
ABSTRACT
Heterotrimeric G proteins are signal transduction proteins involved in regulating numerous signaling events. In particular, previous studies have demonstrated a role for G-proteins in regulating β-catenin signaling. However, the link between G-proteins and β-catenin signaling is controversial and appears to depend on G-protein specificity. We describe a detailed analysis of a link between specific G-alpha subunits and β-catenin using G-alpha subunit genetic knockout and knockdown approaches. The Pasteurella multocida toxin was utilized as a unique tool to activate G-proteins, with LiCl treatment serving as a β-catenin signaling agonist. The results show that Pasteurella multocida toxin (PMT) significantly enhanced LiCl-induced active β-catenin levels in HEK293T cells and mouse embryo fibroblasts. Evaluation of the effect of specific G-alpha proteins on the regulation of β-catenin showed that Gq/11 and G12/13 knockout cells had significantly higher levels of active and total β-catenin than wild-type cells. The stimulation of active β-catenin by PMT and LiCl was lost upon both constitutive and transient knockdown of G12 and G13 but not Gq. Based on our results, we conclude that endogenous G-alpha proteins are negative regulators of active β-catenin; however, PMT-activated G-alpha subunits positively regulate LiCl-induced β-catenin expression in a G12/13-dependent manner. Hence, G-alpha subunit regulation of β-catenin is context dependent.
INTRODUCTION
The heterotrimeric G proteins represented by the Gs, Gi/o, Gq/11, and G12/13 families serve as essential links between the large number of G-protein-coupled receptors (GPCRs) that respond to many agonists and the activation of several defined intracellular signaling pathways (Citation1Citation–Citation3). Each G-protein family is characterized based on specific alpha subunits and is classically associated with a specific signaling pathway. Thus, Gs stimulation activates adenylate cyclase, whereas Gi stimulation inhibits adenylate cyclase activity (Citation4). Activation of Gq/11 stimulates phospholipase C (PLC) and subsequently protein kinase C and calcium-linked signaling (Citation5, Citation6), whereas the activation of the G12/13 family promotes the activity of Rho and cytoskeleton rearrangements (Citation7Citation–Citation11). Although each of the G-protein families is associated with specific signaling activation, there is some evidence demonstrating the interregulation of G-alpha subunits and cross-activation of signaling pathways. For instance, Gq, which stimulates PLC, can also activate Rho signaling proteins, which are classically assigned to G12/13 signaling (Citation12Citation–Citation16). The levels of G-alpha subunits have also been shown to have some degree of interregulation. For example, the short interfering RNA (siRNA) knockdown of Gq resulted in an upregulation of Gi subunits, leading to an activation of Gi-mediated signaling events (Citation17). As well as this interaction among G-protein signaling pathways, G-proteins also impinge on other signaling pathways. In particular, G-proteins are known to interact with and regulate the β-catenin signaling pathway.
β-Catenin is a multifunctional protein that can exhibit cell membrane, cytoplasmic, and nuclear localization to interact with a multitude of signaling cascades and transcription factors (Citation18Citation–Citation20). Interactions between β-catenin and G-proteins have been studied largely in the context of canonical Wnt signaling, an evolutionarily conserved pathway which involves the translocation of β-catenin into the nucleus, where it activates gene transcription (Citation21). In the absence of Wnt ligands, the level of cytoplasmic β-catenin is regulated by the phosphorylation, ubiquitination, and proteosomal degradation mediated by a destruction complex consisting of axin, adenomatous polyposis coli (APC), and glycogen synthase kinase 3 (GSK3) (Citation21Citation–Citation25). Studies on the cross talk between G-proteins and Wnt/β-catenin signaling have revealed complex interactions. Activation of β-catenin signaling following stimulation of the canonical Wnt/Frizzled pathway has been shown to be dependent in part on Gq through inhibition of GSK3β, suggesting that some G-alpha subunits positively regulate the canonical Wnt pathway (Citation26Citation–Citation29). Meigs et al. reported that in cells lacking APC, β-catenin-mediated transcriptional activation is upregulated by expression of activated G12 or G13 (Citation30). Go, a member of the Gi/o family, interacts with the Wnt signaling mediator Dishevelled and plays an essential role in Wnt3a-mediated activation of the Jun N-terminal kinase (Citation31Citation–Citation34). In contrast to the findings described above, studies on fibrous dysplasia showed that activated Gq, G11, G12, and G13 proteins had no significant roles in regulating β-catenin, while only activated Gs was shown to stimulate the Wnt signaling pathway (Citation35). In the broader view of β-catenin signaling independent of Wnt signaling, these studies indicate that the abilities of specific G-alpha subunits to regulate β-catenin signaling are variable and context dependent. Indeed, G-protein and β-catenin signaling cross talk has often been studied by considering each individual G-alpha subunit in isolation. However, as levels of one G-protein family are known to affect the expression and function of other G-protein families, the interrelation between these pathways could be quite complex. Moreover, the role of endogenously activated G-proteins in β-catenin signaling in the absence of exogenous ligand stimulation is poorly understood.
In this work, we have investigated the role of basal and activated Gq/11 and G12/13 families in the regulation of active β-catenin. In this regard, the Pasteurella multocida toxin (PMT) provides a novel tool to dissect these pathways. PMT is a potent intracellularly acting toxin which activates three families of heterotrimeric G-proteins: Gq/11, G12/13, and Gi/o (Citation36Citation–Citation41). PMT acts to deamidate a key glutamine (Q) to glutamic acid (E) in the target G-alpha subunits involved in GTP hydrolysis, leading to chronically activated G-protein function (Citation41Citation–Citation43); these PMT-modified G-alpha subunits can be detected specifically using an anti-QE antibody that recognizes PMT-modified G-alpha subunits (Citation44). As PMT treatment stimulates the activation of various G-protein-mediated downstream events, it offers a unique opportunity to explore the differences between the effects of basal and activated G-proteins on β-catenin signaling in the absence of exogenous stimulation. We have investigated in both fibroblastic and epithelial cell model systems whether PMT treatment leads to an activation of β-catenin signaling and, further, which G-protein families, if any, are likely to be responsible. In this report, we have used the GSK3 inhibitor LiCl as a direct activator of β-catenin signaling independently of the Wnt pathway to focus on how differential activation of specific G-alpha protein affects active β-catenin expression. Short-term activation of G-proteins using PMT enhanced the LiCl-induced active β-catenin levels in a G12/13-dependent manner. In contrast, cells with genetic inactivation of Gq/11 and G12/13 showed upregulated β-catenin signaling. Together, our findings suggest that the link between G-alpha subunits and β-catenin signaling is purely dependent on G-protein abundance and activity.
