view of Bicc1 KH domains distributed along the surface of the central SAM polymer.
To assess the stability of this complex, we performed a molecular dynamics (MD) simulation in water. For 10 ns, the backbone root mean square deviation (RMSD) was 2.05 Å. Electrostatic and hydrophobic interactions and minimal root mean square fluctuation (RMSF) of most of the residues persisted, except at the unconstrained NH2 and COOH extremities, indicating that the complex remained stable (see Fig. S6A and B in the supplemental material). The number of intermolecular H bonds increased during the dynamic from 5 to an average of 8.9 H bonds per time frame, indicating significant stabilization (see Fig. S6C in the supplemental material). This result validates the confidence of our dimeric model. To directly confirm the potential to self-polymerize, we imaged a GST fusion of recombinant mouse Bicc1 SAM. Negative-staining EM of GST-Bicc1 SAM revealed ovoid structures that were, on average, 28.8 ± 3.9 nm long and 22.8 ± 3.4 nm wide (see Fig. S7 in the supplemental material). This size resembles that of human Bicc1 SAM fused to enhanced GFP (EGFP) (Citation42), corresponds to approximately 40 units of the GST-SAM fusion protein, and, therefore, likely represents SAM polymers. Overall, our results confirm that the Bicc1 SAM interacts with itself in higher-order structures large enough to be detected in vitro.
To obtain a 3D model of a Bicc1 SAM domain polymer, we docked multiple dimers to each other via their EH and ML surfaces (). Polymerization of an arbitrary number of SAM units in silico formed a left-handed helix with a diameter of 65 Å and a pitch of 44 Å encompassing 6 SAM units. As observed in related SAM polymers (Citation40), the N and C termini were positioned peripherally, indicating that a central SAM polymer in full-length Bicc1 is surrounded by the N-terminal KH domains, with the intervening regions forming a linker to the SAM, and by the short C termini (see Fig. S1A in the supplemental material). To evaluate the potential impact of SAM polymerization on RNA binding, we also modeled the structures of the Bicc1 KH and KHL domains using the Caenorhabditis elegans GLD-3 (ceGLD-3) X-ray structure as a template (Citation58) (). We found that all three RNA-binding pockets are predicted to reside on the same surface of a large globular domain. For optimal solvent exposure and access to RNA, the KH domains of polymeric full-length Bicc1 would thus have to reside around and peripheral to the central helix organized by the SAM domain ( and ), consistent with their N-terminal positioning relative to the SAM domain (). Each helical turn might thus present as many as 25 KH domains at the surface of the macromolecular assembly within a distance of only 44 Å. While the exact configuration of such a multimer awaits experimental validation, we note that the regulation of its length and helical pitch by SAM polymerization thus likely determines the number of RNA-binding sites as well as their spacing along the longitudinal axis ().
Identification of polymerization-defective Bicc1 mutants.
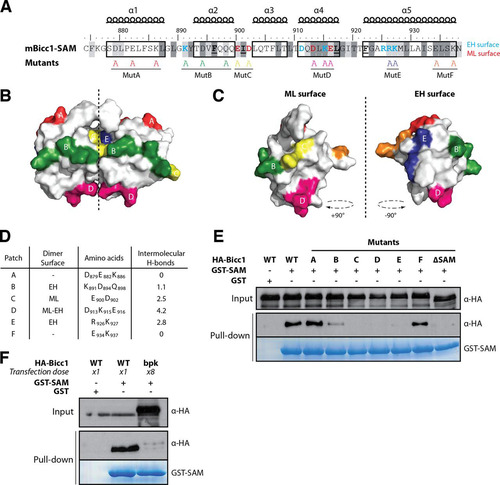
To test the predictive power of our structural model of dimeric Bicc1 SAM, we individually replaced six surface-exposed patches of electrostatic amino acids within or outside the SAM-SAM interface by alanines (). Four of these groups of mutations (those in mutB, mutC, mutD, and mutE) should affect intermolecular H bonds, while two others (those in mutA and mutF) were deliberately introduced outside the predicted dimerization interface ( to ). To screen for polymerization defects, we transfected HA-tagged Bicc1 mutants A to F into HEK293T cells and assessed their retention on glutathione-Sepharose beads coated with recombinant GST-SAM fusion protein as a bait. We found that wild-type HA-Bicc1 efficiently bound to GST-SAM-coated beads but not to beads coated with GST alone (). In contrast, the Bicc1 mutant lacking SAM (HA-Bicc1ΔSAM) failed to bind GST-SAM beads, suggesting that GST-SAM specifically interacts with the SAM domain of full-length Bicc1. Moreover, a comparison with HA-Bicc1 mutants showed that while the mutations in mutants C, D, and E abolished the SAM-SAM interaction, the mutations in mutants A and F did not, a result that concurs with that of our structure model. The mutation in mutant B exhibited an intermediate effect. The mutation in mutant B affected three amino acids at the periphery of the EH surface, and only one of them (K891) is predicted to contribute to the SAM-SAM interaction (). According to theoretical predictions, this electrostatic patch is involved in only 1.1 intermolecular H bonds, on average, explaining why its mutation is insufficient to fully disrupt dimerization (). Altogether, these results strongly corroborate our structure model and provide new tools to specifically probe the consequences of the loss of SAM-SAM interactions in vivo.
FIG 4 Screen for Bicc1 SAM polymer mutants. (A) Bicc1 SAM mutant collection. Shading is as for . Individual electrostatic patches (patches A to F) at the protein surface were replaced by alanines. (B, C) Positions of mutations in the Bicc1 SAM dimer model (B) and on the ML surface and EH surface (C). (D) Table summarizing, for each amino acid patch, the average number of H bonds per time frame during the MD. (E and F) Pulldown of the WT, point mutants, or 8-fold excess bpk mutant HA-tagged Bicc1 from HEK293T cell extracts by glutathione-Sepharose beads coated with a recombinant GST control or GST-Bicc1 SAM. Five percent of total cell extracts were loaded as input.
Polymerization governs Bicc1 clustering and localizes associated mRNA in cytoplasmic puncta.
