
ABSTRACT
Recombination activating gene 1 (RAG1) and RAG2 are critical enzymes for initiating variable-diversity-joining [V(D)J] segment recombination, an essential process for antigen receptor expression and lymphocyte development. The BCL11A transcription factor is required for B cell and plasmacytoid dendritic cell (pDC) development, but its molecular function(s) in early B cell fate specification and commitment is unknown. We show here that the major B cell isoform, BCL11A-XL, binds directly to the RAG1 promoter as well as directly to regulatory regions of transcription factors previously implicated in both B cell and pDC development to activate RAG1 and RAG2 gene transcription in pro- and pre-B cells. We employed BCL11A overexpression with recombination substrates to demonstrate direct consequences of BCL11A/RAG modulation on V(D)J recombination. We conclude that BCL11A is a critical component of a transcriptional network that regulates B cell fate by controlling V(D)J recombination.
PUBLISHER'S NOTE
The American Society for Microbiology and Molecular and Cellular Biology would like to inform readers that this article is a corrected and republished version of https://doi.org/10.1128/MCB.00987-12, which was retracted (https://doi.org/10.1128/MCB.00358-17). The original article was found to have image duplications in Fig. 6E and 7A. The underlying data for these figures were no longer available; thus a correction was not possible and the article was retracted. However, these figures were not critical to the overall conclusions of the article. Therefore, the authors were allowed to submit a revised manuscript for consideration.
INTRODUCTION
Recombination activating gene (RAG) endonuclease catalyzes the cleavage phase of V(D)J recombination (Citation1 Citation– Citation4). RAG is encoded by two adjacent genes, RAG1 and RAG2, whose products must interact to form an active endonuclease (Citation5). Both RAG1 and RAG2 are essential for subsequent B and T lymphocyte development (Citation4, Citation6). In cell-free systems, RAG1 and RAG2 are sufficient to cleave recombination substrates (Citation7, Citation8), but in vivo a number of additional factors are required for chromatin accessibility of V(D)J segments and for appropriate completion of the V(D)J joints (Citation1 Citation– Citation3).
RAG expression occurs at two distinct points during B cell development. The first phase results in the assembly of the immunoglobulin heavy chain (IgH) in pro-B cells, whereas the second catalyzes Ig light chain (L) assembly in pre-B cells. RAG expression is tightly regulated at both the posttranscriptional and transcriptional levels (Citation2). In addition to the individual promoters for RAG1 and RAG2, the RAG locus contains at least 5 distal enhancer elements: the RAG enhancer (Erag), the proximal enhancer (Ep), the distal enhancer (Ed), and two recently discovered regions, termed Irag1 and Irag2 (Citation9 Citation– Citation18) (). Erag is the strongest enhancer, as demonstrated by a 5- to 10-fold reduction in RAG expression and a partial block at the pro-B-to-pre-B transition following targeted deletion of Erag in mice (Citation12). A number of transcription factors (TFs) bind to their corresponding DNA motifs within single or multiple regions within the RAG locus (), and several of these interactions have been shown to activate RAG transcription (Citation9, Citation10, Citation12, Citation16, Citation19, Citation20).
FIG 1 Schematic representation of the RAG locus and the BCL11A superfamily. (A) Transcriptional regulators and binding regions. The human RAG locus is shown, with positions of previously described enhancers (blue arrows), promoters (blue boxes), and exons (black boxes) given; the transcriptional polarities of Rag1 and Rag2 are indicated with black arrows. Positions of DNA binding sites for TFs determined previously (Citation9, Citation10, Citation13, Citation14, Citation16, Citation19, Citation20) or here (BCL11A-XL) to bind within these regions are indicated by vertical lines. (B) BCL11A superfamily of TFs involved in hematological malignancy. Each member has a highly conserved N terminus, MSRRK, shown in this study to be essential for BCL11A-XL transcriptional activity. This is followed by a single, canonical C2HC zinc finger, which is followed by one or more single, double, or triple zinc fingers of the C2H2 type. The BCL11A and BCL11B genes, as well as the early hematopoietic zinc finger (EHZF) gene and the friend-of-GATA hematopoietic transcription regulator FOG1 and FOG2 genes, all encode zinc finger proteins with these conserved features, and several have been implicated in malignancy (Citation23, Citation25, Citation37, Citation41, Citation55).
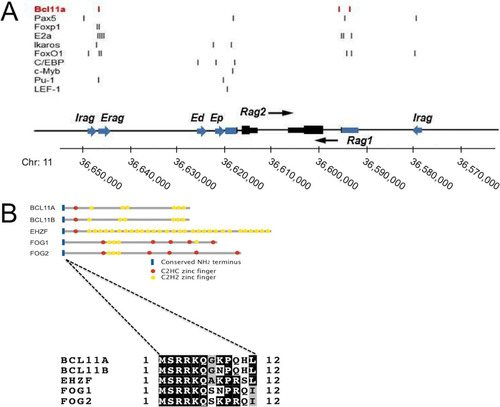
We originally discovered B cell lymphoma/leukemia 11A (BCL11A) as the gene disrupted by t(2;14)(p13;q32) translocation in unusually aggressive cases of B cell chronic lymphocytic leukemia (Citation21 Citation– Citation23). Subsequently, BCL11A was identified as an oncogene in numerous lymphoid malignancies (Citation24 Citation– Citation30). More recently, we found BCL11A to be essential for development of plasmacytoid dendritic cells (pDCs) (Citation31 Citation– Citation33) and fetal hemoglobin (Citation27, Citation34 Citation– Citation36). Among the five BCL11A isoforms (Citation23, Citation37), the extralong isoform (BCL11A-XL; NCBI accession number AJ404611 ) is expressed far more abundantly in hematopoietic lineages (Citation23, Citation37) (). All isoforms share a conserved N terminus and an atypical C2HC Kruppel-like zinc finger, defining a “superfamily” of 5 genes essential to myeloid/lymphoid (EHZF), megakaryocytic (FOG1 and FOG2), T-lymphoid (BCL11B), and B-lymphoid (BCL11A) development (Citation37) ().
Targeted deletion of Bcl11a in the mouse indicated that Bcl11a is selectively required for progression at the earliest stage (pre-pro-B) of B cell progenitor commitment, prior to the RAG-dependent formation of pro-B cells (Citation24, Citation31, Citation33), although the effect of its loss on T and NK lineages remains controversial (Citation31, Citation33). While resolution of this conflict remains unknown, it is clear from these and other studies that BCL11A expression commences prior to that of the RAG transactivators shown in , whose expression is lost or highly reduced in Bcl11a knockouts (Citation31, Citation33; data reported here). What is not clear is whether their loss is an indirect result of the strong progenitor block in Bcl11a-deficient common lymphoid progenitors (CLPs) or whether these factors are direct transcriptional targets of Bcl11a.
Here we identify RAG1 as a direct target of BCL11A-XL. BCL11A-XL binds within the RAG1 promoter and the Erag enhancer to activate RAG1 and RAG2 transcription in pre-B cells while repressing RAG1 promoter activity in epithelial and fibroblast-derived cell lines. Overexpression of BCL11A-XL in a V(D)J recombination-competent pre-B cell line induces RAG expression and V(D)J recombination. We show that BCL11A-XL regulates additional RAG activators, both directly and indirectly, as well as activators of Vκ-Jκ locus accessibility. We propose that in addition to its earlier hematopoietic progenitor role, BCL11A is essential for the pro-B-to-pre-B transition, at least in part from direct loss of V(D)J recombination.
RESULTS
BCL11A-XL modulates RAG expression.
As an initial approach, we used microarrays to identify genes that are deregulated by BCL11A-XL overexpression in mature B (RAJI, Ramos, OCI-LY7, and BJAB) and pre-B (NALM6) human cell lines. B cell lines were transduced with pXY-puro (mock control) or the same virus containing an N-terminally Flag-tagged, full-length BCL11A-XL cDNA. Among the 17,856 clones tested, 43 clones representing 39 genes showed alterations of at least 2-fold in all four cell lines. As expected from previous studies of BCL11A function, most transcripts were downregulated (data not shown). Among the upregulated genes, RAG1 was consistently present ().
FIG 2 BCL11A-XL regulates RAG expression. (A) Microarray identification of RAG1 upregulation. The indicated human B cell lines were stably transduced with a BCL11A-XL (XL) retrovirus or with pXY-puro empty vector. Expression profiling was performed in multiple (n > 4) independent experiments tracked by multiple probe elements spotted per microarray. Data shown are for several transcripts upregulated at least 2-fold relative to those in the vector control in all B cell lines. (B) Overexpression of BCL11A-XL upregulates RAG. Human NALM6 pre-B cells were stably transduced with BCL11A-XL (XL) or the pXY-puro virus control (V). (Left) Western blot. Flag BCL11A, anti-Flag detection of viral BCL11A-XL expression; total BCL11A, anti-BCL11A-XL MAb detection; β-tubulin, anti-β-tubulin control. (Right) RT-PCR demonstrating upregulation of RAG1 and RAG2. Serial template dilution (3-fold; indicated by triangles) confirmed that RT-PCR was semiquantitative. GAPDH was used as a loading control. (C) Inducible shRNA knockdown of BCL11A-XL reduces RAG expression. BJAB B cells were first stably transduced with a retrovirus expressing the bacterial tetracycline repressor (TETR) and the blasticidin resistance gene and then with a pRSMX-PG retrovirus containing shRNA targeted to exon 2 of the BCL11A-XL gene under the control of a modified H1 promoter containing binding sites for TETR (Citation68). Significant downregulation of BCL11A-XL protein (left) and transcript (right) with concomitant downregulation of RAG1 and RAG2 was observed 24 h following addition of the tetracycline analog doxycycline (Dox).
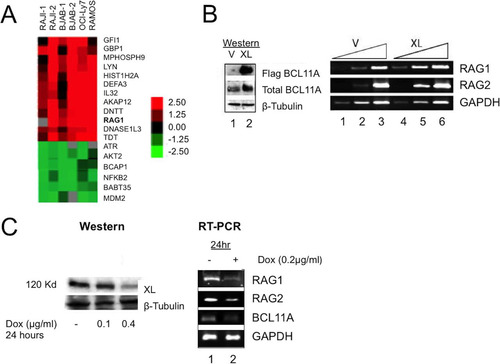
We created a stable BCL11A-XL-overexpressing line of NALM6 pre-B cells and checked RAG1 and RAG2 expression by semiquantitative reverse transcription-PCR (RT-PCR). As shown in , both RAG genes were significantly upregulated. Inducible short hairpin RNA (shRNA) interference was used to target a region (exon 2) common to all BCL11A isoforms. As shown in , we observed a robust knockdown of BCL11A transcripts as well as a corresponding reduction in RAG1 and RAG2 transcripts after 24 h of induction.
BCL11A is recruited to the RAG1 promoter and the Erag enhancer in vivo and in vitro.
Chromatin immunoprecipitation (ChIP) was employed to test whether BCL11A-XL is recruited to the RAG promoter or enhancer loci in human pre-B (NALM6) and mature B cell (RAJI) chromatin. Primers were designed within the upstream 1-kb region of each promoter and within the ∼0.8-kb first third of the Erag enhancer. ChIP-PCR demonstrated that the RAG1 promoter and Erag, but not the RAG2 promoter or the Ep or Ed enhancer, were enriched in pulldown assays with anti-BCL11A but not with a control antibody ( and data not shown). Thus, BCL11A-XL is recruited either directly or indirectly to the proximal promoter of RAG1 and to the RAG enhancer in vivo.