RESULTS
PMT activates G-alpha subunits and enhances LiCl-induced β-catenin signaling.
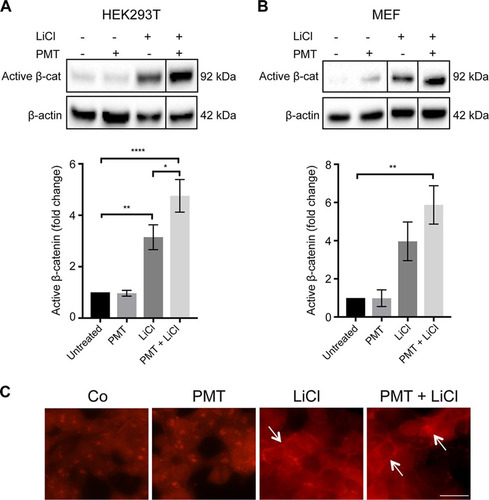
We used PMT to dissect the role of G-proteins in the regulation of active β-catenin. Western blotting using an anti-QE antibody (Citation44) confirmed that PMT treatment caused the expected Q > E modification in G-alpha proteins (). The effect of PMT on β-catenin signaling was evaluated in HEK293T cells and wild-type mouse embryonic fibroblasts (MEFs) by Western blotting using a specific antibody against nonphosphorylated β-catenin, referred to here as active β-catenin. LiCl, a classical GSK inhibitor, was used to activate β-catenin signaling (Citation45, Citation46). PMT treatment alone did not induce significant changes in the levels of active β-catenin in either HEK293T cells or wild-type MEFs. In contrast, PMT was shown to significantly enhance the LiCl-induced stimulation of active β-catenin after 24 h ( and ) but not 6 h (data not shown). Immunofluorescence staining confirmed the increase in active β-catenin expression in cells treated with both PMT and LiCl, including distinct membrane-associated expression of β-catenin, indicating engagement with cadherins (). The synergy with LiCl stimulation of β-catenin suggests that PMT-modified G-proteins further regulate the levels of active β-catenin by interacting with components of the β-catenin destruction complex.
FIG 1 PMT activates G-alpha subunits. (A) Schematic representation of general G-protein signaling and downstream signaling pathways showing PMT activation of G-alpha subunits by specific glutamine deamidation (Q > E) (red). Western blot analysis of MG63 and U2OS cells left unstimulated or stimulated with PMT (40 ng/ml, 24 h) confirms the PMT modification of G-alpha subunits using a specific anti-QE antibody. A similar induction was observed in HEK293T cells (not shown). β-Actin expression was used as a loading control.
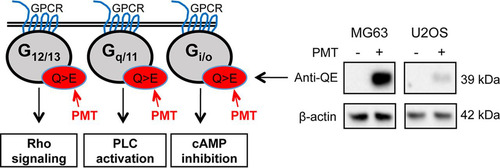
FIG 2 PMT enhances LiCl-induced β-catenin signaling. (A and B) Western blot analysis of active β-catenin levels in HEK293T cells (A) and wild-type MEFs (B) treated with PMT (HEK, 40 ng/ml; MEF, 20 ng/ml), LiCl (25 mM), either alone or together, or left untreated for 24 h. Levels of active β-catenin (Active β-cat) were evaluated using a specific β-catenin antibody on whole-cell extracts. β-Actin was used as a loading control. The histograms represent densitometric analysis of pooled data from three independent experiments normalized to their respective untreated control cells. The data represent the means ± standard errors of the means (SEM). Statistical significance was determined by Tukey’s test (n = 3): P < 0.05 (*), P < 0.001 (**), and P < 0.0001 (****). (C) Immunofluorescence analysis of active β-catenin expression. Representative images of HEK293T cells treated with PMT (40 ng/ml) and LiCl (25 mM), either alone or in combination for 24 h or left untreated (Co). The cells were stained with an antibody specific for active β-catenin, showing membrane-associated and nuclear staining (arrows) and imaged as described in Materials and Methods. Scale bar, 20 μm.
G-proteins negatively regulate the levels of active β-catenin.
Since PMT was found to be a potent activator of G-proteins and enhanced the levels of LiCl-induced active β-catenin, we next investigated the role of specific G-alpha subunits by using MEFs depleted for either Gq/11 or G12/13 (Citation47). Western blotting using specific G-alpha subunit antibodies confirmed the absence of specific G-alpha proteins in the relevant Gq/11−/− and G12/13−/− MEFs (). Interestingly, basal levels of Gi-1 were upregulated ∼2-fold in Gq/11−/− but not in G12/13−/− MEFs compared to those of wild-type cells (). This suggests that Gq/11 is a negative regulator of Gi-1, consistent with our previous observations (Citation42). The levels of Gs were not different among these three cell lines (data not shown).
FIG 3 Gq/11−/− and G12/13−/− cells show upregulated β-catenin signaling. (A) Characterization of Gq/11−/− and G12/13−/− cells using Western blotting. Whole-cell extracts were prepared from wild-type (MEF), Gq/11−/− (q/11 -/-), and G12/13−/− (12/13 -/-) MEFs. Levels of specific G-alpha subunits were evaluated using specific antibodies as indicated. Representative β-actin expression was used as a loading control. (B) Western blot analysis of active β-catenin (Active β-cat) and total β-catenin (Total β-cat) content in MEF, q/11−/−, and 12/13−/− MEFs. The histograms represent densitometric analysis of pooled data from three independent experiments for active β-catenin and two independent experiments for qPCR analysis of axin2 expression compared to that of the wild-type MEFs, as indicated. The respective band intensities for Western blotting and fold change values for qPCR analysis were normalized to the respective β-actin levels and to untreated controls. The data represent the means ± SEM. Statistical significance was determined by Tukey’s test: **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001.
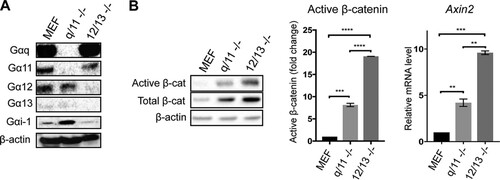
Measurement of β-catenin in these three cell lines showed that cells lacking Gq/11 or G12/13 subunits had ∼8-fold or 20-fold higher levels of active β-catenin, respectively, than wild-type MEFs (). Interestingly, increases in basal total β-catenin levels were also observed (). Quantitative PCR (qPCR) analysis of Axin2 gene expression provided a readout of active β-catenin-dependent transcription and showed that Gq/11−/− MEFs had ∼5-fold and G12/13−/− MEFs had ∼10-fold upregulated expression of Axin2 compared to that of the wild-type MEFs, correlating well with the changes in active β-catenin levels (). These results demonstrate that endogenous levels of Gq/11 and G12/13 negatively regulate β-catenin signaling and that G12/13 appears to inhibit the pathway significantly more than Gq/11.
