
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Senescence is a state of long-term cell cycle arrest that arises in cells that have incurred sublethal damage. While senescent cells no longer replicate, they remain metabolically active and further develop unique and stable phenotypes that are not present in proliferating cells. On one hand, senescent cells increase in size, maintain an active mTORC1 complex, and produce and secrete a substantial amount of inflammatory proteins as part of the senescence-associated secretory phenotype (SASP). On the other hand, these progrowth phenotypes contrast with the p53-mediated growth arrest typical of senescent cells that is associated with nucleolar stress and an inhibition of rRNA processing and ribosome biogenesis. In sum, translation in senescent cells paradoxically comprises both a global repression of translation triggered by DNA damage and a select increase in the translation of specific proteins, including SASP factors.
INTRODUCTION
Cells encountering sublethal damage that cannot be repaired may enter a state of senescence, wherein they exit from the cell cycle and display an antiapoptotic phenotype. In young organisms, senescence is often considered a protective mechanism against tumorigenesis (Citation1Citation–Citation3), although it has also been shown to function in wound healing (Citation4), cellular plasticity (Citation5), and embryonic development (Citation6). However, the accumulation of senescent cells in tissues of older organisms is believed to have a detrimental effect, given that senescent cells alter organ function, promote inflammatory and proangiogenic states, and favor tumorigenesis (Citation7Citation–Citation9). The negative impact of senescent cells in aging has been evidenced in recent studies that link the pharmacological or genetic depletion of senescent cells with improved health in older mice (Citation10Citation–Citation12). Accordingly, the interest in understanding and eliminating senescent cells to improve aging outcomes has escalated in recent years.
Several types of senescence have been identified. Replicative senescence (RS) arises following multiple rounds of cell division that shorten telomeres progressively, triggering a DNA damage response (DDR); stress-induced senescence (SIS) is triggered by exposure to acute stresses that activate the DDR; and oncogene-induced senescence (OIS) is elicited by activation of certain oncogenes. The different senescence types have distinct features depending on the trigger, the cell type undergoing senescence, and the time elapsed since the senescent program was initiated (Citation13, Citation14). However, there are many shared phenotypes, including (i) an enlarged and flattened morphology; (ii) increased expression of cyclin-dependent kinase (CDK) inhibitors including p16 (which prevents phosphorylation of the tumor suppressor RB) and p21 (an effector of the tumor suppressor protein p53); (iii) lysosomal expansion as evidenced by increased activity of a senescence-associated β-galactosidase (SA-βGal) function at pH 6; (iv) the presence of DNA segments with chromatin alterations reinforcing senescence (DNA-SCARS); (v) persistent DNA damage with arrest of proliferation; and (vi) a senescence-associated secretory phenotype (SASP) whereby cells produce and secrete large amounts of proinflammatory proteins, growth factors, and metalloproteases (Citation15Citation–Citation19) (). Among these traits, the SASP has emerged as one of the most clinically relevant phenotypes of senescent cells in vivo (Citation20).
FIG 1 Senescent cell traits including changes in the translation machinery. Classic markers of senescent cells (red boxes) and translation-related markers of senescence (black boxes) are indicated. Traditional markers include the presence of nuclear foci enriched for DDR proteins (DNA Scars); the complete arrest of cell proliferation; the expression of stress response proteins, such as p16, Rb, p53, and p21; positive staining for senescence-associated β-galactosidase at pH 6 (SA-βGal) associated with lysosomal expansion; an enlarged and flattened shape; and the heightened production and secretion of SASP factors. Markers associated with altered translation include the presence of enlarged and stressed nucleoli due to rRNA maturation defects and decreases in components of the translation machinery that may result in decreased protein synthesis.
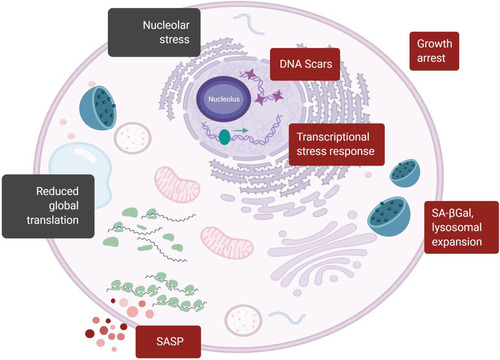
The adoption of these phenotypes by senescent cells requires both active mitochondria (Citation21) and the translation of proteins (Citation22), similar to what would be expected in a proliferating cell. However, senescent cells have shown an overall reduction in protein synthesis (Citation23Citation–Citation25), as well as inhibition of ribosome biogenesis and reductions in the expression and function of translation factors. In this review, we explore the coexistence of translationally active phenotypes of senescence and evidence of global translational suppression, focusing specifically on changes in ribosome biogenesis, translation initiation, and translation elongation. These two paradoxical directions of translation in senescence mirror the selective translation seen in other stress responses.