Among our panel of Bicc1 mutations, the mutation in mutant D disrupted the largest number of intermolecular H bonds at the SAM-SAM interface ( and ). To distinguish whether this interface mediates Bicc1 dimerization or the formation of higher-order assemblies, we compared the sizes of wild-type Bicc1 and mutant D in transfected HEK293T cells by sucrose gradient fractionation. Analysis of polymeric HA-Bicc1 complexes in HEK293T cells revealed a broad size distribution, with wild-type HA-Bicc1 extending beyond the fractions marked by ribosomal protein S6 (RPS6) (Citation59), indicating that wild-type Bicc1 congregates in molecular assemblies larger than ribosomes (). A similar distribution was observed for endogenous Bicc1 in extracts of mIMCD3 cells (see Fig. S4B in the supplemental material). In contrast, HA-Bicc1 mutant D was concentrated in fractions with significantly lower molecular weights in three independent experiments. These data suggest that at least the largest Bicc1 assemblies in such cell extracts likely depend on SAM polymerization. However, despite this marked shift to lower-molecular-weight fractions, HA-Bicc1 mutD levels in the first three fractions did not increase, suggesting that Bicc1 likely fails to stably accumulate as a free monomer.
FIG 5 SAM polymerization is required for Bicc1 clustering. (A) Density fractionation of WT and polymerization mutant Bicc1 on a sucrose gradient. HEK293T cell extracts containing HA-tagged Bicc1 were fractionated on a continuous 15 to 60% sucrose gradient and analyzed by anti-Bicc1 Western blotting. The migration direction from the top to the bottom of the tube is indicated. RPS6 and γ-tubulin (γ-Tub) were used as internal controls. The graph below the gels shows the percentage of Bicc1 compared to the total Bicc1 signal for each fraction. Results represent mean values from 3 independent experiments, and error bars show SEMs. (B) Bicc1 polymer mutants fail to accumulate in cytoplasmic foci. The results of indirect immunofluorescence staining of the HA-Bicc1 WT, mutD, the ΔSAM mutant, or the bpk mutant and the P-body marker GFP-Dcp1a overexpressed in COS-1 cells are shown. Bars, 5 μm. (C) Comparative 3D rendering of the HA-Bicc1 WT and mutD by Imaris software. From the original image (center), z-stacks in two directions (z1 and z2, top and right, respectively) and 3D reconstruction (bottom) are given. Bars, 2 μm. (D) Localization by indirect immunofluorescence staining of the Luc-AC6-MS2×27 reporter mRNA and HA-Bicc1 in COS-1 cells and comparison with that of HA-Bicc1 mutD. Bars, 5 μm.
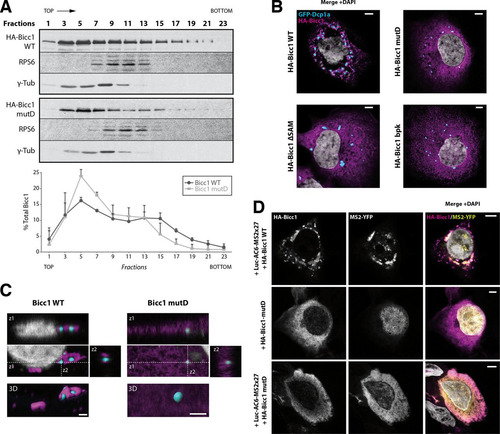
To assess the impact of polymerization on Bicc1 function, we tested whether Bicc1 mutant D forms cytoplasmic puncta. As described previously, immunostaining of COS-1 cells and reconstruction of 3D surfaces detected HA-Bicc1 in a dense network of corpuscles outside and around P-bodies marked by GFP-Dcp1a (Citation35). In contrast, Bicc1 mutD localized more diffusely throughout the cytoplasm, similar to the findings for Bicc1ΔSAM ( and ). Importantly, mutant D also failed to localize the Luc-AC6-MS2×27 reporter in cytoplasmic foci, even though mRNA binding was still detected (; see also Fig. S8 in the supplemental material). Taken together, these results suggest that both Bicc1 and associated mRNAs are concentrated in cytoplasmic foci by specific residues in the SAM domain that mediate polymerization.
SAM polymerization is essential for Bicc1 stabilization and to silence mRNAs but not to inhibit the Wnt pathway.
Since the Bicc1 mutant D apparently did not freely accumulate as a monomer in density fractionation gradients, we asked whether polymerization influences the protein half-life. Translation inhibition by cycloheximide (CHX) followed by a time course analysis by immunoblotting showed that wild-type HA-Bicc1 remained stable over the entire chase period (56 h). In contrast, the half-lives of the HA-Bicc1 mutD and Bicc1ΔSAM proteins were reduced to 34 and 29 h, respectively (), suggesting that SAM polymerization increases Bicc1 stability. Therefore, in all subsequent assays evaluating their functions, we doubled the dose of transfected DNA for Bicc1 mutants in order to reach expression levels comparable to those of wild-type HA-Bicc1 (). In particular, to determine whether polymerization is necessary for mRNA silencing, we compared the potential of wild-type HA-Bicc1 and mutant D to silence the 3′ UTR sequences of AC6 and PKIα mRNAs in luciferase reporter assays. HA-Bicc1 significantly repressed the expression of these luciferase reporters, as described previously (Citation27). In contrast, both HA-Bicc1ΔSAM and HA-Bicc1 mutD failed to repress these targets even at an elevated dosage ( and ), indicating that the silencing activity of Bicc1 directly depends on SAM polymerization.
FIG 6 SAM polymerization is required for Bicc1 accumulation and silencing activity. (A) Time course analysis of HA-Bicc1 WT and mutants expressed in HEK293T cells after CHX treatment. HA-Bicc1 protein levels were compared with γ-tubulin levels by Western blotting at 0, 8, 24, 32, 48, and 56 h. The relative level of each protein is presented in the graph at the bottom, and the estimated half-life is given on the right. AU, arbitrary units. (B) Level of the HA-Bicc1 WT and mutants upon transfection with a single dose (×1) or a double dose (×2) of DNA encoding HA-Bicc1. A double dose of transfected DNA encoding HA-Bicc1 mutD, the ΔSAM mutant, or the bpk mutant is required to obtain a protein level comparable to that of the HA-Bicc1 WT. The relative percentage of WT Bicc1 is indicated for each condition. γ-Tubulin was used for normalization. (C and D) Silencing of AC6 and PKIα 3′ UTR luciferase reporters by WT or polymerization mutant Bicc1 in HEK293T cells. β-Galactosidase was used as a control for normalization. Error bars show SEMs. *, P < 0.005. (E) Induction of the TOPflash reporter of Wnt signaling by Dishevelled 2 (Dvl2) in transfected HEK293T cells is inhibited by both WT and polymerization mutant Bicc1. β-Galactosidase was cotransfected for signal normalization. Error bars show SEMs. *, P < 0.005.