PMT enhancement of β-catenin expression is lost in Gq/11 and G12/13 knockout cells.
As PMT enhanced the LiCl-induced active β-catenin levels, we next investigated if this was dependent on a specific G-alpha protein family. We stimulated the G-alpha-deficient cells with PMT and LiCl and quantified the levels of active β-catenin by Western blotting. Τhe results showed that in Gq/11−/− MEFs, neither PMT nor LiCl treatment alone or in combination for 6 or 24 h caused a significant stimulation of active β-catenin compared to that of the untreated cells or wild-type MEFs ( and ). This did not appear to be due to indirect effects of intracellular calpain, which is an established Gq effector that inhibits β-catenin (Citation48), as treatment with the calpain inhibitor, calpeptin, had no effects on PMT-stimulated β-catenin ().
FIG 4 PMT enhancement of active β-catenin expression is lost in Gq/11−/− and G12/13−/− cells. (A to C) Western blot analysis of active β-catenin expression in wild-type (A), Gq/11−/− (B), and G12/13−/− (C) MEFs. Cells were stimulated with PMT (20 ng/ml) and LiCl (25 mM), either alone or in combination, or left untreated for 6 h or 24 h as indicated. Levels of active β-catenin (Active β-cat) were evaluated using a specific β-catenin antibody on whole-cell extracts. β-Actin was used as a loading control. The histograms represent densitometric analysis of pooled data from three independent experiments. The data represent the means ± SEM. n = 3. *, P < 0.05 by Tukey’s test. (C) Treatment of G12/13−/− cells with both PMT and LiCl showed cytotoxicity and the quantification of active β-catenin was not possible. (D) Inhibition of calpain does not affect the PMT induction of active β-catenin. Western blot analysis of active β-catenin levels in HEK293T cells treated with PMT (40 ng/ml) and calpeptin (15 μM), either alone or in combination, or left untreated for 6 h, as indicated. Active β-catenin and β-actin were evaluated using specific antibodies described for panels A to C. The data are representative of three independent experiments.
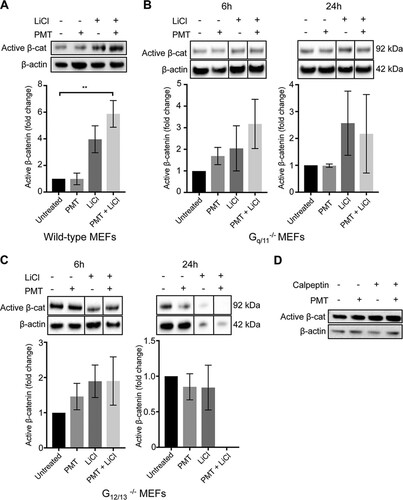
Cells lacking G12/13 were more severely affected. In G12/13−/− cells, PMT did not affect the levels of active β-catenin after 6 h, and after 24 h the levels of active β-catenin were severely compromised, being significantly decreased compared to those of the untreated cells (). LiCl treatment alone did not alter the levels of active β-catenin at 6 h, but after 24 h some cytotoxicity was seen (data not shown). Combined treatment of G12/13−/− MEFs with PMT and LiCl also resulted in further (∼90%) cell cytotoxicity (data not shown). Since both Gq/11−/− and, in particular, G12/13−/− null cells have high endogenous levels of active β-catenin (), it is possible that any further induction of active β-catenin using an agonist would compromise cell viability. Nevertheless, these results suggest that the PMT enhancement of LiCl-induced β-catenin is dependent on its ability to activate G-alpha subunits.
PMT-enhanced active β-catenin is G12/13 dependent.
The dramatic differences in PMT- and LiCl-regulated β-catenin signaling between wild-type MEFs and the G-alpha subunit null MEFs show the importance of the individual G-alpha subunits in limiting the activation of the β-catenin pathway in a time-dependent manner. In order to further understand the role of the individual alpha subunits, we used a transient genetic knockdown approach using specific short interfering RNAs (siRNAs) against Gq, G12, and G13 subunits. Western blotting demonstrated over ∼70% knockdown efficiency of each G-alpha subunit ().
FIG 5 G-alpha subunit knockdown affects PMT and LiCl induction of active β-catenin. (A) Western blot analysis of G-alpha subunit protein expression in cells with the indicated G-protein knockdown. HEK293T cells were transfected with a specific siRNA against GNAQ (Gαq), GNA12 (Gα12), and GNA13 (Gα13) or with a nontargeting control, as described in Materials and Methods. The knockdown efficiency of each siRNA was measured using specific G-alpha subunit antibodies on whole-cell extracts. β-Actin was used as a loading control. (B to D) Western blot analysis of active β-catenin expression in Gq (B), G12 (C), and G13 (D) knockdown HEK293T cells. Cells were transiently transfected with either siRNA against the indicated G-alpha subunits or with nontargeting oligonucleotides (Co). Transfected cells were treated with PMT (40 ng/ml) and LiCl (25 mM), either alone or in combination, or left untreated for 24 h. Levels of active β-catenin (Active β-cat) were evaluated using a specific β-catenin antibody on whole-cell extracts. β-Actin was used as a loading control. Band intensity of active β-catenin was normalized against its respective β-actin. The fold changes were generated for each group compared to the respective untreated controls. The histograms represent densitometric analysis of pooled data from three independent experiments. The data represent the means ± SEM. Statistical significance was determined by Tukey’s test (n = 3): *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001.
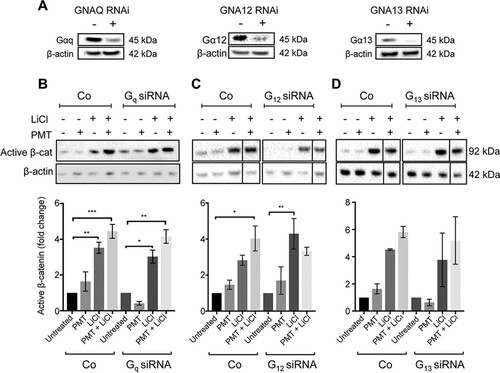
The roles of Gq, G12, and G13 in the regulation of active β-catenin were assessed individually. First, in contrast to the targeted null cells shown earlier (), transient knockdown of individual G-alpha subunits did not affect basal β-catenin levels (). Upon stimulation, there were no significant differences observed with PMT and LiCl treatments in Gq knockdown cells compared to cells transfected with a nontargeting siRNA (). In contrast, in G13 knockdown cells it was found that while PMT-treated cells showed no alterations in active β-catenin levels, treatment with LiCl showed a decrease in the levels of active β-catenin compared to the level for nontargeted controls (). More importantly, PMT-mediated enhancement of LiCl signaling was lost in the absence of either G12 or G13 ( and ). These results suggest that short-term, transient knockdown of basal G13 negatively regulates the LiCl activation of β-catenin signaling and that G12 and G13 signaling is essential for regulation of LiCl-induced β-catenin signaling by PMT-activated G-proteins.