FIG 3 BCL11A is recruited to the RAG1 promoter and the Erag enhancer in vivo and in vitro. (A) ChIP-PCR identified the RAG1 promoter and Erag enhancer as targets for BCL11A binding. A rabbit polyclonal antibody (BL1797; Bethyl) specific to the BCL11A-XL C terminus (XL Ab) was used to immunoprecipitate NALM6 pre-B cell chromatin. The samples were reverse cross-linked, purified, and analyzed by semiquantitative PCR, using primers that amplify regions within the proximal RAG1 and RAG2 promoters and within the Erag enhancer. Specific enrichment was observed for the RAG1 promoter and the Erag enhancer but not for the RAG2 promoter (lanes 3). PCRs with 1% input were used as positive controls (lanes 1). Purified rabbit IgG antibodies (con Ab) did not immunoprecipitate target DNA significantly (lanes 2). (B) BCL11A binds to the Erag enhancer in vitro. A 196-bp probe extending from the 5′ boundary (KpnI site) to the end of region A of Erag (Citation13) was labeled with 32P and then incubated with nuclear extracts prepared from the RAJI B cell line. Specificity of binding was tested by further incubation of the indicated probe-nuclear extract mixtures with anti-BCL11A-XL C-terminal (BL1797) polyclonal (C-Term Ab) and monoclonal (MAb Ab123) antibodies, monoclonal anti-PU.1 (α-PU.1 Ab), or control polyclonal (α-IgG Ab) and monoclonal (α-Tubulin Ab) antibodies employed at the same concentrations. Cold competitions employed double-stranded 30-mers of wild-type (cold oligo) and mutated (cold mutant oligo) sequences spanning the putative BCL11A binding site at nt 121 to 131. Triangles represent molar excesses of 10, 50, and 100 relative to the amount of labeled probe. Complexes were resolved in 6% native gels, with specific binding highlighted by a box. (C and D) BCL11A binds to a consensus site within the distal region (positions −426 to −416) of the RAG1 promoter. The probe was a 300-bp KpnI-XhoI fragment. (E) BCL11A binds to a consensus site within the proximal region (positions −207 to −197) of the RAG1 promoter. The probe was a 263-bp PCR product spanning positions −228 to +35. (F) Alignment of BCL11A binding sites determined in this study with the consensus for the top 500 peaks determined by ENCODE ChIP-seq for a human B cell lymphoma (GM12878) (Citation38).
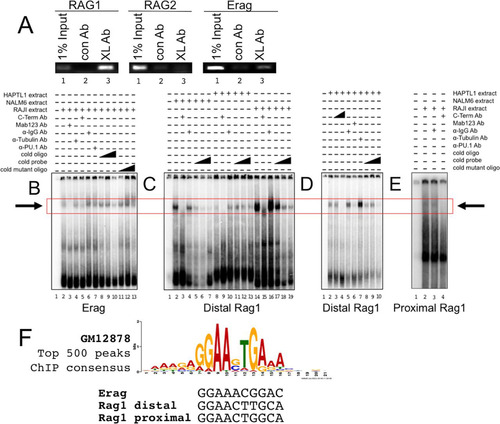
Electrophoretic mobility shift assays (EMSAs) were performed to validate and extend the results shown in . A 196-bp probe extending from the 5′ end to the 3′ end of region A of Erag (Citation13) was incubated with nuclear extracts prepared from RAJI B cells, and several complexes were observed (). A slowly migrating species (boxed in to ) was abolished by pre- or postincubation with an anti-BCL11A monoclonal antibody (MAb) (Ab123) or a polyclonal antibody specific for the BCL11A-XL C terminus (lanes 3 and 5) but not with control antibodies (lanes 4 and 6). Specific double-stranded oligonucleotide competition (compare lanes 8 to 10 to lanes 11 and 12) identified a BCL11A binding site that closely matched the BCL11A consensus determined by genome-wide ChIP sequencing (ChIP-seq) derived publically from the human B cell lymphoma GM12728 (Citation38) (). This site is overlapped from positions −2 to +4 with a consensus PU.1 binding site (GAGGAA), an essential myeloid/lymphoid transactivator which was shown to bind to the Ep element () (Citation39). Accordingly, we observed partial ablation of the BCL11A complex with anti-PU.1 (, lane 7), suggesting that these factors act in concert or opposition at this site. Using a similar approach with nuclear extracts prepared from pre-B (NALM6) (, lanes 2 to 7), pro-B (HAPTL1) (, lanes 8 to 13, and D, lanes 2 to 10), and mature B (RAJI) (, lanes 14 to 19, and E, lanes 2 to 4) cell lines, we validated two similarly migrating BCL11A complexes within the distal (nucleotides [nt] −426 to −416) and proximal (nt −207 to −197) regions of the RAG1 promoter ( to ). Ablation as opposed to supershifting suggested that both anti-BCL11A antibodies interfere with DNA binding. Both proximal and distal BCL11A-XL binding sites are well within the ChIP region determined for . Further consistent with our ChIP-PCR results, no binding to RAG2 promoter probes was observed (data not shown), nor did we detect specific binding when in vitro-translated BCL11A-XL, -L, or -XS was employed (data not shown). This suggested that BCL11A-XL binding requires a complex provided by another factor(s), potentially PU.1.
BCL11A-XL transactivates RAG1 and Erag-RAG2 transcription in pre-B cells.
Transformation of murine fetal liver or bone marrow (BM) B cell progenitors by the Abelson murine leukemia virus arrests cells at the early (large) pre-B cell stage, with low to negligible Rag1 and Rag2 expression (Citation2). One such line, A70 (Citation40), expresses negligible levels of BCL11A (). Stable BCL11A-XL overexpression in A70 cells by retroviral transduction induced high levels of endogenous Rag1 and Rag2 (). These cells (A70-BCL11A-XL) were compared to the parental BCL11A-XL-negative A70 line following transient transfection with luciferase (Luc) reporter constructs previously shown (Citation12, Citation18) to recapitulate activities of the proximal Rag1 (−243-R1p-Luc) and Rag2 (−279-R1p-Luc) promoters in the absence or presence of the full, 2.3-kb Erag enhancer. A70-BCL11A-XL firefly luciferase activities were normalized to those of cotransfected Renilla luciferase and plotted as fold activation over that of a promoterless luciferase control.
FIG 4 RAG transcriptional reporter activity is activated in pre-B cells but repressed in non-B cells following BCL11A-XL overexpression. (A) Establishment of stable overexpression. A70 pre-B cells were transduced with pXY-IRES-puro (V) or pXY-BCL11A-XL (XL) and selected with 1 μg/ml of puromycin for 6 days. Triangles represent 5-fold dilutions of cDNA. (B) BCL11A-XL transactivates RAG1 promoter- and Erag-RAG2 promoter-driven transcription in A70 pre-B cells but represses RAG promoter-driven activity in 293T epithelial cells. A70 and 293T cells stably transduced with pXY vector only, BCL11A-XL (XL), or BCL11A-XL mutated at an essential N-terminal K5 residue (XL-K5/A) were transiently transfected with the indicated firefly luciferase constructs and the Renilla luciferase control as described in Materials and Methods. Cells were harvested 40 to 48 h later, and firefly luciferase values were normalized to Renilla luciferase values. The ratios were expressed as fold changes by arbitrary assignment of a value of 1.0 to the empty vector (pXY-IRES-puro for pre-B cells and pCMV10 for non-B cells). Fold changes are plotted on the y axis, with the magnitude of activation denoted by increasing positive values and repression by negative values. Error bars indicate standard deviations for 3 to 5 independent experiments. (C) Wild-type BCL11A-XL and the BCL11A-K5/A mutant localize indistinguishably within the nuclear matrix of paraspeckles in pre-B and non-B cells. (Top) Fixed mounts of GFP-BCL11A-XL and GFP-BCL11A-XL-K5/A were imaged following transient transfection into 293T cells. Ectopically transduced A70-BCL11A-XL cells were stained with anti-BCL11A MAb Ab123. (Bottom) Biochemical fractionation of transiently transfected 293T cells with the indicated BCL11A-XL wild-type or mutant construct. Equivalent amounts of cytoplasmic (C), soluble nuclear (NP), chromatin (C), and nuclear matrix (NM) fractions were subjected to SDS-PAGE (Citation26) and then Western blotted with antibodies specific for BCL11A or for the NM marker lamin B.
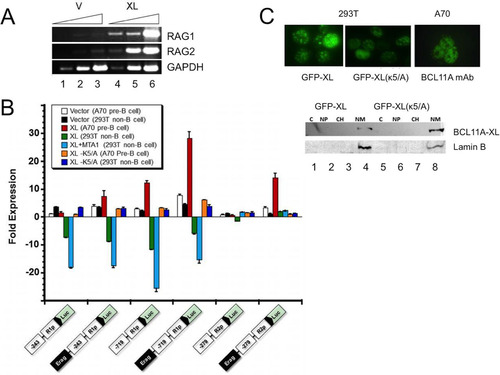
As shown in , BCL11A-XL only modestly increased Rag1 proximal promoter-driven transcription (1.6-fold, on average). Appending Erag to the reporter provided an ∼4-fold enhancement in A70 cells, which was doubled in the A70-BCL11A-XL transductants. When the RAG1 promoter was extended to include the more distal BCL11A-XL binding region located at position ∼−640 (−719-R1p-Luc), we observed an ∼3-fold increase above that observed for the proximal Rag1 promoter alone and an ∼12-fold increase in the presence of Erag. Consistent with the EMSA and ChIP data, no effect of BCL11A-XL overexpression was observed on Rag2 proximal promoter-luciferase reporter activity alone. However, inclusion of Erag resulted in an ∼3-fold increase in the absence of BCL11A-XL and an ∼14-fold increase in Rag2 reporter activity in the presence of BCL11A-XL overexpression ().
The N termini of several members of the BCL11A superfamily (Citation37) constitute the transactivation domain, and K5 was shown to be essential in several cases (Citation25, Citation41) (). Activation of all reporter activities by BCL11A-XL was abrogated by K5/A substitution (). Collectively, the data indicate that BCL11A-XL acts at the Rag1 promoter, primarily through a distal binding site, whereas BCL11A-XL activates Rag2 transcription exclusively via Erag binding.
Enforced expression of BCL11A in non-B cells represses RAG1-mediated transcription.
The above results prompted us to test the cell type specificity of BCL11A-mediated RAG transactivation. BCL11A-XL overexpression in 293T cells led to strong repression (∼5- to 6-fold) of both proximal (−243-R1p) and distal (−719-R1p) Rag1 promoter-driven activities, whereas Rag2 and Erag luciferase activities were unaffected (), suggesting that the proximal BCL11A binding site within the Rag1 promoter is sufficient for targeting maximal repression. Similar results were observed in additional non-B cell lines (data not shown).
BCL11B, the highly similar paralogue of BCL11A (Citation23), was shown to interact with the nucleosome remodeling and histone deacetylase (NuRD) complex (Citation42). The NuRD component metastasis tumor antigen 1 (MTA1) was shown to interact with and enhance BCL11B repression synergistically, while MTA2 had no effect (Citation42). We observed significant augmentation of BCL11A-XL repression following cotransfection with MTA1 () but not after cotransfection with MTA2 (data not shown). Conversely, enforced overexpression of BCL11A-XL with MTA1 in A70 pre-B cells had no effect on activation of Rag1 or Rag2 constructs (data not shown).