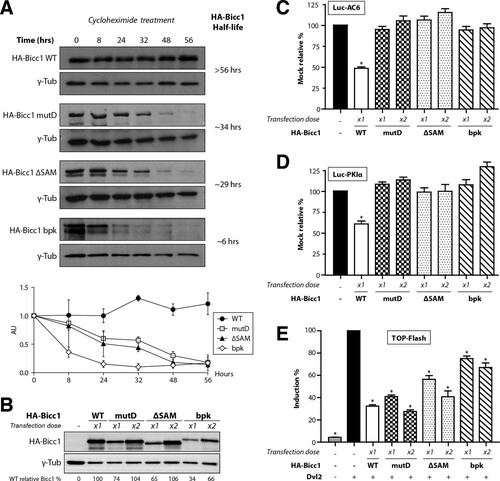
Previously, we have shown that Bicc1 can also block the induction of the TOPflash reporter of canonical Wnt signaling at the level of Dishevelled 2. This inhibition of Dishevelled signaling is independent of the mRNA-binding KH domains but is potentiated by the Bicc1 SAM domain (Citation35). To distinguish whether inhibition of Dishevelled 2 activity directly or indirectly depends on Bicc1 polymerization, we cotransfected wild-type or mutant HA-Bicc1 with TOPflash and a vector expressing Dvl2. As described previously (Citation35), HA-Bicc1ΔSAM inhibited the induction of TOPflash by Dvl2 less efficiently than wild-type Bicc1. Interestingly, this difference disappeared when the dosage of either HA-Bicc1ΔSAM or mutant D was doubled to reach the level of wild-type HA-Bicc1 expression ( and ), suggesting that SAM domain polymerization is not essential for Wnt signal inhibition.
Inhibition of SAM polymerization and Bicc1 function by the bpk mutation.
Our model of the SAM domain raised the possibility that elongation of the C terminus in the Bicc1 bpk mutant inhibits a polymeric SAM helix by overcrowding the surrounding space (see Fig. S1B in the supplemental material). To test this hypothesis, we cloned the bpk mutant in a mammalian expression vector. Similar to our engineered mutant D, the bpk mutant displayed a drastically reduced half-life compared to that of wild-type HA-Bicc1 ( and ). We therefore used pulldown assays to evaluate whether the bpk mutation also inhibits SAM-SAM interactions. To compensate for reduced protein stability, the HA-Bicc1 bpk mutant expression vector was transfected in large excess. Nevertheless, the HA-Bicc1 bpk mutant failed to bind recombinant GST-SAM fusion protein in pulldown assays (). HA-Bicc1 bpk also failed to cluster around P-bodies () and to silence the AC6 and PKIα 3′ UTR luciferase reporters ( and ). In contrast, mRNA binding was still detected (see Fig. S8 in the supplemental material). In proportion to its expression levels, HA-Bicc1 bpk also still inhibited Dvl2 signaling in TOPflash assays ( and ). Overall, these data show that the C-terminal elongation of the Bicc1 bpk mutant abrogates SAM polymerization and mRNA silencing without disrupting the potential to inhibit Dishevelled 2.
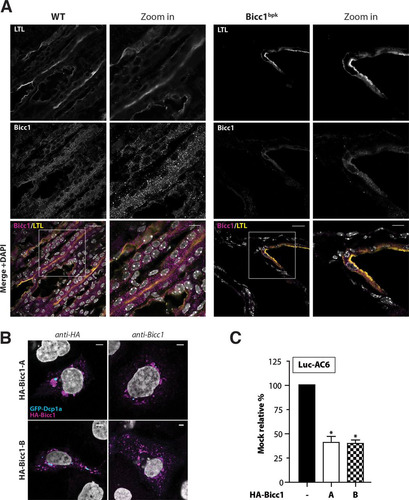
To validate a potential influence of the bpk mutation on polymeric Bicc1 in vivo, cryosections of kidneys obtained from bpk mice postnatally were colabeled using anti-Bicc1 antibody and LTL to mark proximal tubules. Compared to the findings for wild-type Bicc1, where Bicc1 was concentrated in cytoplasmic foci of both LTL-positive and LTL-negative renal tubule cells, the cystic kidneys of bpk mutant animals showed considerably weaker and more diffuse Bicc1 staining that was confined to LTL-positive structures (). This result concurs with the effects of bpk on Bicc1 protein stability and cytoplasmic clustering observed ex vivo. However, despite the abnormally diffuse distribution, some Bicc1 foci remained clearly detectable. To evaluate whether these residual foci could arise from the alternatively spliced transcript B that is unaffected by the bpk mutation (Citation17, Citation22) (see Fig. S1C in the supplemental material), we overexpressed a synthetic cDNA of this splice variant in transfected cells. Similar to the findings for full-length Bicc1 encoded by transcript A, transfected isoform B was readily detected by anti-Bicc1 antibody in cytoplasmic foci, and it efficiently silenced the Luc-AC6 reporter mRNA ( and ). It is likely, therefore, that the bpk mutant phenotype is caused at least in part by the failure of the affected Bicc1 isoform A to polymerize. In addition, these results are consistent with the notion that isoform B remains functional and thus likely accounts for residual hypomorphic Bicc1 activity in bpk mutants. Alternatively, or in addition, cystic growth in bpk mutants may also be attenuated by polymerization-independent Bicc1 functions, e.g., during Wnt signaling.