DISCUSSION
G-proteins are an important class of signal transduction proteins that play an essential role in development, maintaining cellular homeostasis, and in cancer (Citation49, Citation50). Many studies have shown the regulation of their classical signaling pathways mediated by the specific G-alpha subunits, although there has been little discussion regarding any cross talk between these G-alpha subunits or with other pathways, such as β-catenin signaling. The regulation of β-catenin by G-proteins seems to be controversial. Some studies indicate that G-proteins positively regulate β-catenin, while others demonstrate their negative effect. We hypothesized here that this is due to differential effects of individual G-alpha subunits either in their basal state or in response to specific activators. Hence, to understand this potential cross talk better, we have dissected the role of basal and activated G-alpha subunits using genetic loss-of-function approaches to assess their effects on β-catenin signaling and have proposed a model to demonstrate these interactions ().
FIG 6 Model hypothesizing the potential regulation of β-catenin by G-alpha subunits and PMT. The classical G-protein targets for G12/13 (cadherins and Rho/ROCK) and Gq/11 (Rho/ROCK and calpain) proteins and their stimulation of active β-catenin are indicated. (A) Regulation of β-catenin by PMT-modified G-alpha subunits. LiCl stimulates β-catenin signaling through inhibition of GSK. Proposed PMT-stimulated pathways are indicated in red. PMT-modified G-proteins (Q > E) enhance LiCl stimulation of active β-catenin levels (red arrows). This effect could be mediated through the known interactions between G-alpha subunits and either GSK3 or the RGS sequence in axin2. PMT might also affect G-protein interactions with cadherins and Rho/ROCK to induce β-catenin (red dashed arrows). Our data suggest that the Gq-calpain pathway is not involved in PMT action (black dashed arrows). (B) Complete deletion of G-alpha subunits results in significant enhancement of β-catenin signaling in the absence of LiCl treatment (boldface black arrows), compromising cell viability (red dagger). PMT treatment (Q > E) of cells with transient knockdown (KD) of G12 and G13 (shaded) results in decreased β-catenin levels only via the Wnt destruction complex. See the text for details.
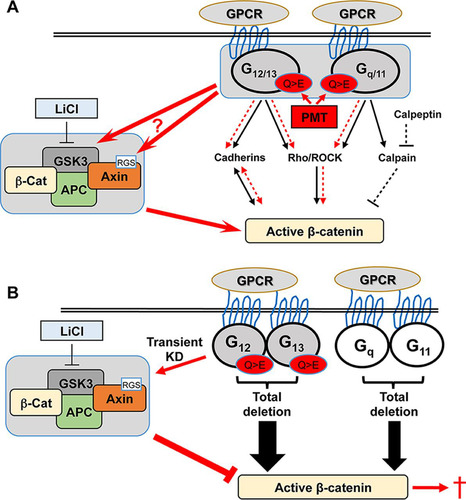
We have analyzed β-catenin signaling in MEFs and HEK293T cells using PMT as a unique tool to activate G-protein function in the absence of GPCR stimulation and have found that PMT enhanced LiCl-stimulated active β-catenin levels. The mechanisms underlying this synergistic effect are not completely understood, although previous studies have demonstrated both direct and indirect interactions between G-alpha subunits and components of the β-catenin signaling pathway. The clear membrane expression of active β-catenin following PMT and LiCl treatment suggests engagement with cadherins. Since G12 and G13 are known to interact with cadherins and release β-catenin from the cadherin complex (Citation30, Citation51), our data suggest that PMT-activated G12/13 is responsible, at least in part, for the increase in active β-catenin. Alternatively, PMT could contribute to β-catenin activation through Rho/Rho kinase signaling. We and others have shown that PMT activates the Rho/Rho kinase pathway through both G12/13 and Gq/11 (Citation40, Citation52), and Rho kinase can stimulate β-catenin (Citation53). Finally, active Gq has also been shown to inhibit GSK3β, leading to the accumulation of β-catenin (Citation26). Thus, while G-alpha activation by PMT synergizes with LiCl to stimulate active β-catenin, it is difficult to identify the specific PMT targets that mediate this, as PMT activates all three G-alpha subunits in wild-type cells.
Cells harboring targeted G-alpha subunit knockouts showed upregulation of endogenous active β-catenin signaling in the absence of exogenous stimulation of either G-protein or Wnt/β-catenin signaling, and G12/13-deficient MEFs showed significantly higher basal β-catenin levels than Gq/11−/− MEFs. These findings suggest that endogenous G-alpha subunits are negative regulators of β-catenin signaling and, in particular, that G12/13 has greater effect in controlling β-catenin than Gq/11. The precise mechanisms that regulate total versus active β-catenin protein levels are not clear at this time, as they are likely to differ between the different G-alpha subunits; thus, we focused our studies on active β-catenin. It is important to note, however, that changes in basal active β-catenin were observed only in the context of stable long-term inactivation of the G-alpha proteins using null MEFs, as transient knockdown of individual G-alpha subunits using siRNA did not affect basal active β-catenin levels. Thus, besides demonstrating clear regulation of active β-catenin by G-alpha proteins, our data suggest additionally that there are important differences in downstream signaling between transient versus long-term deletion of G-alpha subunits.
In MEFs deficient for either Gq/11 or G12/13, which we showed have high endogenous β-catenin activity, additional PMT treatment did not affect β-catenin levels, and the enhancing effect of PMT on LiCl-induced β-catenin signaling was not observed. Longer treatment of G12/13−/− but not Gq/11−/− MEFs with LiCl and/or PMT appeared to compromise cell viability. The reasons for this are not yet clear, but inhibition of GSK3β as well as high levels of β-catenin have been documented to stimulate apoptosis (Citation54Citation–Citation56). Our observed upregulation of Gi in Gq/11 knockout cells is consistent with what we (Citation42) and others (Citation17) have previously reported, and the levels of individual G-alpha subunits can also affect cell survival (Citation57). Thus, the effects of altered levels of G-protein subunit activation, and indeed of GSK3β and β-catenin activity on cell survival, can vary widely in a cell context-dependent manner. Nevertheless, in the context of PMT and LiCl activation, our data clearly demonstrate that in the G-protein-null MEFs harboring constitutive loss of G-alpha subunits, activation of β-catenin is dependent upon functional G12/13 subunits. Interestingly, dissecting individual G-alpha subunit function by transient siRNA knockdown confirmed that G12 and G13, but not Gq, play an essential role in agonist-mediated enhancement of β-catenin. Taken together, these data imply that G12 and G13 subunits protect cells from chronic activation of β-catenin that could cause pathological consequences, such as developmental defects and cancer (Citation20, Citation58).