We previously observed that in B cells, exogenous or ectopically overexpressed BCL11A-XL accumulated primarily within nuclear matrix paraspeckles (Citation37). As with those results, we observed localization indistinguishable from that following ectopic delivery of GFP-BCL11A-XL or GFP-BCL11A-XL-K5/A into 293T cells (, upper panels). Biochemical subcellular fractionations (, lower panels) indicated that, as in B cells (Citation37), wild-type and mutant BCL11A-XL forms accumulated preferentially within the nuclear matrix.
These results suggest that BCL11A-XL acts in a context-dependent, cell type-specific mode to activate or repress RAG1 transcription. The data further suggest that repressive BCL11A-XL complexes differ from activation complexes by the inclusion/exclusion of the MTA1 corepressor but are indistinguishable in their subcellular localization.
BCL11A-XL induces ectopic V(D)J recombination in A70-INV pre-B cells.
The A70-INV Abl-transformed pre-B cell line retains the properties of the parental A70 line, but it has been engineered for inducible V(D)J recombination analysis by integration of a recombination substrate, pMX-INV, which upon inversion activates expression of green fluorescent protein (GFP) (Citation40). A70 and A70-INV cells also constitutively express a Bcl2-Eμ transgene that enables them to survive apoptotic signaling for several days (Citation40). We transduced A70-INV cells with empty virus (pXY-puro) or pXY-BCL11A-XL and then selected bulk (uncloned) cells by use of puromycin. As with A70 parental cells, BCL11A protein was barely detectable in untransduced or mock-transduced A70-INV cells (). Transduced cells expressed significant levels of BCL11A-XL, resulting in robust induction of Rag1 and Rag2 ().
FIG 5 Enforced expression of BCL11A-XL in A70-INV pre-B cells induces V(D)J recombination. (A and B) Establishment of stable overexpression in A70-INV pre-B cells. Transductions and drug selection were performed as described in the legend to . (A) Anti-Flag Western blot detection of ectopic BCL11A-XL and anti-BCL11A immunoblot detection of total expression of BCL11A-XL. β-Tubulin served as a loading control. (B) Transduction of A70-INV cells with BCL11A-XL upregulated RAG1 and RAG2 mRNAs relative to those in mock-transduced cells. GAPDH was the loading control. (C) Schematic diagram of recombination signal sequence (RSS; black and white triangles)-mediated inversion within the integrated retroviral RAG substrate in A70-INV pre-B cells. Primers used to amplify the inversion are denoted by arrows. (D) Flow cytometry detection of enhanced V(D)J recombination substrate inversion in A70-INV pre-B cells overexpressing BCL11A-XL (XL) or treated with the Abelson kinase inhibitor STI571. Cells were transduced as described in previous legends and then incubated in the absence (STI −) or presence (STI +) of 3 μM STI571 for 72 h. Inversion of pMX-INV resulted in GFP expression, which was quantified by fluorescence-activated cell sorter (FACS) analysis following exclusion of dead cells by forward and side scatter characteristics. x axis, GFP fluorescence intensity; y axis, cell numbers. The percentage of GFP-positive cells is noted in each plot. (E) PCR validation of RSS-mediated inversion of pMX-INV substrates. Genomic DNA was isolated from the indicated cell lines for PCR following the strategy and primers shown in panel C. STI571 (STI) treatment (3 μM, 48 h) or transduction with BCL11A-XL retrovirus induced inversion of GFP, allowing PCR amplification (lanes 2 and 4). PCR amplification of the endogenous Rag1 gene (which is unaffected by RSS-mediated recombination) provided a DNA loading control.
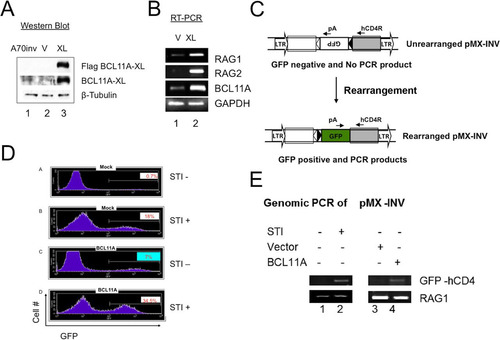
The strategy used to detect the inversion of pMX-INV and V-J recombination of the endogenous κ light chain locus () was described previously (Citation40). As a positive control, strong induction of recombination was achieved by treatment of A70-INV cells with the Abl kinase inhibitor STI571 (Citation43). STI571 causes differentiation of early pre-B cells to a late pre-B-cell-like state (Citation43). Strong Rag1 and Rag2 expression ensues, followed by Vκ-Jκ recombination and, in A70-INV cells, inversion of the integrated pMX-INV substrate via cleavage at the flanking recombination signal sequences (RSSs) () (Citation6). As shown in , panels A and B, mock viral transduction produced 0.7% and 18.1% GFP-positive cells without and with STI571 treatment, respectively. This level of background GFP expression is similar to previously published data (Citation40). BCL11A-XL-transduced cells produced 7% and 34.5% GFP-positive cells without and with STI571 treatment, respectively (, panels C and D).
The ∼10-fold increase in recombination substrate inversion by BCL11A-XL in the absence of STI571 treatment suggested that BCL11A-XL acts independently. Consistent with this interpretation, the level of substrate inversion by BCL11A-XL in the presence of STI571 was additive. BCL11A-XL transduction was accompanied by no alteration in cell cycling (data not shown).
To verify that GFP expression resulted from inversion of pMX-INV, we carried out semiquantitative PCR on genomic DNA by using a 3′ primer complementary to hCD4—a component of the pMX-INV vector that can be used to detect inverted GFP. pMX-INV inversion allows successful PCR product formation by providing the reverse primer with the correct orientation of GFP cDNA (). As shown in , an increase in the intensity of inverted GFP-hCD4 PCR products was observed in STI571-treated or BCL11A-XL-transduced cells compared to that in STI571-untreated or mock-transduced cells.
Reduction of BCL11A expression in transformed pre-B or normal murine pre-B cells impairs endogenous V(D)J recombination.
The pMX-INV reporter locus may not display a heterochromatin structure equivalent to that of the endogenous light chain loci in pre-B cells. To assess whether the endogenous loci can also be induced to rearrange by BCL11A-XL overexpression, genomic DNA was analyzed by PCR, using a “universal” Vκ forward primer and the Jκ2-1 reverse primer (, upper panel). Southern blotting with a Jκ2-2 oligonucleotide probe (, bottom panel) detected increased levels of Jκ rearrangement in both STI571-treated (lanes 4 to 6) and BCL11A-XL-transduced (lanes 10 to 12) cells compared to those for controls.
FIG 6 Modulation of BCL11A expression modulates V(D)J rearrangement. (A) BCL11A-XL overexpression induces Vκ-Jκ recombination in A70-INV pre-B cells. A degenerate Vκ consensus primer (Vcon) and a Jκ2-1 reverse primer (top panel) were used to amplify rearrangements from genomic DNA diluted in 3-fold increments (triangles) to ensure nonsaturating semiquantitative PCR. The strategy predicts products of 190 bp (VJκ2) and 540 bp (VJκ1). Amplified DNAs were resolved in agarose gels, transferred to nylon membranes, and then visualized by autoradiography following hybridization to the Jκ2-2 probe (top panel). (B) Flow cytometric analysis of Bcl11aL/L bone marrow cells following primary infection with the MSCV-Bcl-xL retrovirus and secondary superinfection with a Thy-1.1-expressing MIT or MIT-Cre retrovirus. Cells were stained with antibodies directly conjugated to phycoerythrin (PE-B220) and fluorescein isothiocyanate (FITC–Thy-1.1). (C) Flow cytometric analysis of DNA content/cell cycle progression of Bcl11aL/L bone marrow cells following the retroviral infection protocol detailed for panel B. Detergent-permeabilized cells were stained with propidium iodide and analyzed as described in Materials and Methods, using CellQuest software to calculate percentages of cells in the G1 (M1), G2/M (M2), and S (M1-M2 interval) phases. (D) Cre recombinase-mediated deletion of loxP (L)-modified Bcl11a alleles (Bcl11aL/L ) blocks Vκ-Jκ recombination in pre-B/IL-7 bone marrow (BM) cultures. Following retroviral infection as detailed for panel B, semiquantitative and real-time PCRs of genomic DNAs were performed on samples serially diluted 5-fold (triangles). DNAs amplified semiquantitatively were gel fractionated and either directly visualized by ethidium bromide staining (top and bottom panels) or visualized following hybridization as described for panel A (middle panel). PCRs of Cμ served as loading controls. Real-time PCR values, shown as fold reductions (Fold ↓) below corresponding lanes, were determined for each DNA dilution by dividing each MIT control value by the corresponding MIT-Cre value. The analyses are representative of 4 independent experiments performed with 4 separate BM isolates.
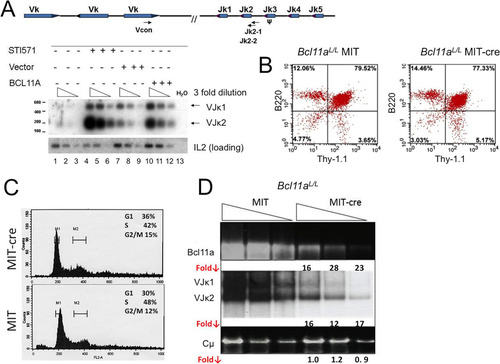
To address the issue in normal B cells, we employed mice with loxP (L) sites flanking exon 1 of the Bcl11a gene (Bcl11aL /L ) (Citation27, Citation31, Citation35). Bone marrow cultures (Citation10, Citation44) were enriched by use of beads for B220+ cells in culture with interleukin-7 (IL-7). The cells were then infected with either empty vector (MIT; mouse stem cell virus–internal ribosomal entry site–Thy-1.1) or an equivalent retrovirus which expresses Cre recombinase (MIT-Cre). Within 3 days, the MIT-Cre-infected cells underwent apoptosis and died (data not shown). To overcome this and to allow for sufficient numbers of viable cells to be harvested, we transduced the cells with a retrovirus (MSCV-Bcl-xL) that encodes the antiapoptotic Bcl-xL factor and cultured these cells for 2 days prior to superinfection with either MIT or MIT-Cre. Proliferation was rescued in the MIT-Cre cultures, and cells were collected 3 days after secondary infection for analysis.
Taking advantage of the Thy-1.1 marker carried by both vectors, we observed by flow cytometry that control and Cre-deleted B220+ B cell progenitors showed comparable transduction efficiencies (>75%) (). Since Cre-MIT-infected cells required Bcl-xl expression to bypass apoptosis, it was crucial to check their proliferative status, particularly since both V(D)J recombination and RAG2 expression are regulated at the G1 phase of the cell cycle (Citation45). As shown in , comparable cell cycle progressions, within normal limits for diploid cells, were observed for control and Cre-deleted cultures.
To quantify the extent of Cre-mediated Bcl11aL/L deletion and VJκ reduction at the DNA level, we employed 5-fold dilutions of genomic DNA analyzed by both semiquantitative and real-time PCR (). Normalized real-time fold reduction values (Fold ↓) given under corresponding ethidium bromide-stained, semiquantitative PCR gels indicate that VJκ rearrangement was strongly reduced (∼15-fold) in direct correlation with Bcl11a deletion (∼22-fold), whereas the Cμ control was unaffected.
BCL11A modulates transcription critical to V(D)J recombination.