FIG 7 Impaired localization of Bicc1 in bpk mutant mouse kidney. (A) Frozen sections of WT and bpk mutant mouse kidneys labeled with anti-Bicc1 antibodies and with the proximal tubule marker LTL on postnatal day 11. Boxed areas are magnified in the panels to the right. Bars, 20 μm (large views) and 10 μm (magnified views). (B) Bicc1 isoform B (Bicc1-B) accumulates in cytoplasmic foci. The results of indirect immunofluorescence staining of HA-Bicc1 isoforms A and B and the P-body marker GFP-Dcp1a in transfected COS-1 cells are shown. Bars, 5 μm. (C) Silencing of AC6 3′ UTR luciferase reporter by HA-Bicc1 isoform A or B in HEK293T cells. β-Galactosidase was cotransfected for signal normalization. Error bars show SEMs. *, P < 0.005.
DISCUSSION
By combining structure modeling and mutagenesis with functional analysis, we identified a self-polymerization interface in the C-terminal SAM domain of Bicc1 and showed that it stabilizes full-length Bicc1 in higher-order complexes to regulate its subcellular distribution in the cytoplasm. We provide the first evidence that Bicc1 also localizes a reporter mRNA that binds the N-terminal KH domains and that this process is regulated by SAM clustering. The helical conformation of the polymeric SAM implies that RNA-binding sites are hyperconcentrated in orderly arrays in the surrounding space. The loss of SAM polymerization may explain why the Bicc1 function is impaired in the bpk mouse model of autosomal recessive polycystic kidney disease. Regulation of mRNA localization and silencing by SAM polymerization to our knowledge has not been reported previously.
Structural model of the SAM-SAM interface.
SAM domains are found in many eukaryotic proteins with diverse functions. Among 72 known human SAM domains that have previously been analyzed in isolation for their potential to multimerize in vitro, 47% form polymers (e.g., TEL, Ph, Shank3, DGKδ1, and ANKS3), 10% can oligomerize (e.g., ANKS6), and 43% are monomeric (e.g., Smaug/Vts1p) (Citation42). A purified EGFP fusion protein of the human BICC1 SAM was shown by EM to self-polymerize (Citation42). Here, EM analysis of an analogous mouse Bicc1 SAM fusion with GST revealed the presence of similar structures of approximately 40 nm. Extending this observation, we used GST pulldown and sucrose gradient fractionation assays to show that the capacity to self-interact in higher-order structures is preserved in full-length Bicc1. The high sequence conservation (31% identity and 54% similarity) between the Bicc1 and DGKδ1 SAM domains allowed us to derive a homology-based 3D model of dimeric Bicc1 SAM. In this model, the EH surface from one Bicc1 SAM domain interacts with the ML surface of another through a large electrostatic network that is stabilized by 8 hydrogen bonds during molecular dynamic simulation and by Phe922 interacting with a hydrophobic pocket comprising residues L917, L909, I901, and F896. Confirming the robustness of our model, alanine scanning mutagenesis of 6 distinct surface-exposed electrostatic clusters showed that only the mutations at the predicted SAM-SAM interface impaired polymerization. The determinants mediating this interaction are conserved in homologous SAM domains, consistent with an important biological function (see Fig. S1D in the supplemental material).
The SAM domain organizes a helical Bicc1 polymer that is inhibited by the bpk mutation.
A model of polymerized Bicc1 via a SAM-SAM interface built in silico led intrinsically to the formation of a left-handed head-to-tail helix with 6 SAM units per turn. Since the N and C termini of each SAM subunit protrude from a helical polymer toward the periphery, attached domains occupy the surrounding space in regular arrays ( to ). The putative RNA-binding GXXG signatures that distinguish the N-terminal KH from KH-like domains are thus likely displayed at the periphery and linked to the central SAM scaffold by the intervening spacer region. This organization suggests that the space near the center may be too crowded to accommodate bulky additions at the C terminus or other factors in a polymer. Direct evidence in support of such a model comes from our analysis of the Bicc1 bpk mutant, where replacement of the short wild-type C terminus by a sequence approximately five times larger was sufficient to disrupt the ability of Bicc1 to self-interact. The simplest possible explanation is that the C-terminal extension of Bicc1bpk sterically hinders SAM polymerization.
Bicc1 SAM polymerization is not essential to inhibit Dvl activity but is necessary to stabilize cytoplasmic foci and to locally concentrate target mRNA.
A previous analysis suggested that a C-terminal fragment encompassing the SAM domain localizes Bicc1 in cytoplasmic puncta, but the underlying mechanism remained elusive (Citation35). Here, immunostaining of cryosections revealed that endogenous Bicc1 forms similar clusters in renal tubule cells and in liver cholangiocytes. We propose that clustering in cytoplasmic foci is mediated by SAM polymerization, since both the engineered polymerization mutant D and Bicc1bpk were diffusely distributed in transfected cells compared to the distribution of wild-type Bicc1. In good agreement with that finding, clustering of Bicc1 in cytoplasmic foci was also impaired in the kidneys of mice with the bpk mutation in vivo. In addition, failure to polymerize reduced the Bicc1 protein half-life, suggesting that polymerization may influence some Bicc1 functions indirectly. Confirming this prediction, polymerization-deficient Bicc1ΔSAM, Bicc1bpk, and engineered mutant D all inhibited the induction of the TOPflash reporter by Dvl2 less potently than wild-type Bicc1, but this defect was rescued when the dosage of mutant proteins was adapted to restore normal expression levels. Polymeric Bicc1 therefore likely facilitates the inhibition of Dvl2 activity indirectly by increasing Bicc1 stability.
Our findings show that SAM polymerization also influences the localization or binding of Bicc1-associated RNAs. Previous experiments established that Bicc1 coimmunoprecipitates AC6 and PKIα mRNAs and specific miRNAs independently of the SAM domain, whereas deletion of KH domains blocked these interactions (Citation27). In good agreement with those findings, here Bicc1 also bound an AC6-3′ UTR luciferase reporter that was tagged with MS2 hairpins, and this interaction was not overtly altered by either bpk or the mutation engineered in mutant D or upon deletion of the entire SAM domain. Imaging of the reporter mRNA using an MS2-YFP fusion protein revealed extensive colocalization with wild-type Bicc1 in cytoplasmic foci, whereas in cells lacking Bicc1, it was diffusely distributed throughout the cytoplasm. Interestingly, the reporter mRNA also colocalized with the engineered SAM polymerization mutant D, albeit it did so diffusely throughout the cytoplasm. We conclude that SAM polymerization is not essential for RNA binding but is essential to concentrate associated transcripts in cytoplasmic foci.