The mechanisms underlying G-alpha protein regulation of β-catenin are not clear. Our data suggest that one proposed mechanism involving Gq-calpain interactions does not have a significant role. However, the observed increase in Axin2 expression in both Gq/11 and G12/13 null cells suggests that there is G-alpha subunit interaction with the destruction complex of Wnt signaling, as axin2 has a regulator of the G-protein signaling (RGS) site for G-alpha proteins, in particular for activated G12 (Citation59). In some contexts, activated G13 and Gq have also been shown to bind to axin2 (Citation35); thus, there is likely to be competition for axin2 binding among G-alpha subunits. Whether PMT treatment causes subcellular distribution of G-alpha proteins and binding to axin2 is not yet known, but this response would vary depending on which G-alpha proteins are present given the transient versus constitutive gain- or loss-of-function setting described here. Our data have pointed to a novel relationship between G-proteins and β-catenin signaling, namely, that the endogenous G-proteins serve to mediate a supervised activation of agonist-stimulated β-catenin signaling. However, where G-alpha subunit content has been altered, β-catenin signaling evades this control from G-proteins and is elevated. Hence, β-catenin regulation differs between the nonmodified cells and those with manipulated G-alpha subunits.
In summary, using the potent toxin PMT, which activates three of the four families of heterotrimeric G-proteins, and genetic deletions of G-alpha subunits, we have confirmed a link between G-alpha subunit activation and β-catenin signaling in the absence of canonical Wnt ligand stimulation. PMT provides an extremely useful tool to predict and test the effects of sustained activation of G-alpha subunits, which provides additional insights into the transient overexpression of constitutively activated subunits commonly reported. Indeed, our preliminary evidence suggests that PMT activates G-alpha subunits, especially G12, with kinetics different from those of other G-alpha subunits (C. Thompson, A. E. Grigoriadis, and A. J. Lax, unpublished data), and that long-term chronic treatment of cells with PMT continuously over months changes their endogenous G-alpha subunit content (A. Banu, A. E. Grigoriadis, and A. J. Lax, unpublished data). Our data also show that cells lacking G-proteins in the long term show potentially deleterious levels of β-catenin. However, short-term inhibition of G-proteins has no direct effect on β-catenin levels but was shown to alter agonist-mediated β-catenin signaling in a specific G-alpha-dependent way. Studying specific activated G-alpha subunits using PMT has revealed a specific role for G12 and G13 in cooperating with β-catenin signaling. Given the established roles of altered G12/13 proteins and β-catenin in tumorigenesis, it is tempting to speculate that PMT-induced G-protein fine-tuning of β-catenin signaling supports the procarcinogenic signature of chronic PMT exposure that we have previously hypothesized (Citation60). Our findings suggest that the relative role of both basal and activated G-protein subunit levels will have to be understood before the full repertoire of their cellular function can be elucidated.
MATERIALS AND METHODS
Cell lines and preparation of whole-cell lysates.
Wild-type mouse embryonic fibroblasts (wild-type MEFs), Gq/11−/− MEFs, and G12/13−/− MEFs were obtained from Stefan Offermanns, (Department of Pharmacology, Max Planck Institute for Heart and Lung Research, Bad Nauheim, Germany [Citation47]). MEFs and HEK293T, MG63, and U2OS cells were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum (FBS), 50 units/ml of penicillin, 50 μg/ml of streptomycin, and l-glutamine, incubated at 37°C in a humidified atmosphere of 5% CO2. Equal numbers of cells were cultured in 6-well plates and grown until confluence. The cells were then incubated with the respective compounds as indicated in the figure legends for either 6 h or 24 h. After the respective treatment, cells were washed with ice-cold phosphate-buffered saline (PBS) and incubated with radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitor cocktail (1:100 dilution) (P2714-1BTL; Sigma-Aldrich, Poole, UK) and phosphatase inhibitor cocktail (1:100 dilution) (40010; Active Motif, Carlsbad, CA) for 5 min on ice. Cells were removed using cell scrapers, and the whole-cell lysates were transferred to a microcentrifuge. The tubes were centrifuged at a relative centrifugal force of 14,000 for 15 min, and the supernatant was aliquoted and stored at −80°C. For anti-QE antibody preparation, hybridoma cells (a kind gift from Yasuhiko Horiguchi, Osaka University, Japan) were cultured in hybridoma-SFM medium supplemented with recombinant human interleukin-6 (IL-6) and purified as described previously (Citation44).
Western blotting.
The proteins were separated on 4 to 12% gradient gels and electrotransferred onto nitrocellulose membrane. The following antibodies were used: anti-active β-catenin antibody (8814S; New England Biolabs, Hitchin, UK) and total anti-β-catenin antibody (E5; Insight Biotechnology, Wembley, UK). All of the G-protein antibodies were purchased from Santa Cruz Biotechnology (Heidelberg, Germany): anti-Gα12 (sc- 409), anti-Gα13 (sc-410), anti-Gαq (sc-393), anti-Gα11 (sc-394), anti-Gαs (sc-823), anti-Gαi-1 (sc-391), anti-Gαi-2 (sc-7276), anti-Gαi-3 (sc-262), and anti-Gαo (sc-387). Bands were analyzed using the ChemiDoc MP imaging system from Bio-Rad. The band intensity was measured using Image Lab software (version 5.2.1). The band intensity of each protein was normalized against its β-actin housekeeping gene (Citation61).
Immunofluorescence.
Cells (5 × 104 cells/ml) were cultured on 12-mm collagen-coated coverslips under standard conditions as described above and treated with the respective compounds. Cells were then fixed with 4% paraformaldehyde (PFA) for 1 h at room temperature (RT) and permeabilized with 0.2% Triton X-100 in PBS for 15 min at 4°C. They were then incubated with blocking buffer containing 1% bovine serum albumin in PBS for 30 min at RT. The primary anti-active β-catenin antibody and secondary anti-rabbit fluorescent antibodies were diluted in blocking buffer. The cells were incubated with the primary antibody (1:100) at 4°C overnight, washed with PBS, and incubated with secondary antibody (1:500) for 1 h at RT. The cells were washed with PBS three times and mounted using Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (Vector Laboratories, Peterborough, UK). The imaging was carried out using an ApoTome Zeiss fluorescence microscope, and the images were captured using an AxiCAm MRm Zeiss camera and processed on AxioVision Rel 4.8 software.