V(D)J recombination initiates at the pro-B cell stage and is completed at the pre-B cell stage. We employed STI571 treatment of stably pXY-BCL11A-XL-Puro-transformed pro-B (220-8) and pre-B (A70) murine Abelson cell lines to measure the consequences of BCL11A overexpression on RAG1/2 as well as on factors previous implicated in V(D)J recombination. As shown in , STI571 treatment led to the anticipated strong induction of RAG1/2 at the anticipated pre-B cell stage (lane 8) but also showed their robust induction in pro-B cells (lane 4). Both interferon regulatory factor 4 (IRF4) and IRF8 were upregulated strongly in pre-B cells but only marginally in pro-B cells, consistent with previous observations that these TFs are essential for light chain recombination (Citation46). Reciprocally, Spi-B, which is modestly upregulated by BCL11A in pro-B cells, was strongly downregulated in pre-B cells. Perhaps BCL11A-XL downregulation of Spi-B, which along with IRF4 is sufficient to induce Igκ germ line transcripts (Citation43), was rate limiting. Expression of PAX5, FOXO1, and FOXP1, key transcriptional regulators of V(D)J recombination (Citation9, Citation47, Citation48), was upregulated by BCL11A-XL in pre-B cells. Germ line transcription of gVk, which reflects chromatin accessibility of the Jκ locus (Citation9), or of the λ5 chain of the pre-B cell receptor (pre-BCR) was not affected by BCL11A-XL overexpression. The other pre-BCR component, Vpre-B, was modestly reduced.
FIG 7 Comparison of BCL11A-XL overexpression levels following induction of V(D)J recombination-associated genes and following isolation of RNA from lymphoid cell types. Pro-B (220-8) (left) and pre-B (A70) (right) cell lines were either treated with STI571 (3 μM for 20 h) or stably transduced with BCL11A-XL (XL) or empty virus (M) as described in the legend to and then subjected to semiquantitative RT-PCR analysis. cDNAs were amplified under linear conditions, gel fractionated, and visualized by ethidium bromide staining. GAPDH was the loading control.
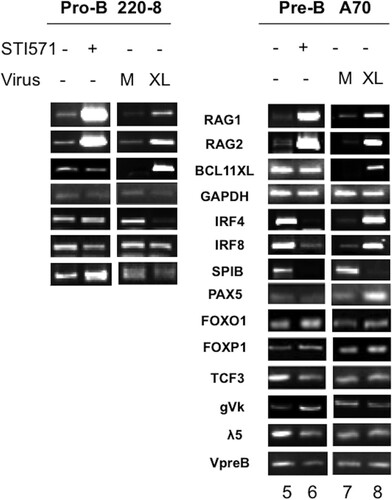
Transcript modulation following STI571 treatment correlated only modestly with that induced by BCL11A-XL overexpression, emphasizing the different mechanisms of these two RAG activators. For example, and as observed in other Abelson pre-B cell lines (Citation43), STI571 induced expression of Igκ germ line transcription (). But in contrast to a previous report (Citation43), we observed robust STI571 repression of IRF4 and IRF8 in A70 cells but only modest effects in pro-B 200-8 cells (Citation43). In our hands, massive cell death occurred within 24 h of STI571 treatment of 200-8 transfectants (data not shown), limiting our ability to collect data from these cells (). However, constitutive expression of the Bcl2 transgene in A70 cells (Citation40) allowed us to analyze events even 4 to 6 days following STI571 treatment.
Genome-wide BCL11A target genes include previously established regulators of B cell and pDC development.
Genome-wide ChIP-seq confirmed BCL11A binding to the RAG1/2 locus but also identified in NALM6 pre-B and RAJI B cells a strongly bound region within the single RAG1 exon ( and ). Over an ∼1-kb stretch centered by ChIP-seq, we identified 8 repeats of 8 to 12 bp that shared strong similarity with one another () as well as with the BCL11A-XL binding site consensus deduced from the EMSA data shown in . The Rag1 internal site, as with the promoter site, contained a perfect consensus PU.1 motif (). It is probable that the enhanced BCL11A avidity for this region was provided by this tandem array. Further studies are required to determine whether this is a bona fide enhancer.
FIG 8 ChIP-seq analysis of genome-wide BCL11A target binding in human lymphoid cell lines. (A) ChIP-seq analysis of BCL11A was performed in the human pre-B cell line NALM6 and the human Burkitt's lymphoma line RAJI (all peak scores were ≥10). A target gene was defined by a binding site occurring within 50 kb upstream of and within the body of the gene. Analysis of the false-discovery rate and associated q values was performed using Benjamini-Hochberg statistics. BCL11A directly bound RAG1 and genes for TFs known to control RAG1/2 expression, including IKZF1/IKAROS, FOXO1, PAX5, TCF3, SPIB, and IRF4. BCL11A also directly bound the surrogate light chain VPREB1 promoter region. (B) Alignment of BCL11A-bound RAG1 target sites by ChIP-seq. Over an ∼1-kb stretch, we identified 8 repeats of 10 to 12 bp that shared strong similarity with one another as well as with the BCL11A-XL binding site consensus deduced from the EMSA data shown in . The Rag1 internal site, as with the promoter site, contained a perfect consensus PU.1 motif. (C) Microarray analysis of human peripheral blood showing high expression of BCL11A transcripts in B cells (B) and plasmacytoid dendritic cells (pDCs) but not in monocytes (mono), NK cells, or T cells (CD4 or CD8). The microarray profile corresponds directly to what has been observed by analyzing BCL11A protein expression using the anti-BCL11A Ab123 monoclonal antibody (Citation31, Citation33). Two BCL11A target genes expressed highly in pDCs (Spi-B and IRF-8) have been shown to regulate the development of pDCs (Citation31, Citation33).
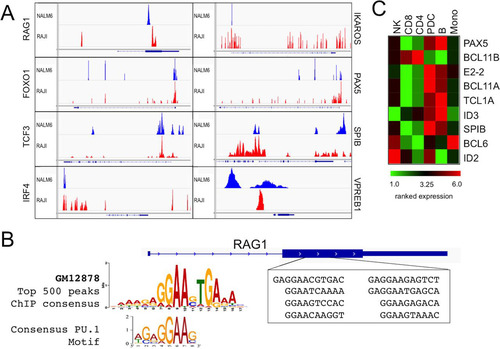
ChIP-seq also identified gene-proximal regions for several key TFs (), including FOXO1, IKZF1/IKAROS, TCF3/E2A, SPI-B, VPREB1, IRF4, and PAX5 (; discussed further below).
DISCUSSION
BCL11A was identified as a proto-oncogene implicated in multiple B cell malignancies (Citation24 Citation– Citation30) and represents one of ∼200 “ultraconserved” genes in the human genome (Citation20). We initially identified RAG1 as a potential target gene of the major BCL11A isoform (BCL11A-XL) by microarray analysis (). Both it and RAG2 were confirmed as targets by a combination of BCL11A-XL overexpression and shRNA knockdown ( and ). BCL11A-XL might regulate RAG either directly, via targeting cis-regulatory elements, or indirectly, by affecting other pathways. We observed by both BCL11A chromatin occupancy and in vitro DNA binding approaches that BCL11A was recruited to chromatin, within the RAG1 promoter and the Erag enhancer but not within the RAG2 promoter (). Transcriptional reporter studies in mouse pre-B cells suggested that two BCL11A binding sites within the Rag1 promoter synergize to activate transcription (). The more promoter-distal BCL11A binding site conveyed much stronger (>4-fold) Erag enhancement (). Furthermore, BCL11A-XL failed to drive Rag2 transcription unless Erag was appended to the construct ().
Genome-wide ChIP-seq of sequences derived from human pre-B and mature B cell lines confirmed BCL11A binding of the RAG locus but also identified strong binding to a tandem array of 8 putative BCL11A-XL sites (), whose consensus was established by EMSA and ChIP-PCR ( and and ). Genome-wide ChIP data (Citation38) indicate that ∼500 of the strongest BCL11A sites within the genome overlap PU.1 sites (). Similarly, equivalent genome-wide ENCODE data for PU.1 ChIP-seq (Citation38) indicated with high statistical significance (P < 1.0−11) that a large proportion of PU.1 peaks are in the close vicinity of BCL11A peaks. Together, these data show that it is highly probable that BCL11A and PU.1 broadly collaborate or antagonize transcription via closely spaced or even overlapping composite sites.
Unexpectedly, we observed strong repression of RAG1 promoter-driven transcription in epithelial and fibroblast-derived cell lines, with the more promoter-proximal BCL11A-XL binding site being sufficient to target the activity ( and data not shown). Potential physiologic relevance is provided by the observations that the RAG1 promoter is not lymphoid cell restricted (Citation11, Citation14) and that RAG expression is observed in several nonlymphoid malignant cell types (Citation45, Citation48 Citation– Citation50) as well as in plasmacytoid dendritic cells (pDCs) (Citation51, Citation52). In addition, Bcl11a expression within T lineages is limited to DN2 thymocytes, the stage at which V(D)J recombination of the TcRβ locus initiates (Citation53). In DN3 thymocytes, Bcl11a is extinguished, and expression of its highly similar paralogue, Bcl11b, ensues (Citation54). Although Bcl11b has not previously been implicated in Rag control, Bcl11b −/− thymocytes are impaired in Vβ-Dβ recombination (Citation39).
The domain responsible for both non-B cell repression and pre-B cell activation of BCL11A-XL maps to the N-terminal 80 amino acids. In this region, a single point mutation (K5/A) within the highly conserved MSRRK motif () abrogated both transcriptional activities while retaining normal BCL11A-XL subnuclear localization ( and and data not shown). This motif defines a superfamily of TFs crucial to the development of several hematopoietic lineages (Citation23, Citation25, Citation37, Citation41, Citation55) (). The N terminus, extended to include the equally conserved C2HC zinc finger (), is required for BCL11A-XL to homodimerize with all BCL11A isoforms (Citation37) and with several corepressor complexes, including NuRD, LSD1/CoREST, NCoR/SMRT, and SIN3 (Citation42, Citation48). In support of this, we observed enhanced BCL11A-XL repression of RAG1 following coexpression with MTA1, a single member of the 12-membered NuRD complex, which we suspect to be limiting under our conditions ( and data not shown). Collectively, the results suggest that the N-terminal domain of BCL11A-XL is employed in a context-dependent fashion for assembly of both well-characterized corepressor complexes and coactivation complexes whose members remain to be defined.
We observed modulation of V(D)J recombination proportional to BCL11A-XL overexpression in cultured 220-8 pro- and A70 pre-B cells (, , and ) or following Cre-mediated knockdown in cultured explants of Bcl11aL/L bone marrow-derived pre-B cells ( and ). The robustness of the effect, particularly in the normal pre-B cells that dominate IL-7 cultures, may derive both from direct transcriptional activation of RAG and by upregulation of additional TFs that either perform a redundant function or act to promote chromatin accessibility of V(D)J. This hypothesis was buttressed by both BCL11A-XL overexpression/RT-PCR and BCL11A-XL-mediated ChIP-seq studies ( and ). We observed that BCL11A-XL directly upregulated two members of the forkhead box TF family. Both FOXP1 and FOXO1 directly transactivate RAG via binding to Erag (Citation9, Citation10, Citation13, Citation56). The observation that BCL11A directly targets PAX5 has consequences for V(D)J recombination. PAX5 upregulates the direct BCL11A target, IKZF1/IKAROS (Citation47), to silence transcription of the surrogate light chain (Citation57). This in turn results in reduction of the pre-BCR complex and, ultimately, to a complete block of B cell differentiation at the pre-B stage (Citation58). Further, as shown in , BCL11A upregulates (either via PAX5 or in parallel) expression of both IRF8 and IRF4 (Citation46, Citation59)—direct targets of pre-BCR signaling (Citation60)—to allow progression through the pre-B cell stage (Citation46). Although BCL11A-XL bound strongly within the TCF3 regulatory region, the effect on expression was modest. TCF3-deficient CLPs fail to express RAG1 and several other lymphoid cell-restricted genes (Citation59).