The SAM-SAM interface is required for silencing of Bicc1 target mRNAs.
Previous work established that truncated Bicc1 lacking the SAM domain still binds target mRNAs and miRNAs yet fails to induce RNA-induced silencing complex loading and silencing (Citation27). Here, we showed that silencing is also abrogated by mutations that specifically disrupt the SAM-SAM interface (mutant D) or its potential to polymerize (bpk mutant). These observations provide strong evidence that the SAM domain promotes silencing by mediating Bicc1 polymerization. In contrast, polymerization was not essential for mRNA binding, indicating that Bicc1 likely binds target mRNAs before entering polymers. Since mRNA binding to polymerization-deficient Bicc1 was not sufficient for silencing, the transition to a polymeric configuration may be the molecular switch that turns off mRNA translation. Such a model predicts that polymerization is likely regulated. Interestingly, a heterozygous mutation in human BICC1 that associates with pediatric renal cystic dysplasia replaces the conserved residue E932 in the SAM domain by glycine. Since E932 is outside the SAM-SAM interface and dispensable for cytoplasmic clustering and Bicc1 silencing activity (Citation19), we speculate that it controls access to a steric inhibitor of Bicc1 polymerization. The extent or dynamics of SAM polymerization could also be regulated by other SAM domain proteins, such as ANKS6. ANKS6 binds and colocalizes with Bicc1 (Citation60) and is needed to suppress renal cysts (Citation61, Citation62). Although ANKS6 was initially considered to self-polymerize (Citation42), a more recent study showed that it inhibits polymerization of the related SAM domain protein ANKS3, which can also interact with Bicc1 (Citation63, Citation64).
Between the KH domains and SAM, Bicc1 contains an intervening sequence of 461 residues. Interestingly, the homologous sequence of Xenopus Bic-C is sufficient to mediate translational repression when tethered as an MS2 fusion protein to a luciferase reporter mRNA containing MS2 hairpins, and addition of the SAM domain potentiated this activity (Citation65). In contrast, tethering of KH or SAM domains alone had no effect. These findings support our model that SAM primarily serves to cluster and stabilize Bicc1 in polymers. They also concur with our earlier finding that mRNA binding to Bicc1 KH domains alone is not sufficient for translational repression (Citation27). Additional factors may include miRISC components (Citation27) or binding of unknown factors to the region between KH and SAM domains, depending on the context (Citation65). How these or other factors cooperate with SAM polymerization to repress translation will be a fascinating novel area of future investigation.
How does polymerization influence Bicc1 silencing activity?
One possible mechanism to promote translational repression by Bicc1 clustering involves the local enrichment of target mRNAs in microdomains that privilege the access to specific silencing factors through molecular crowding. Crowding influences the thermodynamic properties of cellular components, and hyperconcentration, especially of macromolecules, can alter reaction equilibrium constants by increasing the probability that substrates will meet and by decreasing their conformational entropy (Citation66). In a catalytic model, polymeric Bicc1 may thus thermodynamically favor interactions with silencing factors by locally increasing the concentration of associated mRNAs and/or by presenting them in an orderly manner, creating, in effect, a catalytic surface for translational repression.
An alternative, mutually nonexclusive hypothesis is that Bicc1 acts as an RNA chaperone. A chaperone model takes into account the structural dimension and striking degree of symmetry of the Bicc1 polymer. The pitch of helical SAM polymers arranges Bicc1 KH domains such that each individual KH domain n is aligned with the related n + 6 domain. Six rows of a given KH domain thus cover the polymer along its length. With 3 KH domains per Bicc1 molecule that can bind RNA, a polymer will present these domains in 18 regularly spaced rows at the surface. Compared to the binding of monomeric Bicc1, the surface of a polymer is more likely to bind several RNA molecules simultaneously and/or at multiple anchoring points and, thus, to favor alternative secondary RNA structures that expose or mask specific binding sites of cognate miRNAs or regulatory proteins. The accessibility of miRNA seed sequences is essential for silencing and can be regulated by specific RNA-binding proteins, such as Pumilio (Citation67Citation–Citation69). In a chaperone model, the conformation of an mRNA that is initially captured by monomeric Bicc1 will change its equilibrium under the influence of nearby KH domains and possibly other RNAs on the surface of multimeric Bicc1 after SAM-SAM polymerization. The identity of RNA recognition motifs, their number, and their relative positioning in space could thus govern the silencing by Bicc1 by a complex code.
Our results demonstrate that the SAM domain mediates the polymerization of full-length Bicc1. We found that this process is inhibited in the spontaneously mutated Bicc1 bpk allele and needed to enrich Bicc1 and associated mRNA in cytoplasmic clusters. SAM-dependent polymerization of the unrelated protein polyhomeotic-like 2 induces clustering of the polycomb group-repressive complex 1 on chromatin to repress the transcription of Hoxb and other target genes (Citation70). SAM-SAM interactions also control the activity of the transcriptional repressor Yan (Citation71). Extending this portfolio, our data reveal that SAM domain polymerization can also be used by an RNA-binding protein to localize target mRNAs. This feature might become useful to manipulate other RNA-binding proteins that normally contain no SAM domains. Further studies and new experimental approaches are also warranted to further investigate the influence of polymerization on the spatial positioning of the RNA-binding KH domains or on the conformation of bound RNAs and its potential regulation by Bicc1-interacting SAM domain proteins, such as ANKS6 or ANKS3 (Citation60, Citation64).
tmcb_a_12275756_sm0001.pdf
Download PDF (5.8 MB)ACKNOWLEDGMENTS
We thank Paul Guichard and Pierre Gönczy Pierre (EPFL) and Xavier Manival (Université Lorraine-Nancy) for their advice on negative-staining EM and for comments on the manuscript and Jessica Dessimoz and Arne Seitz (EPFL) and their technical staff for assistance with histology and imaging. Computations were performed at Vital-IT (http://www.vital-it.ch) of the SIB Swiss Institute of Bioinformatics.
This work was generously supported by grants from the Stiftung für Wissenschaftliche Forschung Zürich (to B.R.) and the Gebert-Rüf Stiftung (to L.L.-E.) and by a Swiss National Science Foundation Sinergia grant to D.B.C.