G-protein knockdown.
siRNAs developed against GNAQ, GNA12, and GNA13 were purchased from GE Healthcare Dharmacon, Ltd. (Lafayette, CO): ON-TARGET plus human GNAQ (L-008562-00-0005), GNA12 (L-008435-00-0005), GNA13 (L-009948-00-0005), and nontargeting pool (D-001810-10-05). The transfection protocol was performed according to the manufacturer’s instructions. Briefly, HEK293T cells (2 × 104 cells/well) were plated in a 24-well plate in medium containing no antibiotics and cultured overnight under standard conditions. Specific siRNAs and the transfection reagent were diluted in media without antibiotics in separate tubes. The contents of each tube were gently mixed and incubated for 5 min and then mixed and incubated for a further 20 min. Media were added into the above-described mix to obtain a final siRNA concentration of 25 nM. The cells were incubated with medium containing each siRNA for 72 h. Cells with each respective G-protein knockdown were analyzed by Western blotting for decreased expression and then used for further experiments. Significant knockdown of GNA11 expression was not achieved and was not pursued further.
qPCR analysis.
For qPCR analysis, cells were cultured until confluence, and RNA was extracted using TRIzol (Invitrogen/ThermoFisher, Paisley, UK). Reverse transcription was performed using an RTnanoscript2 kit (Primer Design). The following primers were used: β-actin, 3′-GGCTGTATTCCCCTCCATCG-5′ and 5′-CCAGTTGGTAACAATGCCTGT-3′; axin2, 3′-TGACTCTCCTTCCAGATCCCA-5′ and 5′-TGCCCACACTAGGCTGACA-3′. The ΔΔCT method (where CT is threshold cycle) was used for data analysis.
Statistical analysis.
All experiments were repeated at least three independent times. One-way analysis of variance and Tukey’s test were used to assess the difference between treatments using GraphPad Prism software, version 7.
ACKNOWLEDGMENTS
We thank Yasuhiko Horiguchi (Osaka University, Japan) for the kind gift of the anti-QE hybridoma and Jon Robbins (King’s College London) for critical reading of the manuscript.
This work was supported in part by the King’s College London Dental Institute Studentship Fund, the King’s College London Graduate Scholarship Scheme and BBSRC grant BB/1021922/1 (K.J.L.).
We have no conflicts of interest to declare.
A.B., K.J.L., A.J.L., and A.E.G. were responsible for conception and design, data analysis and interpretation, and writing, reviewing, and editing the manuscript.
REFERENCES
- McCudden CR, Hains MD, Kimple RJ, Siderovski DP, Willard FS. 2005. G-protein signaling: back to the future. Cell Mol Life Sci 62:551–577. https://doi.org/10.1007/s00018-004-4462-3.
- Strathmann M, Simon MI. 1990. G protein diversity: a distinct class of alpha subunits is present in vertebrates and invertebrates. Proc Natl Acad Sci U S A 87:9113–9117. https://doi.org/10.1073/pnas.87.23.9113.
- Simon MI, Strathmann MP, Gautam N. 1991. Diversity of G proteins in signal transduction. Science 252:802–808. https://doi.org/10.1126/science.1902986.
- Gilman AG. 1984. G proteins and dual control of adenylate cyclase. Cell 36:577–579. https://doi.org/10.1016/0092-8674(84)90336-2.
- Sánchez-Fernández G, Cabezudo S, García-Hoz C, Benincá C, Aragay AM, Mayor F, Ribas C. 2014. Galphaq signalling: the new and the old. Cell Signal 26:833–848. https://doi.org/10.1016/j.cellsig.2014.01.010.
- Wang T, Pentyala S, Elliott JT, Dowal L, Gupta E, Rebecchi MJ, Scarlata S. 1999. Selective interaction of the C2 domains of phospholipase C-beta1 and -beta2 with activated Galphaq subunits: an alternative function for C2-signaling modules. Proc Natl Acad Sci U S A 96:7843–7846. https://doi.org/10.1073/pnas.96.14.7843.
- Fukuhara S, Murga C, Zohar M, Igishi T, Gutkind JS. 1999. A novel PDZ domain containing guanine nucleotide exchange factor links heterotrimeric G proteins to Rho. J Biol Chem 274:5868–5879. https://doi.org/10.1074/jbc.274.9.5868.
- Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, Bollag G, Sternweis PC. 1998. p115 RhoGEF, a GTPase activating protein for Galpha12 and Galpha13. Science 280:2109–2111. https://doi.org/10.1126/science.280.5372.2109.
- Kourlas PJ, Strout MP, Becknell B, Veronese ML, Croce CM, Theil KS, Krahe R, Ruutu T, Knuutila S, Bloomfield CD, Caligiuri MA. 2000. Identification of a gene at 11q23 encoding a guanine nucleotide exchange factor: evidence for its fusion with MLL in acute myeloid leukemia. Proc Natl Acad Sci U S A 97:2145–2150. https://doi.org/10.1073/pnas.040569197.
- Bian D, Mahanivong C, Yu J, Frisch SM, Pan ZK, Ye RD, Huang S. 2006. The G12/13-RhoA signaling pathway contributes to efficient lysophosphatidic acid-stimulated cell migration. Oncogene 25:2234–2244. https://doi.org/10.1038/sj.onc.1209261.
- Wang D, Tan YC, Kreitzer GE, Nakai Y, Shan D, Zheng Y, Huang XY. 2006. G proteins G12 and G13 control the dynamic turnover of growth factor-induced dorsal ruffles. J Biol Chem 281:32660–32667. https://doi.org/10.1074/jbc.M604588200.
- Bodmann EL, Rinne A, Brandt D, Lutz S, Wieland T, Grosse R, Bunemann M. 2014. Dynamics of Galphaq-protein-p63RhoGEF interaction and its regulation by RGS2. Biochem J 458:131–140. https://doi.org/10.1042/BJ20130782.
- Rojas RJ, Yohe ME, Gershburg S, Kawano T, Kozasa T, Sondek J. 2007. Galphaq directly activates p63RhoGEF and Trio via a conserved extension of the Dbl homology-associated pleckstrin homology domain. J Biol Chem 282:29201–29210. https://doi.org/10.1074/jbc.M703458200.