Elevation and concerted action of these and similar factors that upregulate RAG may be sufficient for the Vκ-Jκ inversion of the exogenous recombination target, pMX-INV ( and ). IRF4 and IRF8 are required for chromatin accessibility of the endogenous V(D)J locus (Citation46). V(D)J recombination and B cell development are blocked in IRF4/IRF8 double-knockout mice at the pre-B cell stage (Citation46). Accessibility and recombination of Vκ-Jκ have been correlated with selected posttranslational modifications of nucleosomal histone tails (Citation61 Citation– Citation63). Ectopic expression of IRF4 in IRF4 −/− IRF8 −/− pre-B cells was shown to increase acetylation of H3K9 and H4K14 as well as trimethylation of H3K4—marks associated with active chromatin (Citation46). However, induction of Jκ-Cκ germ line transcription, typically associated with locus accessibility (Citation64), was not observed following BCL11A-XL overexpression (). Perhaps direct BCL11A repression of Spi-B ( and ), which along with IRF4 has been reported to be sufficient for inducing Igκ germ line transcripts (Citation44), is rate limiting under these conditions. Resolution will require a careful assessment of BCL11A-mediated germ line transcription and histone epigenetic alterations across this locus. We did observe BCL11A-mediated direct transcriptional upregulation of the pre-B antigen receptor chain, Vpre-B, but not its pre-BCR partner, λ5 ( and ). While the λ5 protein alone is unable to bind IgM heavy chains, VpreB alone is capable of forming this critical signaling component (Citation65).
Consistent with the work of Yu et al. (Citation33), we observed significantly reduced proliferation and increased apoptosis relative to those of controls in floxed cultures of Bcl11aL/L B220+ bone marrow pre-B cells ( and data not shown). In further agreement (Citation33), prior infection of Bcl11aL/L cells with a Bcl-xL-expressing virus bypassed Cre-mediated apoptosis and proliferation. Bcl11a−/− fetal liver hematopoietic progenitors suffer a severe pre-pro-B cell block (Citation39) as a consequence of a proliferative defect in IL-7 receptor (IL-7R) signaling or a defect in V(D)J recombination (Citation2). Observations of ours and others (Citation24, Citation33, Citation53, Citation66) that Bcl11a expression initiates at the hematopoietic stem cell (HSC) stage suggest that the initial D-J recombination step of antigen receptor assembly should be impaired by conditional Bcl11a deficiency. Indeed, FOXP1, a target of BCL11A-XL ( and ), activates this earliest RAG-mediated step of V(D)J joining (Citation13).
While a requirement for BCL11A in T and NK lineage development is controversial, its status as an essential regulator of the pDC lineage is indisputable (Citation31 Citation– Citation33). pDCs, which coordinate both innate and adaptive antiviral immunity, share characteristics with B cells, including RAG1 expression, RAG1/2-mediated but nonproductive DJ rearrangements, prototypic B cell markers (e.g., B220), and, most relevant here, critical TFs conventionally associated with B lymphocyte development (Citation31, Citation51). We and others (Citation31, Citation32) previously demonstrated that BCL11A is critical to pDC development and function. We also confirmed that DCs and B cells share critical TFs () regulated indirectly or directly by BCL11A. These results have prompted us to hypothesize a central developmental route for these two hematopoietic lineages. Further work is essential to deconvolute the potential origin of a lymphoid cell-derived pDC subset in vivo and to evaluate a requirement for BCL11A in its transcriptional specification.
MATERIALS AND METHODS
Mice.
C57BL/6 mice with loxP sites flanking exons 1 and 2 of Bcl11a were generated and genotyped as previously described (Citation31). Mice were bred and housed in the University of Texas animal research facility. All experiments were approved by the IACUC. Four- to 6-month-old mice were used for bone marrow cultures.
Cell culture.
Human 293T, human Phoenix, mouse NIH 3T3, and human HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (GibcoBRL) containing 10% fetal bovine serum (FBS) (HyClone), 100 U/ml penicillin, and 100 μg/ml streptomycin. Human NALM6 pre-B and mouse A70 pre-B cell lines were maintained in RPMI 1640 medium (GibcoBRL) containing 10% FBS (HyClone), 50 μM β-mercaptoethanol, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Pre-B cells were cultured as described previously (Citation44). B220+ cells were isolated from mouse bone marrow (BM) by use of B220-labeled magnetic beads (Miltenyi Biotech). Purified cells were overlaid on an irradiated S17 stromal cell layer. The cells were maintained in pre-B cell medium, i.e., Opti-MEM (Gibco) medium containing 15% FBS, 50 μM β-mercaptoethanol, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 5 ng/ml interleukin-7 (IL-7) (R&D Systems). Cells were passaged every 3 days onto a new S17 stromal cell layer.
Retroviruses and transductions.
Human BCL11A-XL cDNA was subcloned into the pXY-IRES-puro (pXY-puro) vector, which was a kind gift from Louis Staudt, National Cancer Institute (Citation67). Mouse stem cell-based retroviruses (MIT; mouse stem cell virus–internal ribosomal entry site–Thy-1.1) encoding the Cre recombinase (MIT-Cre) or the parental vector (MIT) (Citation10) were gifts of Robert Rickert, Burnham Institute. A mouse stem cell virus encoding the antiapoptotic factor Bcl-xL was obtained from Toku-E Pharm (MSCV-Bcl-xLPuro; HCD57). Viral DNAs were transfected into the Phoenix-eco cell packaging line by use of Fugene (Roche) to produce retroviral particles. Viral particles were harvested 48 h after transfection and centrifuged to remove cell debris. The viral supernatants were ultracentrifuged at 20,000 rpm for 2 h and resuspended in a proper amount of serum free Opti-MEM medium (Invitrogen).
For pXY-puro viruses, titers were established by infecting NIH 3T3 cells and counting the numbers of puromycin-resistant cells. Viral particles were incubated with 10 μg/ml of DOTAP (Roche) for 10 min. Various human pre-B and B cell lines were transduced in 24-well culture plates at 25°C by spin inoculation at 1,000 × g for 90 min at 30°C. Forty-eight hours after transduction, cells were selected in the presence of 0.5 to 2 μg/ml of puromycin (Sigma).
MIT viruses were preincubated with Polybrene (8 μg/ml) and then diluted 2-fold with Opti-MEM. Following culture (for less than 5 passages) as described above, B220+ BM pre-B cells were plated at a density of ∼1 × 106 cells/ml and 1 day later were transduced with MSCV-Bcl-xL by spin inoculation as described above. Following incubation for 1 h at 37°C, cells were collected, resuspended in pre-B cell medium, and then cultured for an additional 48 h. Cells were then infected at a high multiplicity of infection (MOI) with MIT or MIT-Cre. Culture was continued with IL-7 for 3 days, and then cultures were collected for PCR and RT-PCR experiments.
Inducible shRNA silencing of BCL11A.
Two complementary shRNA template oligonucleotides targeting exon 2 of BCL11A were synthesized and cloned into the pRSMX-PG vector (Citation68) by use of HindIII and BglII sites. The pRSMX-PG-BCL11A-shRNA retrovirus was produced in Phoenix cells as described above and then used to infect a Tet-on human B cell line, BJAB, which expresses the Tet repressor. Transduced BJAB cells were selected in the presence of puromycin for 6 days. BCL11A-specific shRNA expression was induced with different concentrations of doxycycline, and samples were tested at regular intervals for 48 h. Levels of BCL11A expression were measured by RT-PCR and Western blot analysis. The following oligomers were used for the BCL11A shRNA template: forward, 5′-GATCACCCCAGCACTTAAGCAAATTCAAGAGATTTGCTTAAGTGCTGGGGTTTTTTTGGAAA-3′; and reverse, 5′-AGCTTTTCCAAAAAAACCCAGCACTTAAGCAAATCTCTTGAATTTGCTTAAGTGCTGGGGTG-3′. BglII and HindIII sites were used for cloning.
Plasmid construction.
CMV10-BCL11A-XL and CMV10-BCL11A-S were constructed as follows. BCL11A-XL and BCL11A-S were amplified by PCR with the same forward primer (5′-GCCAAAGCTTATGTCTCGCCGCAAGCAAGGC-3′) and different reverse primers (for the BCL11A-XL isoform, 5′-CCCAGGATCCTATTCAGTTTTTATATCATTATTCAAC-3′; and for the S isoform, 5′-CCCAGGATCCTCAAATTTTCTCAGAACTTAAG-3′). The PCR products were digested with BamHI and HindIII and cloned into CMV10 (Sigma), which contains a sequence for a 3×Flag tag. The pXY-BCL11A-XL plasmid was constructed using CMV10-BCL11A-XL as a template and the following PCR primers: forward, 5′-CCGGGATCCATGGACTACAAAGACCATGAC-3′; and reverse, 5′-GGACGCGGCCGCTATTCAGTTTTTATATCATTATTC-3′. The PCR products were digested with BamHI and NotI and cloned into the pXY-IRES-puro plasmid by use of BglII and NotI sites. pEGFPC1-BCL11A and BCL11A-XL-K5N were generated by site-directed mutagenesis using a QuikChange site-directed mutagenesis kit and protocol from Stratagene. The following primers were used: forward, 5′-CCATGTCTCGCCGCAACCAAGGCAAACCCCCAGC-3′; and reverse, 5′-GCTGGGGTTTGCCTTGGTTGCGGCGAGACATGG-3′. A deletion mutant, pEGFPC1-BCL11ABCL11A-XLdelN80, was constructed by PCR with the following primer pair: forward, 5′-GGCGTCGACAGCGAACACGGAAGTCCC-3′; and reverse, 5′-CCCAGGATCCTATTCAGTTTTTATATCATTATTCAAC-3′. The PCR products were digested using SalI and BamHI and cloned into the plasmid vector pEGFPC1. The final clones were verified by sequencing.
ChIP and EMSA.
Chromatin immunoprecipitation (ChIP) was performed according to the instructions for a ChIP kit (Upstate Biotechnology). Human pre-B and B cell lines (Nalm6 and RAJI) were cross-linked by incubation at room temperature for 8 min with a final concentration of 1% formaldehyde. Fixed cells were washed twice with ice-cold phosphate-buffered saline (PBS) and lysed in SDS lysis buffer (1% SDS, 10 mM EDTA, and 50 mM Tris, pH 8.1). Lysates were sonicated for 0.5 min six times to a modal distribution of ∼1,000 bp. Sonicated chromatin was diluted 10-fold in a dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl) and then immunoprecipitated with purified rabbit IgG or with an anti-BCL11A-XL rabbit polyclonal antibody (BL1797; Bethyl). Antibody-chromatin complexes were washed with a low-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 150 mM NaCl), a high-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl), a LiCl wash buffer (0.25 M LiCl, 1% IGEPALCA630, 1% deoxycholic acid [sodium salt], 1 mM EDTA, 10 mM Tris, pH 8.1) and a TE wash buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA). These complexes were eluted in elution buffer (1% SDS, 0.1 M NaHCO3) and reverse cross-linked at 65°C for 6 to 12 h in the presence of 0.2 M NaCl. After proteinase K treatment for 1 h at 45°C, DNA was recovered by use of PCR purification columns (Invitrogen) and then used for PCR analysis. The RAG1 and RAG2 promoters and the Erag enhancer region were detected using the following primers: RAG1 forward, 5′-CATTCTCAGGGAGGGAACTG-3′; RAG1 reverse, 5′-GGAGGGCTAACCACAAATGA-3′; RAG2 forward, 5′-GTGGTCTCTGCTTCAGGACA-3′; RAG2 reverse, 5′-AGCAACAATGGCAACACAAT-3′; Erag forward, 5′-GCACTGCAAATGGCCTGTGAAC-3′; and Erag reverse, 5′-GAGACCAGAGGGCTTAACATTG-3′. ChIP-PCR of RAG1, RAG2, and Erag produced 161-bp, 180-bp, and 196-bp products, respectively. Thermal cycling conditions for the RAG1 and RAG2 promoters were 95°C for 5 min, 35 cycles of 95°C for 10 s, 55°C for 15 s, and 72°C for 10 s, and then 72°C for 7 min. The PCR conditions for the Erag enhancer were 95°C for 5 min, 35 cycles of 95°C for 1 min, 57°C for 1 min, and 72°C for 1 min, and then 72°C for 7 min.