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00341-15.
REFERENCES
- Marchand V, Gaspar I, Ephrussi A. 2012. An intracellular transmission control protocol: assembly and transport of ribonucleoprotein complexes. Curr Opin Cell Biol 24:202–210. http://dx.doi.org/10.1016/j.ceb.2011.12.014.
- Kim-Ha J, Smith JL, Macdonald PM. 1991. oskar mRNA is localized to the posterior pole of the Drosophila oocyte. Cell 66:23–35. http://dx.doi.org/10.1016/0092-8674(91)90136-M.
- Ephrussi A, Dickinson LK, Lehmann R. 1991. oskar organizes the germ plasm and directs localization of the posterior determinant nanos. Cell 66:37–50. http://dx.doi.org/10.1016/0092-8674(91)90137-N.
- Ephrussi A, Lehmann R. 1992. Induction of germ cell formation by oskar. Nature 358:387–392. http://dx.doi.org/10.1038/358387a0.
- Kim-Ha J, Kerr K, Macdonald PM. 1995. Translational regulation of oskar mRNA by bruno, an ovarian RNA-binding protein, is essential. Cell 81:403–412. http://dx.doi.org/10.1016/0092-8674(95)90393-3.
- Markussen FH, Michon AM, Breitwieser W, Ephrussi A. 1995. Translational control of oskar generates short OSK, the isoform that induces pole plasma assembly. Development 121:3723–3732.
- St Johnston D. 2005. Moving messages: the intracellular localization of mRNAs. Nat Rev Mol Cell Biol 6:363–375. http://dx.doi.org/10.1038/nrm1643.
- Ghosh S, Marchand V, Gaspar I, Ephrussi A. 2012. Control of RNP motility and localization by a splicing-dependent structure in oskar mRNA. Nat Struct Mol Biol 19:441–449. http://dx.doi.org/10.1038/nsmb.2257.
- Kim G, Pai CI, Sato K, Person MD, Nakamura A, Macdonald PM. 2015. Region-specific activation of oskar mRNA translation by inhibition of Bruno-mediated repression. PLoS Genet 11:e1004992. http://dx.doi.org/10.1371/journal.pgen.1004992.
- Kugler JM, Lasko P. 2009. Localization, anchoring and translational control of oskar, gurken, bicoid and nanos mRNA during Drosophila oogenesis. Fly 3:15–28. http://dx.doi.org/10.4161/fly.3.1.7751.
- Bouvrette DJ, Price SJ, Bryda EC. 2008. K homology domains of the mouse polycystic kidney disease-related protein, Bicaudal-C (Bicc1), mediate RNA binding in vitro. Nephron Exp Nephrol 108:e27–e34. http://dx.doi.org/10.1159/000112913.
- Hollingworth D, Candel AM, Nicastro G, Martin SR, Briata P, Gherzi R, Ramos A. 2012. KH domains with impaired nucleic acid binding as a tool for functional analysis. Nucleic Acids Res 40:6873–6886. http://dx.doi.org/10.1093/nar/gks368.
- Mahone M, Saffman EE, Lasko PF. 1995. Localized Bicaudal-C RNA encodes a protein containing a KH domain, the RNA binding motif of FMR1. EMBO J 14:2043–2055.
- Saffman EE, Styhler S, Rother K, Li W, Richard S, Lasko P. 1998. Premature translation of oskar in oocytes lacking the RNA-binding protein Bicaudal-C. Mol Cell Biol 18:4855–4862.
- Kugler JM, Chicoine J, Lasko P. 2009. Bicaudal-C associates with a Trailer Hitch/Me31B complex and is required for efficient Gurken secretion. Dev Biol 328:160–172. http://dx.doi.org/10.1016/j.ydbio.2009.01.024.
- Chicoine J, Benoit P, Gamberi C, Paliouras M, Simonelig M, Lasko P. 2007. Bicaudal-C recruits CCR4-NOT deadenylase to target mRNAs and regulates oogenesis, cytoskeletal organization, and its own expression. Dev Cell 13:691–704. http://dx.doi.org/10.1016/j.devcel.2007.10.002.
- Cogswell C, Price SJ, Hou X, Guay-Woodford LM, Flaherty L, Bryda EC. 2003. Positional cloning of jcpk/bpk locus of the mouse. Mamm Genome 14:242–249. http://dx.doi.org/10.1007/s00335-002-2241-0.
- Tran U, Pickney LM, Ozpolat BD, Wessely O. 2007. Xenopus Bicaudal-C is required for the differentiation of the amphibian pronephros. Dev Biol 307:152–164. http://dx.doi.org/10.1016/j.ydbio.2007.04.030.
- Kraus MR, Clauin S, Pfister Y, Di Maio M, Ulinski T, Constam D, Bellanne-Chantelot C, Grapin-Botton A. 2012. Two mutations in human BICC1 resulting in Wnt pathway hyperactivity associated with cystic renal dysplasia. Hum Mutat 33:86–90. http://dx.doi.org/10.1002/humu.21610.
- Flaherty L, Messer A, Russell LB, Rinchik EM. 1992. Chlorambucil-induced mutations in mice recovered in homozygotes. Proc Natl Acad Sci U S A 89:2859–2863. http://dx.doi.org/10.1073/pnas.89.7.2859.
- Nauta J, Ozawa Y, Sweeney WE, Jr, Rutledge JC, Avner ED. 1993. Renal and biliary abnormalities in a new murine model of autosomal recessive polycystic kidney disease. Pediatr Nephrol 7:163–172. http://dx.doi.org/10.1007/BF00864387.
- Ryan S, Verghese S, Cianciola NL, Cotton CU, Carlin CR. 2010. Autosomal recessive polycystic kidney disease epithelial cell model reveals multiple basolateral epidermal growth factor receptor sorting pathways. Mol Biol Cell 21:2732–2745. http://dx.doi.org/10.1091/mbc.E09-12-1059.
- Kotsis F, Boehlke C, Kuehn EW. 2013. The ciliary flow sensor and polycystic kidney disease. Nephrol Dial Transplant 28:518–526. http://dx.doi.org/10.1093/ndt/gfs524.