- Slice LW, Han SK, Simon MI. 2003. Galphaq signaling is required for Rho-dependent transcriptional activation of the cyclooxygenase-2 promoter in fibroblasts. J Cell Physiol 194:127–138. https://doi.org/10.1002/jcp.10195.
- Sriwai W, Zhou H, Murthy KS. 2008. G(q)-dependent signalling by the lysophosphatidic acid receptor LPA(3) in gastric smooth muscle: reciprocal regulation of MYPT1 phosphorylation by Rho kinase and cAMP-independent PKA. Biochem J 411:543–551. https://doi.org/10.1042/BJ20071299.
- Ueda H, Morishita R, Narumiya S, Kato K, Asano T. 2004. Galphaq/11 signaling induces apoptosis through two pathways involving reduction of Akt phosphorylation and activation of RhoA in HeLa cells. Exp Cell Res 298:207–217. https://doi.org/10.1016/j.yexcr.2004.04.015.
- Krumins AM, Gilman AG. 2006. Targeted knockdown of G protein subunits selectively prevents receptor-mediated modulation of effectors and reveals complex changes in non-targeted signaling proteins. J Biol Chem 281:10250–10262. https://doi.org/10.1074/jbc.M511551200.
- Everly DN, Kusano S, Raab-Traub N. 2004. Accumulation of cytoplasmic beta-catenin and nuclear glycogen synthase kinase 3beta in Epstein-Barr virus-infected cells. J Virol 78:11648–11655. https://doi.org/10.1128/JVI.78.21.11648-11655.2004.
- López-Knowles E, Zardawi SJ, McNeil CM, Millar EK, Crea P, Musgrove EA, Sutherland RL, O'Toole SA. 2010. Cytoplasmic localization of beta-catenin is a marker of poor outcome in breast cancer patients. Cancer. Epidemiol Biomarkers Prev 19:301–309. https://doi.org/10.1158/1055-9965.EPI-09-0741.
- Valenta T, Hausmann G, Basler K. 2012. The many faces and functions of β-catenin. EMBO J 31:2714–2736. https://doi.org/10.1038/emboj.2012.150.
- Gordon MD, Nusse R. 2006. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J Biol Chem 281:22429–22433. https://doi.org/10.1074/jbc.R600015200.
- Wu G, He X. 2006. Threonine 41 in beta-catenin serves as a key phosphorylation relay residue in beta-catenin degradation. Biochemistry 45:5319–5323. https://doi.org/10.1021/bi0601149.
- He X, Semenov M, Tamai K, Zeng X. 2004. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: arrows point the way. Development 131:1663–1677. https://doi.org/10.1242/dev.01117.
- Kimelman D, Xu W. 2006. Beta-catenin destruction complex: insights and questions from a structural perspective. Oncogene 25:7482–7491. https://doi.org/10.1038/sj.onc.1210055.
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. 2002. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108:837–847. https://doi.org/10.1016/S0092-8674(02)00685-2.
- Salmanian S, Najafi SM, Rafipour M, Arjomand MR, Shahheydari H, Ansari S, Kashkooli L, Rasouli SJ, Jazi MS, Minaei T. 2010. Regulation of GSK-3beta and beta-Catenin by Galphaq in HEK293T cells. Biochem Biophys Res Commun 395:577–582. https://doi.org/10.1016/j.bbrc.2010.04.087.
- Najafi SM. 2009. Activators of G proteins inhibit GSK-3beta and stabilize beta-Catenin in Xenopus oocytes. Biochem Biophys Res Commun 382:365–369. https://doi.org/10.1016/j.bbrc.2009.03.027.
- Liu T, DeCostanzo AJ, Liu X, Wang H, Hallagan S, Moon RT, Malbon CC. 2001. G protein signaling from activated rat frizzled-1 to the beta-catenin-Lef-Tcf pathway. Science 292:1718–1722. https://doi.org/10.1126/science.1060100.
- Liu X, Rubin JS, Kimmel AR. 2005. Rapid, Wnt-induced changes in GSK3beta associations that regulate beta-catenin stabilization are mediated by Galpha proteins. Curr Biol 15:1989–1997. https://doi.org/10.1016/j.cub.2005.10.050.
- Meigs TE, Fields TA, McKee DD, Casey PJ. 2001. Interaction of Galpha 12 and Galpha 13 with the cytoplasmic domain of cadherin provides a mechanism for beta -catenin release. Proc Natl Acad Sci U S A 98:519–524. https://doi.org/10.1073/pnas.021350998.
- Egger-Adam D, Katanaev VL. 2008. Trimeric G protein-dependent signaling by Frizzled receptors in animal development. Front Biosci 13:4740–4755. https://doi.org/10.2741/3036.
- Feigin ME, Malbon CC. 2007. RGS19 regulates Wnt-beta-catenin signaling through inactivation of Galpha(o). J Cell Sci 120:3404–3414. https://doi.org/10.1242/jcs.011254.
- Katanaev VL, Ponzielli R, Sémériva M, Tomlinson A. 2005. Trimeric G protein-dependent frizzled signaling in Drosophila. Cell 120:111–122. https://doi.org/10.1016/j.cell.2004.11.014.
- Jung H, Kim HJ, Lee SK, Kim R, Kopachik W, Han JK, Jho EH. 2009. Negative feedback regulation of Wnt signaling by Gbetagamma-mediated reduction of Dishevelled. Exp Mol Med 41:695–706. https://doi.org/10.3858/emm.2009.41.10.076.
- Regard JB, Cherman N, Palmer D, Kuznetsov SA, Celi FS, Guettier JM, Chen M, Bhattacharyya N, Wess J, Coughlin SR, Weinstein LS, Collins MT, Robey PG, Yang Y. 2011. Wnt/β-catenin signaling is differentially regulated by Gα proteins and contributes to fibrous dysplasia. Proc Natl Acad Sci U S A 108:20101–20106. https://doi.org/10.1073/pnas.1114656108.
- Orth JH, Fester I, Preuss I, Agnoletto L, Wilson BA, Aktories K. 2008. Activation of Galpha (i) and subsequent uncoupling of receptor-Galpha(i) signaling by Pasteurella multocida toxin. J Biol Chem 283:23288–23294. https://doi.org/10.1074/jbc.M803435200.
- Preuss I, Kurig B, Nürnberg B, Orth JH, Aktories K. 2009. Pasteurella multocida toxin activates Gbetagamma dimers of heterotrimeric G proteins. Cell Signal 21:551–558. https://doi.org/10.1016/j.cellsig.2008.12.007.