To construct EMSA probes, the proximal promoter region (positions −243 to +123) of the mouse Rag1 gene (Citation40) was amplified from the plasmid template R1P-Luc by PCR using the following primers: RAG1 forward, 5′-CATTCTCAGGGAGGGAACTG-3′; and RAG1 reverse, 5′-GGCAAAGTGTGTTCTCTGCTC-3′. The Erag enhancer (Citation12) probe was a 196-bp fragment extending from the KpnI site to the end of region A (Citation13). The probe for the distal promoter region of Rag1 (positions −713 to −563) was generated by KpnI-HindIII digestion of plasmid −713-R1P-Luc. Probes were end labeled with 32P by use of polynucleotide kinase and then purified using Bio-Spin columns (Bio-Rad) and 6% native gels. Reaction mixtures consisted of 5- to 10-μg aliquots of nuclear extracts prepared from various B cell lines, probes, and a binding buffer (20 mM HEPES, pH 7.9, 40 mM KCl, 6 mM MgCl2, 1 mM dithiothreitol [DTT], 0.1% NP-40, 3 mg/ml bovine serum albumin, 10% glycerol, 2% Ficoll, 50 μg/ml of sonicated salmon sperm DNA, and protease inhibitor cocktail). Each binding mixture was incubated at room temperature for 30 min. For the antibody binding competition assays, 100 ng of BCL11A polyclonal antibody (BL1797) or monoclonal antibody Ab123 (Citation69) or an equal amount of a control rabbit IgG antibody or an irrelevant tubulin MAb was added at the initiation of each binding reaction. Cold competitions were carried out with increasing molar ratios of wild-type (5′-GGTCAGTCCGTTTCCAACTTCCTCCAGCAG-3′) and mutant (5′-GGTCAGTCCGTGGGTAAGCCCCTCCAGCAG-3′) duplexed, gel-purified oligonucleotides spanning the putative BCL11A binding site at Erag nt 121 to 131. (Mixtures were fractionated in 6.0 to 7.5% nondenaturing acrylamide gels, and the gels were dried and analyzed using a phosphorimager [Molecular Dynamics].)
Luciferase assays.
Pre-B cells (A70, A70-BCL11A-XL, and A70-BCL11A-XL-K5/A) and non-B cells (293T, COS7, and NIH 3T3) were plated at ∼1 × 106 cells per well in 12-well plates. Twenty-four hours later, pre-B cells were transiently transfected with 1 μg of firefly luciferase reporter (detailed in ), 5 ng of Renilla luciferase, and in some cases 1 μg of pFLAG-MTA1 by use of a mouse B cell Nucleofector kit (VPA-1010, program Z-01) and cell line kit (VCA-1003, program M13) according to the manufacturer's recommended protocol. Transfection efficiencies (determined by pEG-GFP transfection in parallel assays) achieved in pre-B cells were 40 to 50%. Empty vector plasmid DNA was used to keep the total amounts of transfected DNA equal under each experimental condition.
Non-B cells were transiently transfected with 1 μg of firefly luciferase reporter construct (detailed in ), 5 ng of Renilla luciferase, and either a BCL11A-XL wild-type or BCL11A-XL-K5/A expression vector over a concentration range of 0.1 to 1.5 ng. An empty pCMV10 construct was then added to each transfection mix to ensure that final DNA concentrations were equal.
Forty-eight hours after transfection, cells were harvested for dual-luciferase reporter assays (Promega) by using cell lysis and luciferase measurements as described in the manufacturer's manual. Values were normalized by determining the ratio of firefly to Renilla luciferase activity for each transfection mixture. To determine fold expression, the firefly/Renilla luciferase activity ratio for experimental samples (i.e., those with BCL11A-XL overexpression, the Renilla vector, and the respective firefly luciferase RAG reporter) was compared to the firefly/Renilla luciferase activity ratio for basal luciferase expression samples (i.e., those without BCL11A-XL overexpression). Western blotting confirmed that BCL11A-XL protein lysate levels corresponded to the concentration of input DNA.
Microarray analysis.
Briefly, 3 μg of mRNAs of various human pre-B and B cell lines was reverse transcribed using aminoallyl UTP and deoxynucleoside triphosphates (dNTPs). Synthesized cDNA was hydrolyzed and column purified (Invitrogen). Each cDNA sample was coupled with Cy3 or Cy5 dye, mixed, and hybridized to full-genome human array slides. Microarray slides were scanned using a GenePix 400a microarray scanner. A detailed protocol has been previously described (Citation73).
Flow cytometry.
A70-INV and A70-INV-BCL11A-XL cells were incubated in the absence or presence of 3 μM STI571 (Novartis) for 3 days. The harvested cells were washed two times with ice-cold PBS and resuspended in PBS at a concentration of ∼1 × 106 cells/ml. A minimum of 5,000 cells per sample were used for each analysis. Flow cytometry (BD FACSCalibur flow cytometry system) was used to analyze the samples for GFP-positive signals. Cell debris and dead cells were excluded from the analysis by forward and side scatter analysis. The parental A70-INV cells were used for GFP gating. Data were analyzed using Cell Quest software. Bcl11aL/L/MIT and Bcl11aL/L/MIT cultured bone marrow cells were costained under similar conditions and stained with fluorescein isothiocyanate (FITC)-conjugated anti-Thy-1.1 (T1-A3; Abcam) and phycoerythrin (PE)-conjugated anti-B220 (RA3-6B2; BD Pharmingen). For DNA content/cell cycle analysis, cells were incubated for 15 min at 20°C in 300 to 500 μl propidium iodide–Triton X-100 staining solution (Roche). Data were acquired as described above.
Genomic DNA isolation and Southern blotting.
A DNeasy tissue kit (Qiagen) was used to isolate genomic DNA from cultured A70-INV cells or bone marrow-derived B220+ pre-B cells following previously published protocols (Citation40, Citation49), with some modifications. For genomic PCR, 100 to 200 ng of genomic DNA was PCR amplified and then subjected to electrophoresis in agarose gels. The scheme for detection of inversion of the ectopic locus by GFP flow cytometry or by PCR is illustrated in . PCR products were transferred to a nylon membrane for Southern blot analysis. The Jκ2-2 oligonucleotide probe was end labeled with T4 polynucleotide kinase. Southern blots were prehybridized overnight and then hybridized with probes for 3 h at 50°C in Ultra-Hyb solution (Ambion). Blots were washed once at room temperature and twice at 50°C in 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate)–0.1% SDS for 30 min each, and signals were detected using a phosphorimager (Molecular Dynamics). The following primers were used for PCR detection of inversion: pA, 5′-CACAACATCGAGGACGG-3′; and hCD4R, 5′-GCACCACTTTCTTTCCCTGA-3′. The primers for endogenous rearrangement and blotting were as follows: Vcon forward, 5′-CCGAATTCGSTTCAGTGGCAGTGGRTCWGGTAC-3′; Jκ2-1 reverse, 5′-GGTTAGACTTAGTGAACAAGAGTTGAGAA-3′; Jκ2-2 probe, 5′-CAAGAGTTGAGAAGACTACTTACGTTTT-3′; and reverse, 5′-TGCCACGTCAACTGATAATGAGCCCTCTC-3′. The amplification conditions were 30 cycles at 94°C, 90 cycles at 60°C, and 60 cycles at 72°C.
Semiquantitative RT-PCR.
RNA purification was performed with TRIzol LS as recommended by the manufacturer's instructions (Invitrogen). Pellets were resuspended in RNase-free water. For first-strand synthesis by reverse transcription, we used a SuperScript II First kit (Invitrogen). Serial dilutions of template cDNA were subjected to 30 cycles of PCR, using previously published amplification conditions (Citation9, Citation10, Citation13, Citation16, Citation20, Citation37, Citation70).
Quantitative PCR.
Either genomic DNA or cDNA transcribed with the Superscript RT III based-protocol described above was analyzed by quantitative real-time PCR performed on an Applied Biosystems ViiA7 thermocycler, using Sybr green chemistry (Applied Biosystems Sybr Green or Quantus Sybr green). All assays were performed in triplicate and analyzed by the standard curve method, with normalization to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) housekeeping gene (Citation71, Citation72).
Oligonucleotide primers.
Primers used for semiquantitative and quantitative PCRs were as follows (5′-to-3′ orientation): RAG1-F, CCAAGCTGCAGACATTCTAGCACTC; RAG1-R, CTGGATCCGGAAAATCCTGGCAATG; RAG2-F, CACATCCACAAGCAGGAAGTACAC; RAG2-R, GGTTCAGGGACATCTCCTACTAAG; IRF4-F, CCACGGACACACCTATGATG; IRF4-R, GGTCTGGAAACTCCTCACCA; IRF8-F, GGGCTGCCTAAGTTGTATG; IRF8-B, ACCACCCTGCTGTCAGGTAG; Spi-B-F, GAGGACTTCACCAGCCAGA; Spi-B-R, TGAGTTTGCGTTTGACCTTG; gVk-F, CCACATGCCTTTCTTCAGGGACAAGTG; gVk-R, TTATGTCGTTCATACTCGTCCTTGGTC; Foxp1-F, AAGGGGCAGTATGGACAGTG; Foxp1-R, CCCAGAGGTTCACTCCATGT; BCL11A-F, CCCAGAGTAGCAAGCTCACC; BCL11A-R, CAGGATCCTATTCAGTTTTTATATCATTATTC; FoxO1-F, CAAAGTACACATACGGCCAATCC; FoxO1-R, CGTAACTTGATTTGCTGTCCTGAA; E2A-F, TTGTGGACATTTTCTAGGCAG; E2A-R, ATGTGTGGTGGCCCACACTTG; Pax5-F, AGACAGGAAGCATCAAGCCTG; Pax5-R, CAGCGAGGCCATGGCTGAATA; Vpre-B-F, GTAGAGGCATGCCAGCCGGTGCT; Vpre-B-R, CTTGAAGCTTTCGAGGGACACGTGT; λ5-F, ACTGTCGGATCCTCGCAGAGCAGG; λ5-R, CAGTCAAGCTTCTATGAACATTCT; GAPDH-F, ACTTTGTCAAGCTCATTTCC; GAPDH-R, TGCAGCGAACTTTATTGATG; VCON-F, CCGAATTCGSTTCAGTGGCAGTGGRTCWGGTAC; and Jκ2-1-R, GGTTAGACTTAGTGAAGAGTTGAGAA.
Biochemistry.
Western blotting was carried out as previously described (Citation37). The following dilutions were used for each antibody: Flag, 1:5,000 (Sigma); β-tubulin, 1:2,000 (Santa Cruz); RAG1, 1:2,000 (554116; Pharmingen); BCL11A-XLc, 1:3,000 (BL1797; Bethyl); BCL11A, 1:2,000 (Ab123; prepared in our laboratory); lamin B, 1:1,000 (Santa Cruz); and cytochrome c, 1:1,500 (556432; Pharmingen).