RIBOSOME BIOGENESIS, NUCLEOLAR STRESS, AND ACTIVATION OF STRESS-RESPONSE PATHWAYS
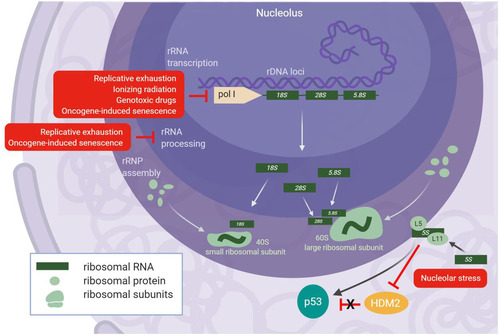
Ribosome biogenesis is an highly complex process that involves hundreds of trans-acting factors. The production of ribosomal subunits occurs in the nucleolus, a nuclear compartment that harbors the rDNA tandem repeats of the genome. The nucleolus is organized into three layers, namely, the fibrillar center, the dense fibrillar component, and the granular component, where rRNA transcription, rRNA processing, and RNP assembly occur, respectively (Citation26). The eukaryotic ribosome is composed of four rRNAs divided between the 40S subunit (containing the 18S rRNA) and the 60S subunit (containing the 28S, 5.8S, and 5S rRNAs), onto which 33 and 47 ribosomal proteins (RPs), respectively, are assembled in humans (Citation27); ribosomal proteins of the small subunit are denoted RPS, while ribosomal proteins of the large subunit are denoted RPL. The 28S, 5.8S, and 18S rRNAs are all transcribed from tandem rDNA repeats by RNA polymerase I into a single transcript (47S in mammals) that undergoes extensive processing by endo- and exonucleases before maturation (Citation28); the 5S rRNA is transcribed separately by RNA polymerase III and undergoes more limited exonucleolytic processing, followed by association with RPL5 and transport into the nucleolus for 60S subunit assembly (Citation29) ().
FIG 2 Senescence causes nucleolar stress and induction of p53 through the IRBC. All of the commonly studied senescent inducers (red boxes) have been shown to cause decreases in rRNA transcription or rRNA processing that result in nucleolar stress. A reduction in fully mature rRNA species prevents proper assembly of the 40S and 60S ribosomal subunits, thus releasing the 5S RNP into the nucleoplasm where it can stabilize p53 by inactivation of HDM2 (bottom right). Of note, OIS accelerates rRNA transcription shortly after induction, which also causes nucleolar stress, but rRNA transcription and processing decrease thereafter.
It was recently found that ribosome biogenesis, particularly at the level of rRNA processing, is severely inhibited in cells undergoing RS and OIS (Citation30, Citation31). While some molecular details differ between RS and OIS, both replicative exhaustion and some oncogenes induce perturbations to rRNA maturation that lead to nucleolar stress, followed by activation of other stress response pathways. In this section, we discuss the research linking stress response pathways to ribosome biogenesis and recent studies that further associate dysfunctional ribosome biogenesis as an important first step toward the establishment of senescence.
NUCLEOLAR STRESS STABILIZES p53 THROUGH THE IMPAIRED RIBOSOME BIOGENESIS CHECKPOINT
Ribosome biogenesis requires a large amount energy and is thus highly responsive to the growth conditions of the cell. As the place where ribosomes assemble, the nucleolus acts as a sensor of cellular stress; exposure to genotoxic damage leads to nucleolar segregation or disruption, in turn inhibiting nucleolar function (Citation32). Proteomic studies of the nucleolus have found that several nucleolar proteins function in the DDR, and some of them may serve roles in both DNA repair and ribosome biogenesis (Citation33). In agreement with these findings, multiple studies have associated the nucleolus with genomic instability, and the rDNA itself is a common site of damage during genotoxic stress (Citation34).