- Wang S, Zhang J, Nauli SM, Li X, Starremans PG, Luo Y, Roberts KA, Zhou J. 2007. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol Cell Biol 27:3241–3252. http://dx.doi.org/10.1128/MCB.00072-07.
- Lian P, Li A, Li Y, Liu H, Liang D, Hu B, Lin D, Jiang T, Moeckel G, Qin D, Wu G. 2014. Loss of polycystin-1 inhibits Bicc1 expression during mouse development. PLoS One 9:e88816. http://dx.doi.org/10.1371/journal.pone.0088816.
- Tran U, Zakin L, Schweickert A, Agrawal R, Doger R, Blum M, De Robertis EM, Wessely O. 2010. The RNA-binding protein bicaudal C regulates polycystin 2 in the kidney by antagonizing miR-17 activity. Development 137:1107–1116. http://dx.doi.org/10.1242/dev.046045.
- Piazzon N, Maisonneuve C, Guilleret I, Rotman S, Constam DB. 2012. Bicc1 links the regulation of cAMP signaling in polycystic kidneys to microRNA-induced gene silencing. J Mol Cell Biol 4:398–408. http://dx.doi.org/10.1093/jmcb/mjs027.
- Gattone VH, II, Wang X, Harris PC, Torres VE. 2003. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 9:1323–1326. http://dx.doi.org/10.1038/nm935.
- Rees S, Kittikulsuth W, Roos K, Strait KA, Van Hoek A, Kohan DE. 2014. Adenylyl cyclase 6 deficiency ameliorates polycystic kidney disease. J Am Soc Nephrol 25:232–237. http://dx.doi.org/10.1681/ASN.2013010077.
- Harris PC, Torres VE. 2014. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest 124:2315–2324. http://dx.doi.org/10.1172/JCI72272.
- Lee DC, Chan KW, Chan SY. 1998. Expression of transforming growth factor alpha and epidermal growth factor receptor in adult polycystic kidney disease. J Urol 159:291–296. http://dx.doi.org/10.1016/S0022-5347(01)64084-9.
- Sweeney WE, Jr, Avner ED. 1998. Functional activity of epidermal growth factor receptors in autosomal recessive polycystic kidney disease. Am J Physiol 275:F387–F394.
- Dell KM, Nemo R, Sweeney WE, Jr, Levin JI, Frost P, Avner ED. 2001. A novel inhibitor of tumor necrosis factor-alpha converting enzyme ameliorates polycystic kidney disease. Kidney Int 60:1240–1248. http://dx.doi.org/10.1046/j.1523-1755.2001.00963.x.
- Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, Flask CA, Novick AC, Goldfarb DA, Kramer-Zucker A, Walz G, Piontek KB, Germino GG, Weimbs T. 2006. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A 103:5466–5471. http://dx.doi.org/10.1073/pnas.0509694103.
- Maisonneuve C, Guilleret I, Vick P, Weber T, Andre P, Beyer T, Blum M, Constam DB. 2009. Bicaudal C, a novel regulator of Dvl signaling abutting RNA-processing bodies, controls cilia orientation and leftward flow. Development 136:3019–3030. http://dx.doi.org/10.1242/dev.038174.
- Pennekamp P, Menchen T, Dworniczak B, Hamada H. 2015. Situs inversus and ciliary abnormalities: 20 years later, what is the connection? Cilia 4:1. http://dx.doi.org/10.1186/s13630-014-0010-9.
- Ponting CP. 1995. SAM: a novel motif in yeast sterile and Drosophila polyhomeotic proteins. Protein Sci 4:1928–1930. http://dx.doi.org/10.1002/pro.5560040927.
- Carroll M, Tomasson MH, Barker GF, Golub TR, Gilliland DG. 1996. The TEL/platelet-derived growth factor beta receptor (PDGF beta R) fusion in chronic myelomonocytic leukemia is a transforming protein that self-associates and activates PDGF beta R kinase-dependent signaling pathways. Proc Natl Acad Sci U S A 93:14845–14850. http://dx.doi.org/10.1073/pnas.93.25.14845.
- Meruelo AD, Bowie JU. 2009. Identifying polymer-forming SAM domains. Proteins 74:1–5. http://dx.doi.org/10.1002/prot.22232.
- Kim CA, Gingery M, Pilpa RM, Bowie JU. 2002. The SAM domain of polyhomeotic forms a helical polymer. Nat Struct Biol 9:453–457.
- Tran HH, Kim CA, Faham S, Siddall MC, Bowie JU. 2002. Native interface of the SAM domain polymer of TEL. BMC Struct Biol 2:5. http://dx.doi.org/10.1186/1472-6807-2-5.
- Knight MJ, Leettola C, Gingery M, Li H, Bowie JU. 2011. A human sterile alpha motif domain polymerizome. Protein Sci 20:1697–1706. http://dx.doi.org/10.1002/pro.703.
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. 1997. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science 275:1784–1787. http://dx.doi.org/10.1126/science.275.5307.1784.
- Schwarz-Romond T, Metcalfe C, Bienz M. 2007. Dynamic recruitment of axin by Dishevelled protein assemblies. J Cell Sci 120:2402–2412. http://dx.doi.org/10.1242/jcs.002956.
- Cougot N, Babajko S, Seraphin B. 2004. Cytoplasmic foci are sites of mRNA decay in human cells. J Cell Biol 165:31–40. http://dx.doi.org/10.1083/jcb.200309008.
- National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC.
- Harada BT, Knight MJ, Imai S, Qiao F, Ramachander R, Sawaya MR, Gingery M, Sakane F, Bowie JU. 2008. Regulation of enzyme localization by polymerization: polymer formation by the SAM domain of diacylglycerol kinase delta1. Structure 16:380–387. http://dx.doi.org/10.1016/j.str.2007.12.017.
- Arnold K, Bordoli L, Kopp J, Schwede T. 2006. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201. http://dx.doi.org/10.1093/bioinformatics/bti770.
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. 2006. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics Chapter 5:Unit 5.6. http://dx.doi.org/10.1002/0471250953.bi0506s15.
- Shen MY, Sali A. 2006. Statistical potential for assessment and prediction of protein structures. Protein Sci 15:2507–2524. http://dx.doi.org/10.1110/ps.062416606.
- Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E. 2013. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29:845–854. http://dx.doi.org/10.1093/bioinformatics/btt055.
- Morse PM, Feshbach H. 1953. Asymptotic series; method of steepest descent, p 434–443. Methods of theoretical physics, part I. McGraw-Hill, New York, NY.
- Mackerell AD, Jr, Feig M, Brooks CL, III. 2004. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem 25:1400–1415. http://dx.doi.org/10.1002/jcc.20065.
- Flaherty L, Bryda EC, Collins D, Rudofsky U, Montgomery JC. 1995. New mouse model for polycystic kidney disease with both recessive and dominant gene effects. Kidney Int 47:552–558. http://dx.doi.org/10.1038/ki.1995.69.
- Bertrand E, Chartrand P, Schaefer M, Shenoy SM, Singer RH, Long RM. 1998. Localization of ASH1 mRNA particles in living yeast. Mol Cell 2:437–445. http://dx.doi.org/10.1016/S1097-2765(00)80143-4.
- Fu Y, Kim I, Lian P, Li A, Zhou L, Li C, Liang D, Coffey RJ, Ma J, Zhao P, Zhan Q, Wu G. 2010. Loss of Bicc1 impairs tubulomorphogenesis of cultured IMCD cells by disrupting E-cadherin-based cell-cell adhesion. Eur J Cell Biol 89:428–436. http://dx.doi.org/10.1016/j.ejcb.2010.01.002.
- Qiao F, Bowie JU. 2005. The many faces of SAM. Sci STKE 2005:re7.
- Nakel K, Hartung SA, Bonneau F, Eckmann CR, Conti E. 2010. Four KH domains of the C. elegans Bicaudal-C ortholog GLD-3 form a globular structural platform. RNA 16:2058–2067. http://dx.doi.org/10.1261/rna.2315010.
- Hofmann I, Casella M, Schnolzer M, Schlechter T, Spring H, Franke WW. 2006. Identification of the junctional plaque protein plakophilin 3 in cytoplasmic particles containing RNA-binding proteins and the recruitment of plakophilins 1 and 3 to stress granules. Mol Biol Cell 17:1388–1398.
- Stagner EE, Bouvrette DJ, Cheng J, Bryda EC. 2009. The polycystic kidney disease-related proteins Bicc1 and SamCystin interact. Biochem Biophys Res Commun 383:16–21. http://dx.doi.org/10.1016/j.bbrc.2009.03.113.
- Hoff S, Halbritter J, Epting D, Frank V, Nguyen TM, van Reeuwijk J, Boehlke C, Schell C, Yasunaga T, Helmstadter M, Mergen M, Filhol E, Boldt K, Horn N, Ueffing M, Otto EA, Eisenberger T, Elting MW, van Wijk JA, Bockenhauer D, Sebire NJ, Rittig S, Vyberg M, Ring T, Pohl M, Pape L, Neuhaus TJ, Elshakhs NA, Koon SJ, Harris PC, Grahammer F, Huber TB, Kuehn EW, Kramer-Zucker A, Bolz HJ, Roepman R, Saunier S, Walz G, Hildebrandt F, Bergmann C, Lienkamp SS. 2013. ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat Genet 45:951–956. http://dx.doi.org/10.1038/ng.2681.
- Taskiran EZ, Korkmaz E, Gucer S, Kosukcu C, Kaymaz F, Koyunlar C, Bryda EC, Chaki M, Lu D, Vadnagara K, Candan C, Topaloglu R, Schaefer F, Attanasio M, Bergmann C, Ozaltin F. 2014. Mutations in ANKS6 cause a nephronophthisis-like phenotype with ESRD. J Am Soc Nephrol 25:1653–1661. http://dx.doi.org/10.1681/ASN.2013060646.
- Leettola CN, Knight MJ, Cascio D, Hoffman S, Bowie JU. 2014. Characterization of the SAM domain of the PKD-related protein ANKS6 and its interaction with ANKS3. BMC Struct Biol 14:17. http://dx.doi.org/10.1186/1472-6807-14-17.
- Yakulov TA, Yasunaga T, Ramachandran H, Engel C, Muller B, Hoff S, Dengjel J, Lienkamp SS, Walz G. 2015. Anks3 interacts with nephronophthisis proteins and is required for normal renal development. Kidney Int 87:1191–1200. http://dx.doi.org/10.1038/ki.2015.17.
- Zhang Y, Cooke A, Park S, Dewey CN, Wickens M, Sheets MD. 2013. Bicaudal-C spatially controls translation of vertebrate maternal mRNAs. RNA 19:1575–1582. http://dx.doi.org/10.1261/rna.041665.113.
- Zhou HX, Rivas G, Minton AP. 2008. Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences. Annu Rev Biophys 37:375–397. http://dx.doi.org/10.1146/annurev.biophys.37.032807.125817.
- Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. 2007. The role of site accessibility in microRNA target recognition. Nat Genet 39:1278–1284. http://dx.doi.org/10.1038/ng2135.
- Kedde M, van Kouwenhove M, Zwart W, Oude Vrielink JA, Elkon R, Agami R. 2010. A Pumilio-induced RNA structure switch in p27-3′ UTR controls miR-221 and miR-222 accessibility. Nat Cell Biol 12:1014–1020. http://dx.doi.org/10.1038/ncb2105.
- Miles WO, Tschop K, Herr A, Ji JY, Dyson NJ. 2012. Pumilio facilitates miRNA regulation of the E2F3 oncogene. Genes Dev 26:356–368. http://dx.doi.org/10.1101/gad.182568.111.
- Isono K, Endo TA, Ku M, Yamada D, Suzuki R, Sharif J, Ishikura T, Toyoda T, Bernstein BE, Koseki H. 2013. SAM domain polymerization links subnuclear clustering of PRC1 to gene silencing. Dev Cell 26:565–577. http://dx.doi.org/10.1016/j.devcel.2013.08.016.
- Qiao F, Song H, Kim CA, Sawaya MR, Hunter JB, Gingery M, Rebay I, Courey AJ, Bowie JU. 2004. Derepression by depolymerization; structural insights into the regulation of Yan by Mae. Cell 118:163–173. http://dx.doi.org/10.1016/j.cell.2004.07.010.