Nuclear subcellular fractionation was carried out as a minor modification (Citation11) of the procedure of Reyes et al. (Citation26). Cytoplasmic contamination in the nuclear fraction will result in contamination within the soluble NP fraction (Citation5). Lamin B signals were used to evaluate contamination of nuclear fractions in the cytoplasmic fractions (Citation26). Cytoplasmic contamination was also assessed by anti-cytochrome c Western blotting.
ChIP followed by ChIP-seq.
ChIP assays for BCL11A were performed and analyzed as described previously (Citation31). ChIP-seq data were analyzed by deep sequencing, using Illumina sequencing technology as described above.
ACKNOWLEDGMENTS
We thank June V. Harriss for expert assistance in the generation of Bcl11a conditional knockout mice, Chhaya Das and Maya Ghosh for help with ChIP experiments and cell culture, and Paul Das for preparation of the manuscript. We thank Mark Schlissel for sharing luciferase constructs. The CAL1 cell line was kindly provided by Takahiro Maeda and Boris Reizis. Library preparation and Illumina ChIP- and RNA-seq assays were performed at the NGS core of the MD Anderson Cancer Center.
This work was supported by Lymphoma Research Foundation fellowship 300463 (to J.D.D.), NIAID, National Institutes of Health, award 1R21AI119368-01A1 (to G.C.I.), NIH grant R01CA31534, Cancer Prevention Research Institute of Texas (CPRIT) grants RP120348 and RP120459, and the Marie Betzner Morrow Centennial Endowment (to H.O.T.).
B.-S.L., J.D.D., and H.O.T. designed research; B.-S.L., J.D.D., B.-K.L., G.C.I., V.R.I., B.P.S., and A.L.S. performed research; B.-K.L. and J.D.D. contributed new reagents/analytic tools; B.-S.L., B.-K.L., V.R.I., A.L.S., G.C.I., J.D.D., and H.O.T. analyzed data; and B.-S.L., J.D.D., and H.O.T. wrote the paper.
Notes
* Present address: Baeck-Seung Lee, Department of Pathology and Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
Citation Lee B-S, Lee B-K, Iyer VR, Sleckman BP, Shaffer AL, III, Ippolito GC, Tucker HO, Dekker JD. 2018. Corrected and republished from: BCL11A is a critical component of a transcriptional network that activates RAG expression and V(D)J recombination. Mol Cell Biol 38:e00362-17. https://doi.org/10.1128/MCB.00362-17.
REFERENCES
- Desiderio S. 2010. Temporal and spatial regulatory functions of the V(D)J recombinase. Semin Immunol 22:362–369. https://doi.org/10.1016/j.smim.2010.09.001.
- Schatz DG. 2004. V(D)J recombination. Immunol Rev 200:5–11. https://doi.org/10.1111/j.0105-2896.2004.00173.x.
- Somasundaram R, Prasad MA, Ungerback J, Sigvardsson M. 2015. Transcription factor networks in B-cell differentiation link development to acute lymphoid leukemia. Blood 126:144–152. https://doi.org/10.1182/blood-2014-12-575688.
- Schatz DG, Ji Y. 2011. Recombination centres and the orchestration of V(D)J recombination. Nat Rev Immunol 11:251–263. https://doi.org/10.1038/nri2941.
- Oettinger MA, Schatz DG, Gorka C, Baltimore D. 1990. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science 248:1517–1523. https://doi.org/10.1126/science.2360047.
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. 1992. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68:869–877. https://doi.org/10.1016/0092-8674(92)90030-G.
- McBlane JF, van Gent DC, Ramsden DA, Romeo C, Cuomo CA, Gellert M, Oettinger MA. 1995. Cleavage at a V(D)J recombination signal requires only RAG1 and RAG2 proteins and occurs in two steps. Cell 83:387–395. https://doi.org/10.1016/0092-8674(95)90116-7.
- van Gent DC, McBlane JF, Ramsden DA, Sadofsky MJ, Hesse JE, Gellert M. 1995. Initiation of V(D)J recombination in a cell-free system. Cell 81:925–934. https://doi.org/10.1016/0092-8674(95)90012-8.
- Amin RH, Schlissel MS. 2008. Foxo1 directly regulates the transcription of recombination-activating genes during B cell development. Nat Immunol 9:613–622. https://doi.org/10.1038/ni.1612.
- Dengler HS, Baracho GV, Omori SA, Bruckner S, Arden KC, Castrillon DH, DePinho RA, Rickert RC. 2008. Distinct functions for the transcription factor Foxo1 at various stages of B cell differentiation. Nat Immunol 9:1388–1398. https://doi.org/10.1038/ni.1667.
- Fuller K, Storb U. 1997. Identification and characterization of the murine Rag1 promoter. Mol Immunol 34:939–954. https://doi.org/10.1016/S0161-5890(97)00000-X.
- Hsu LY, Lauring J, Liang HE, Greenbaum S, Cado D, Zhuang Y, Schlissel MS. 2003. A conserved transcriptional enhancer regulates RAG gene expression in developing B cells. Immunity 19:105–117. https://doi.org/10.1016/S1074-7613(03)00181-X.
- Hu H, Wang B, Borde M, Nardone J, Maika S, Allred L, Tucker PW, Rao A. 2006. Foxp1 is an essential transcriptional regulator of B cell development. Nat Immunol 7:819–826. https://doi.org/10.1038/ni1358.
- Kishi H, Jin ZX, Wei XC, Nagata T, Matsuda T, Saito S, Muraguchi A. 2002. Cooperative binding of c-Myb and Pax-5 activates the RAG-2 promoter in immature B cells. Blood 99:576–583. https://doi.org/10.1182/blood.V99.2.576.
- Lauring J, Schlissel MS. 1999. Distinct factors regulate the murine RAG-2 promoter in B- and T-cell lines. Mol Cell Biol 19:2601–2612. https://doi.org/10.1128/MCB.19.4.2601.
- Ochiai K, Maienschein-Cline M, Mandal M, Triggs JR, Bertolino E, Sciammas R, Dinner AR, Clark MR, Singh H. 2012. A self-reinforcing regulatory network triggered by limiting IL-7 activates pre-BCR signaling and differentiation. Nat Immunol 13:300–307. https://doi.org/10.1038/ni.2210.
- Wei XC, Dohkan J, Kishi H, Wu CX, Kondo S, Muraguchi A. 2005. Characterization of the proximal enhancer element and transcriptional regulatory factors for murine recombination activating gene-2. Eur J Immunol 35:612–621. https://doi.org/10.1002/eji.200425185.
- Wei XC, Kishi H, Jin ZX, Zhao WP, Kondo S, Matsuda T, Saito S, Muraguchi A. 2002. Characterization of chromatin structure and enhancer elements for murine recombination activating gene-2. J Immunol 169:873–881. https://doi.org/10.4049/jimmunol.169.2.873.
- Borghesi L, Aites J, Nelson S, Lefterov P, James P, Gerstein R. 2005. E47 is required for V(D)J recombinase activity in common lymphoid progenitors. J Exp Med 202:1669–1677. https://doi.org/10.1084/jem.20051190.
- Reynaud D, Demarco IA, Reddy KL, Schjerven H, Bertolino E, Chen Z, Smale ST, Winandy S, Singh H. 2008. Regulation of B cell fate commitment and immunoglobulin heavy-chain gene rearrangements by Ikaros. Nat Immunol 9:927–936. https://doi.org/10.1038/ni.1626.
- Fell HP, Smith RG, Tucker PW. 1986. Molecular analysis of the t(2;14) translocation of childhood chronic lymphocytic leukemia. Science 232:491–494. https://doi.org/10.1126/science.3961491.
- Richardson AL, Humphries CG, Tucker PW. 1992. Molecular cloning and characterization of the t(2;14) translocation associated with childhood chronic lymphocytic leukemia. Oncogene 7:961–970.
- Satterwhite E, Sonoki T, Willis TG, Harder L, Nowak R, Arriola EL, Liu H, Price HP, Gesk S, Steinemann D, Schlegelberger B, Oscier DG, Siebert R, Tucker PW, Dyer MJ. 2001. The BCL11 gene family: involvement of BCL11A in lymphoid malignancies. Blood 98:3413–3420. https://doi.org/10.1182/blood.V98.12.3413.
- Liu P, Keller JR, Ortiz M, Tessarollo L, Rachel RA, Nakamura T, Jenkins NA, Copeland NG. 2003. Bcl11a is essential for normal lymphoid development. Nat Immunol 4:525–532. https://doi.org/10.1038/ni925.
- Matthews JM, Kowalski K, Liew CK, Sharpe BK, Fox AH, Crossley M, MacKay JP. 2000. A class of zinc fingers involved in protein-protein interactions: biophysical characterization of CCHC fingers from fog and U-shaped. Eur J Biochem 267:1030–1038. https://doi.org/10.1046/j.1432-1327.2000.01095.x.
- Reyes JC, Muchardt C, Yaniv M. 1997. Components of the human SWI/SNF complex are enriched in active chromatin and are associated with the nuclear matrix. J Cell Biol 137:263–274. https://doi.org/10.1083/jcb.137.2.263.
- Sankaran VG, Xu J, Ragoczy T, Ippolito GC, Walkley CR, Maika SD, Fujiwara Y, Ito M, Groudine M, Bender MA, Tucker PW, Orkin SH. 2009. Developmental and species-divergent globin switching are driven by BCL11A. Nature 460:1093–1097. https://doi.org/10.1038/nature08243.
- Yin J, Zhang F, Tao H, Ma X, Su G, Xie X, Xu Z, Zheng Y, Liu H, He C, Mao ZJ, Wang Z, Chang W, Gale RP, Wu D, Yin B. 2016. BCL11A expression in acute phase chronic myeloid leukemia. Leuk Res 47:88–92. https://doi.org/10.1016/j.leukres.2016.05.018.
- Zhang X, Wang L, Wang Y, Shi S, Zhu H, Xiao F, Yang J, Yang A, Hao X. 2016. Inhibition of FOXQ1 induces apoptosis and suppresses proliferation in prostate cancer cells by controlling BCL11A/MDM2 expression. Oncol Rep 36:2349–2356. https://doi.org/10.3892/or.2016.5018.
- Zhou J, Yang Y, Zhang D, Zhou L, Tao L, Lu LM. 2017. Genetic polymorphisms and plasma levels of BCL11A contribute to the development of laryngeal squamous cell carcinoma. PLoS One 12:e0171116. https://doi.org/10.1371/journal.pone.0171116.
- Ippolito GC, Dekker JD, Wang YH, Lee BK, Shaffer AL III, Lin J, Wall JK, Lee BS, Staudt LM, Liu YJ, Iyer VR, Tucker HO. 2014. Dendritic cell fate is determined by BCL11A. Proc Natl Acad Sci U S A 111:E998–E1006. https://doi.org/10.1073/pnas.1319228111.
- Wu X, Satpathy AT, Kc W, Liu P, Murphy TL, Murphy KM. 2013. Bcl11a controls Flt3 expression in early hematopoietic progenitors and is required for pDC development in vivo. PLoS One 8:e64800. https://doi.org/10.1371/journal.pone.0064800.
- Yu Y, Wang J, Khaled W, Burke S, Li P, Chen X, Yang W, Jenkins NA, Copeland NG, Zhang S, Liu P. 2012. Bcl11a is essential for lymphoid development and negatively regulates p53. J Exp Med 209:2467–2483. https://doi.org/10.1084/jem.20121846.