Nucleolar stress is primarily communicated to the cell through the transcriptional activation of p53 that elicits an antiproliferative response culminating in cell cycle arrest (Citation35). During periods of cellular growth, basal p53 activity remains low through the action of the E3 ubiquitin ligase HDM2 (MDM2 in mice) that both targets p53 for proteasome-mediated degradation and blocks the transcriptional activation domain of p53 (Citation36). During periods of cellular stress requiring p53 activity, the interaction between p53 and HDM2 is disrupted, thereby stabilizing p53 and enabling its transcriptional activity (Citation36, Citation37). The initial connections between ribosome biogenesis and p53 were discovered in experiments showing that RPL11, a component of the 60S subunit, could directly bind and inhibit HDM2 after induction of nucleolar stress (Citation38, Citation39). The stabilization of p53 was further broadened to include RPL5 (Citation40Citation–Citation42) and the 5S rRNA, thus implicating the 5S RNP (Citation43Citation–Citation45) in the activation of p53. Dysregulation of ribosome biogenesis prevents the incorporation of the 5S RNP into nascent ribosomes, and the release of free 5S RNP particles into the nucleoplasm stabilizes and activates p53 (Citation45). This mechanism of p53 activation has been termed the impaired ribosome biogenesis checkpoint (IRBC) (Citation46) ().
TRIGGERS OF SENESCENCE ELICIT CELL CYCLE ARREST THROUGH THE IRBC
A common way to induce the IRBC is by inhibiting rRNA transcription with doses of actinomycin D, a potent repressor of RNA polymerase I (Pol I) (Citation47). Lowering the levels of rRNA inhibits ribosome assembly and thus generates free 5S RNP molecules that trigger the IRBC. Genotoxic stressors, such as DNA-damaging drugs, UV radiation (UV), and ionizing radiation (IR), can inhibit rRNA transcription and generate nucleolar stress (Citation48, Citation49) (). Consistent with their ability to activate the IRBC, genotoxins require components of the 5S RNP for stabilization of p53 (Citation50, Citation51). In turn, p53 further inhibits Pol I-mediated rRNA transcription (Citation52), thus solidifying the IRBC.
Direct involvement of the IRBC in senescence was first reported in a study on the effects of OIS and RS on ribosome biogenesis. An increase in nucleolar size at the onset of senescence was found to be the result of accumulating rRNA, either due to excessive rRNA transcription in the case of OIS, or due to inhibition of rRNA processing in the case of RS (Citation30). While the mechanisms by which OIS and RS generated nucleolar stress were different, in both cases, the IRBC was activated and triggered cell cycle arrest through p53. Furthermore, it was shown that late-passage cells expressed lower levels of rRNA processing factors and that siRNA-mediated reduction of these factors recapitulated much of the senescent phenotypes observed from RS (Citation30). Similarly, an ectopic increase of rRNA transcription through overexpression of transcription intermediary factor 1α (TIFIA) was found to mimic the phenotypes observed during OIS (Citation30). For both OIS and RS, the IRBC was found to be necessary for the activation of the p53/p21 axis, and silencing the 5S RNP components suppressed the ensuing senescent phenotypes (Citation30).
A more recent study of senescence confirmed the presence of dysregulated ribosome biogenesis during OIS and RS. OIS was found to trigger a massive decrease in rRNA transcription and processing 18 to 20 days after induction (Citation31), in contrast to the increase in rRNA levels shortly after OIS (Citation30, Citation53). The decrease in rRNA processing observed in both senescence models corresponded to decreases in the levels of labile rRNA processing factors; silencing such factors similarly induced senescent phenotypes (Citation31). Interestingly, the defects in ribosome biogenesis were attributed to reduced phosphorylation of the Rb tumor suppressor protein, rather than stabilization of p53; it was further shown that the oncoprotein E6, which inhibits p53, was less effective at preventing senescent phenotypes caused by silencing rRNA processing factors than the oncoprotein E7, which blocks the tumor suppressor Rb (Citation31). It was further demonstrated that the Rb-induced senescence was mediated by free RPS14 protein, as unbound RPS14 inhibited CDK activity and decreased Rb phosphorylation, similar to the mechanism utilized by p16. These results were further supported by experiments showing that overexpression of RPS14 alone was sufficient to induce senescence (Citation31).
While the IRBC was first described for the activation of p53 by 5S RNP, the function of the ribosomal protein RPS14 as an activator of the Rb tumor suppressor further expands the antiproliferative signaling that results from impaired ribosome biogenesis. Whether mediated through Rb or p53, the robust association of nucleolar dysfunction with oncogenic stress, genotoxic stress, and replicative exhaustion strongly suggests that impaired ribosome biogenesis is a shared and early feature across senescence types.