- Rozengurt E, Higgins T, Chanter N, Lax AJ, Staddon JM. 1990. Pasteurella multocida toxin: potent mitogen for cultured fibroblasts. Proc Natl Acad Sci U S A 87:123–127. https://doi.org/10.1073/pnas.87.1.123.
- Wilson BA, Zhu X, Ho M, Lu L. 1997. Pasteurella multocida toxin activates the inositol triphosphate signaling pathway in Xenopus oocytes via G(q)alpha-coupled phospholipase C-beta1. J Biol Chem 272:1268–1275. https://doi.org/10.1074/jbc.272.2.1268.
- Orth JH, Lang S, Taniguchi M, Aktories K. 2005. Pasteurella multocida toxin-induced activation of RhoA is mediated via two families of G{alpha} proteins, G{alpha}q and G{alpha}12/13. J Biol Chem 280:36701–36707. https://doi.org/10.1074/jbc.M507203200.
- Orth JH, Fester I, Siegert P, Weise M, Lanner U, Kamitani S, Tachibana T, Wilson BA, Schlosser A, Horiguchi Y, Aktories K. 2013. Substrate specificity of Pasteurella multocida toxin for α subunits of heterotrimeric G proteins. FASEB J 27:832–842. https://doi.org/10.1096/fj.12-213900.
- Babb RC, Homer KA, Robbins J, Lax AJ. 2012. Modification of heterotrimeric G-proteins in Swiss 3T3 cells stimulated with Pasteurella multocida toxin. PLoS One 7:e47188. https://doi.org/10.1371/journal.pone.0047188.
- Orth JH, Preuss I, Fester I, Schlosser A, Wilson BA, Aktories K. 2009. Pasteurella multocida toxin activation of heterotrimeric G proteins by deamidation. Proc Natl Acad Sci U S A 106:7179–7184. https://doi.org/10.1073/pnas.0900160106.
- Kamitani S, Ao S, Toshima H, Tachibana T, Hashimoto M, Kitadokoro K, Fukui-Miyazaki A, Abe H, Horiguchi Y. 2011. Enzymatic actions of Pasteurella multocida toxin detected by monoclonal antibodies recognizing the deamidated α subunit of the heterotrimeric GTPase Gq. FEBS J 278:2702–2712. https://doi.org/10.1111/j.1742-4658.2011.08197.x.
- Hedgepeth CM, Conrad LJ, Zhang J, Huang HC, Lee VM, Klein PS. 1997. Activation of the Wnt signaling pathway: a molecular mechanism for lithium action. Dev Biol 185:82–91. https://doi.org/10.1006/dbio.1997.8552.
- Orme MH, Giannini AL, Vivanco MD, Kypta RM. 2003. Glycogen synthase kinase-3 and Axin function in a beta-catenin-independent pathway that regulates neurite outgrowth in neuroblastoma cells. Mol Cell Neurosci 24:673–686. https://doi.org/10.1016/S1044-7431(03)00229-X.
- Zywietz A, Gohla A, Schmelz M, Schultz G, Offermanns S. 2001. Pleiotropic effects of Pasteurella multocida toxin are mediated by Gq-dependent and -independent mechanisms. J Biol Chem 276:3840–3845. https://doi.org/10.1074/jbc.M007819200.
- Li G, Iyengar R. 2002. Calpain as an effector of the Gq signaling pathway for inhibition of Wnt/beta -catenin-regulated cell proliferation. Proc Natl Acad Sci U S A 99:13254–13259. https://doi.org/10.1073/pnas.202355799.
- Malbon CC. 2005. G proteins in development. Nat Rev Mol Cell Biol 6:689–701. https://doi.org/10.1038/nrm1716.
- O'Hayre M, Vazquez-Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S, Gutkind JS. 2013. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat Rev Cancer 13:412–424. https://doi.org/10.1038/nrc3521.
- Meigs TE, Fedor-Chaiken M, Kaplan DD, Brackenbury R, Casey PJ. 2002. Galpha12 and Galpha13 negatively regulate the adhesive functions of cadherin. J Biol Chem 277:24594–24600. https://doi.org/10.1074/jbc.M201984200.
- Harmey D, Stenbeck G, Nobes CD, Lax AJ, Grigoriadis AE. 2004. Regulation of osteoblast differentiation by Pasteurella multocida toxin (PMT): a role for Rho GTPase in bone formation. J Bone Miner Res 19:661–670. https://doi.org/10.1359/JBMR.040105.
- Li L, Tam L, Liu L, Jin T, Ng DS. 2011. Wnt-signaling mediates the anti-adipogenic action of lysophosphatidic acid through cross talking with the Rho/Rho associated kinase (ROCK) pathway. Biochem Cell Biol 89:515–521. https://doi.org/10.1139/o11-048.
- Jacobs KM, Bhave SR, Ferraro DJ, Jaboin JJ, Hallahan DE, Thotala D. 2012. GSK-3β: A Bifunctional Role in Cell Death Pathways. Int J Cell Biol 2012:930710. https://doi.org/10.1155/2012/930710.
- Kim K, Pang KM, Evans M, Hay ED. 2000. Overexpression of beta-catenin induces apoptosis independent of its transactivation function with LEF-1 or the involvement of major G1 cell cycle regulators. Mol Biol Cell 11:3509–3523. https://doi.org/10.1091/mbc.11.10.3509.
- Ming M, Wang S, Wu W, Senyuk V, Le Beau MM, Nucifora G, Qian Z. 2012. Activation of Wnt/β-catenin protein signaling induces mitochondria-mediated apoptosis in hematopoietic progenitor cells. J Biol Chem 287:22683–22690. https://doi.org/10.1074/jbc.M112.342089.
- Yanamadala V, Negoro H, Denker BM. 2009. Heterotrimeric G proteins and apoptosis: intersecting signaling pathways leading to context dependent phenotypes. Curr Mol Med 9:527–545. https://doi.org/10.2174/156652409788488784.
- Shang S, Hua F, Hu ZW. 2017. The regulation of β-catenin activity and function in cancer: therapeutic opportunities. Oncotarget 8:33972–33989. https://doi.org/10.18632/oncotarget.15687.
- Stemmle LN, Fields TA, Casey PJ. 2006. The regulator of G protein signaling domain of axin selectively interacts with Galpha12 but not Galpha13. Mol Pharmacol 70:1461–1468. https://doi.org/10.1124/mol.106.023705.
- Lax AJ. 2005. Opinion: Bacterial toxins and cancer–a case to answer? Nat Rev Microbiol 3:343–349. https://doi.org/10.1038/nrmicro1130.
- Towbin H, Staehelin T, Gordon J. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A 76:4350–4354.