- Basak A, Sankaran VG. 2016. Regulation of the fetal hemoglobin silencing factor BCL11A. Ann N Y Acad Sci 1368:25–30. https://doi.org/10.1111/nyas.13024.
- Xu J, Peng C, Sankaran VG, Shao Z, Esrick EB, Chong BG, Ippolito GC, Fujiwara Y, Ebert BL, Tucker PW, Orkin SH. 2011. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science 334:993–996. https://doi.org/10.1126/science.1211053.
- Xu J, Sankaran VG, Ni M, Menne TF, Puram RV, Kim W, Orkin SH. 2010. Transcriptional silencing of {gamma}-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev 24:783–798. https://doi.org/10.1101/gad.1897310.
- Liu H, Ippolito GC, Wall JK, Niu T, Probst L, Lee BS, Pulford K, Banham AH, Stockwin L, Shaffer AL, Staudt LM, Das C, Dyer MJ, Tucker PW. 2006. Functional studies of BCL11A: characterization of the conserved BCL11A-XL splice variant and its interaction with BCL6 in nuclear paraspeckles of germinal center B cells. Mol Cancer 5:18. https://doi.org/10.1186/1476-4598-5-18.
- ENCODE. 2004. The ENCODE (Encyclopedia of DNA Elements) Project. Science 306:636–640. https://doi.org/10.1126/science.1105136.
- Inoue J, Kanefuji T, Okazuka K, Watanabe H, Mishima Y, Kominami R. 2006. Expression of TCR alpha beta partly rescues developmental arrest and apoptosis of alpha beta T cells in Bcl11b−/− mice. J Immunol 176:5871–5879. https://doi.org/10.4049/jimmunol.176.10.5871.
- Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, Nuskey B, Sullivan KE, Pandita TK, Bassing CH, Sleckman BP. 2006. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature 442:466–470. https://doi.org/10.1038/nature04866.
- Tsang AP, Visvader JE, Turner CA, Fujiwara Y, Yu C, Weiss MJ, Crossley M, Orkin SH. 1997. FOG, a multitype zinc finger protein, acts as a cofactor for transcription factor GATA-1 in erythroid and megakaryocytic differentiation. Cell 90:109–119. https://doi.org/10.1016/S0092-8674(00)80318-9.
- Cismasiu VB, Adamo K, Gecewicz J, Duque J, Lin Q, Avram D. 2005. BCL11B functionally associates with the NuRD complex in T lymphocytes to repress targeted promoter. Oncogene 24:6753–6764. https://doi.org/10.1038/sj.onc.1208904.
- Muljo SA, Schlissel MS. 2003. A small molecule Abl kinase inhibitor induces differentiation of Abelson virus-transformed pre-B cell lines. Nat Immunol 4:31–37. https://doi.org/10.1038/ni870.
- Ma S, Pathak S, Trinh L, Lu R. 2008. Interferon regulatory factors 4 and 8 induce the expression of Ikaros and Aiolos to down-regulate pre-B-cell receptor and promote cell-cycle withdrawal in pre-B-cell development. Blood 111:1396–1403.
- Lin WC, Desiderio S. 1994. Cell cycle regulation of V(D)J recombination-activating protein RAG-2. Proc Natl Acad Sci U S A 91:2733–2737. https://doi.org/10.1073/pnas.91.7.2733.
- Lu R, Medina K, Lancki D, Singh H. 2003. IRF-4, 8 orchestrate the pre-B-to-B transition in lymphocyte development. Genes Dev 17:1703–1708. https://doi.org/10.1101/gad.1104803.
- Pridans C, Holmes ML, Polli M, Wettenhall JM, Dakic A, Corcoran LM, Smyth GK, Nutt SL. 2008. Identification of Pax5 target genes in early B cell differentiation. J Immunol 180:1719–1728. https://doi.org/10.4049/jimmunol.180.3.1719.
- Chen Z, Xiao Y, Zhang J, Li J, Liu Y, Zhao Y, Ma C, Luo J, Qiu Y, Huang G, Korteweg C, Gu J. 2011. Transcription factors E2A, FOXO1 and FOXP1 regulate recombination activating gene expression in cancer cells. PLoS One 6:e20475. https://doi.org/10.1371/journal.pone.0020475.
- Qiu X, Zhu X, Zhang L, Mao Y, Zhang J, Hao P, Li G, Lv P, Li Z, Sun X, Wu L, Zheng J, Deng Y, Hou C, Tang P, Zhang S, Zhang Y. 2003. Human epithelial cancers secrete immunoglobulin G with unidentified specificity to promote growth and survival of tumor cells. Cancer Res 63:6488–6495.
- Laszkiewicz A, Sniezewski L, Kasztura M, Bzdzion L, Cebrat M, Kisielow P. 2012. Bidirectional activity of the NWC promoter is responsible for RAG-2 transcription in non-lymphoid cells. PLoS One 7:e44807. https://doi.org/10.1371/journal.pone.0044807.
- Pelayo R, Hirose J, Huang J, Garrett KP, Delogu A, Busslinger M, Kincade PW. 2005. Derivation of 2 categories of plasmacytoid dendritic cells in murine bone marrow. Blood 105:4407–4415. https://doi.org/10.1182/blood-2004-07-2529.
- Sathe P, Vremec D, Wu L, Corcoran L, Shortman K. 2013. Convergent differentiation: myeloid and lymphoid pathways to murine plasmacytoid dendritic cells. Blood 121:11–19. https://doi.org/10.1182/blood-2012-02-413336.
- Seita J, Sahoo D, Rossi DJ, Bhattacharya D, Serwold T, Inlay MA, Ehrlich LI, Fathman JW, Dill DL, Weissman IL. 2012. Gene Expression Commons: an open platform for absolute gene expression profiling. PLoS One 7:e40321. https://doi.org/10.1371/journal.pone.0040321.
- Wakabayashi Y, Watanabe H, Inoue J, Takeda N, Sakata J, Mishima Y, Hitomi J, Yamamoto T, Utsuyama M, Niwa O, Aizawa S, Kominami R. 2003. Bcl11b is required for differentiation and survival of alphabeta T lymphocytes. Nat Immunol 4:533–539. https://doi.org/10.1038/ni927.
- Bond HM, Mesuraca M, Carbone E, Bonelli P, Agosti V, Amodio N, De Rosa G, Di Nicola M, Gianni AM, Moore MA, Hata A, Grieco M, Morrone G, Venuta S. 2004. Early hematopoietic zinc finger protein (EHZF), the human homolog to mouse Evi3, is highly expressed in primitive human hematopoietic cells. Blood 103:2062–2070. https://doi.org/10.1182/blood-2003-07-2388.
- Herzog S, Hug E, Meixlsperger S, Paik JH, DePinho RA, Reth M, Jumaa H. 2008. SLP-65 regulates immunoglobulin light chain gene recombination through the PI(3)K-PKB-Foxo pathway. Nat Immunol 9:623–631. https://doi.org/10.1038/ni.1616.
- Thompson EC, Cobb BS, Sabbattini P, Meixlsperger S, Parelho V, Liberg D, Taylor B, Dillon N, Georgopoulos K, Jumaa H, Smale ST, Fisher AG, Merkenschlager M. 2007. Ikaros DNA-binding proteins as integral components of B cell developmental-stage-specific regulatory circuits. Immunity 26:335–344. https://doi.org/10.1016/j.immuni.2007.02.010.
- Heizmann B, Kastner P, Chan S. 2013. Ikaros is absolutely required for pre-B cell differentiation by attenuating IL-7 signals. J Exp Med 210:2823–2832. https://doi.org/10.1084/jem.20131735.
- Jo SH, Schatz JH, Acquaviva J, Singh H, Ren R. 2010. Cooperation between deficiencies of IRF-4 and IRF-8 promotes both myeloid and lymphoid tumorigenesis. Blood 116:2759–2767. https://doi.org/10.1182/blood-2009-07-234559.
- Johnson K, Hashimshony T, Sawai CM, Pongubala JM, Skok JA, Aifantis I, Singh H. 2008. Regulation of immunoglobulin light-chain recombination by the transcription factor IRF-4 and the attenuation of interleukin-7 signaling. Immunity 28:335–345. https://doi.org/10.1016/j.immuni.2007.12.019.
- Fitzsimmons SP, Bernstein RM, Max EE, Skok JA, Shapiro MA. 2007. Dynamic changes in accessibility, nuclear positioning, recombination, and transcription at the Ig kappa locus. J Immunol 179:5264–5273. https://doi.org/10.4049/jimmunol.179.8.5264.
- Goldmit M, Ji Y, Skok J, Roldan E, Jung S, Cedar H, Bergman Y. 2005. Epigenetic ontogeny of the Igk locus during B cell development. Nat Immunol 6:198–203. https://doi.org/10.1038/ni1154.
- Perkins EJ, Kee BL, Ramsden DA. 2004. Histone 3 lysine 4 methylation during the pre-B to immature B-cell transition. Nucleic Acids Res 32:1942–1947. https://doi.org/10.1093/nar/gkh523.
- Angelin-Duclos C, Calame K. 1998. Evidence that immunoglobulin VH-DJ recombination does not require germ line transcription of the recombining variable gene segment. Mol Cell Biol 18:6253–6264. https://doi.org/10.1128/MCB.18.11.6253.
- Seidl C, Donner H, Petershofen E, Usadel KH, Seifried E, Kaltwasser JP, Badenhoop K. 1999. An endogenous retroviral long terminal repeat at the HLA-DQB1 gene locus confers susceptibility to rheumatoid arthritis. Hum Immunol 60:63–68. https://doi.org/10.1016/S0198-8859(98)00095-0.
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB. 2004. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A 101:6062–6067. https://doi.org/10.1073/pnas.0400782101.
- Shaffer AL, Yu X, He Y, Boldrick J, Chan EP, Staudt LM. 2000. BCL-6 represses genes that function in lymphocyte differentiation, inflammation, and cell cycle control. Immunity 13:199–212. https://doi.org/10.1016/S1074-7613(00)00020-0.
- Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, Lam LT, Dave S, Yang L, Powell J, Staudt LM. 2006. A loss-of-function RNA interference screen for molecular targets in cancer. Nature 441:106–110. https://doi.org/10.1038/nature04687.
- Nakamura T, Yamazaki Y, Saiki Y, Moriyama M, Largaespada DA, Jenkins NA, Copeland NG. 2000. Evi9 encodes a novel zinc finger protein that physically interacts with BCL6, a known human B-cell proto-oncogene product. Mol Cell Biol 20:3178–3186. https://doi.org/10.1128/MCB.20.9.3178-3186.2000.
- Verkoczy L, Ait-Azzouzene D, Skog P, Martensson A, Lang J, Duong B, Nemazee D. 2005. A role for nuclear factor kappa B/rel transcription factors in the regulation of the recombinase activator genes. Immunity 22:519–531. https://doi.org/10.1016/j.immuni.2005.03.006.
- Fu WJ, Hu J, Spencer T, Carroll R, Wu G. 2006. Statistical models in assessing fold change of gene expression in real-time RT-PCR experiments. Comput Biol Chem 30:21–26. https://doi.org/10.1016/j.compbiolchem.2005.10.005.
- Yuan JS, Reed A, Chen F, Stewart CN Jr. 2006. Statistical analysis of real-time PCR data. BMC Bioinformatics 7:85. https://doi.org/10.1186/1471-2105-7-85.
- Diaz E, Barisone GA. 2011. DNA microarrays: sample quality control, array hybridization and scanning. J Vis Exp 2011(49):e2546. https://doi.org/10.3791/2546.