TRANSLATION INITIATION: ACTIVATION AND INHIBITION BY SENESCENCE TRIGGERS
In eukaryotes, translation initiation begins with formation of the 43S initiation complex that contains the 40S ribosomal subunit and the ternary complex; the 43S initiation complex binds mRNA loaded with the eukaryotic initiation factor 4F (eIF4F) complex (eIF4G, eIF4A, and eIF4E) and then scans the 5′ untranslated region (UTR) until it reaches a start codon that then triggers both initiation factor displacement and 60S ribosomal subunit joining (Citation54). This multistep process creates several points of regulatory control, as changes in the concentration of 40S ribosomal subunits, ternary complex, and the eIF4F complex can augment or reduce translation efficiency. In this section, we describe data exploring the relationship between senescence and the translation initiation machinery, with a particular focus on signaling through mammalian target of rapamycin complex (mTORC1) and the eukaryotic translation initiation factor 2α (eIF2α; eIF2A).
mTORC1 IS ACTIVE DURING SENESCENCE AND PROMOTES THE SASP
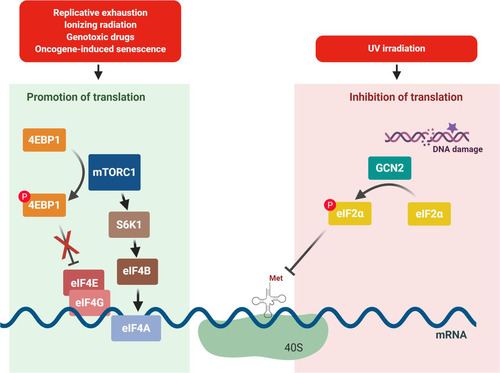
mTORC1 has been reported to increase protein synthesis and to regulate cell metabolism through the serine/threonine protein kinase activity of the mTOR catalytic subunit (Citation55); mTOR also serves as the catalytic subunit of the mTORC2 complex, which has functions relevant to cell survival and proliferation. While there is evidence that mTORC2 performs some functions during senescence (Citation56, Citation57), here we focus on mTORC1 since its role in senescence is clearer at present. mTORC1 promotes protein synthesis by phosphorylating two downstream effectors, namely, the p70S6 kinase 1 (S6K1) and the eIF4E-binding protein 4E-BP. Following phosphorylation, S6K1 becomes activated and in turn increases the function of downstream translation initiation factors, such as eIF4B; at the same time, phosphorylation of 4E-BP renders it unable to sequester the eIF4E component of the eIF4F complex (Citation54, Citation55). Accordingly, the phosphorylation of both S6K1 and 4E-BP promotes formation of the pre-initiation complex and cap-dependent translation, and thus, increased mTORC1 activity is expected to increase translation initiation and ultimately protein synthesis (Citation58) (). Given the progrowth influence of mTORC1, its activation during senescence is somewhat paradoxical but is well supported by the literature. DNA damage from IR can activate mTOR by promoting phosphatidylinositol 3-kinase (PI3K)–AKT signaling (Citation21, Citation59), and oncogenic RAS can inactivate tuberous sclerosis complex (TSC), an upstream inhibitor of mTORC1 (Citation60). At the onset of OIS and genotoxic stress, the phosphorylation of S6K1 and 4E-BP increases, suggesting a rise in translation initiation (Citation61, Citation62). However, senescent cells also exhibit increased autophagy, a process that is typically repressed by mTORC1. The increased autophagy is evidenced by increases in lysosomal content and activity in senescent cells (Citation62), and these observations are consistent with the enhanced SA-βGal phenotype of senescent cells (Citation63). The importance of autophagy in senescence is further underscored by experiments linking reduced expression of autophagy proteins ATG5 and ATG7 to delayed senescent traits during OIS (Citation53).
FIG 3 Senescence inducers may promote or inhibit translation initiation. Left, genotoxic stress, OIS, and replicative exhaustion have all been shown to require the activity of mTORC1 for the onset of senescence. mTORC1 functions to promote translation initiation by inhibiting 4E-BP, which sequesters eIF4E, and also by activating S6K1, which activates other downstream translation factors, including eIF4B. Right, DNA damage caused by UV irradiation activates the eIF2α kinase GCN2, leading to the repression of translation initiation.
It is interesting to note that these typically antagonistic processes, anabolic and catabolic, appear to function cooperatively during senescence. There is a compartmentalized mTORC1-driven increase in nascent protein synthesis during the onset of senescence that was found to colocalize with autolysosomes in a cellular region termed the TOR-autophagy spatial coupling compartment (TASCC) (Citation61). Given the spatial convergence at this compartment of the Golgi apparatus, the rough endoplasmic reticulum, autolysosomes, and mTORC1, the TASCC was suggested to function as a type of “protein-remodeling” center that can selectively enable the implementation of a senescent proteome (presumably including SASP and lysosomal proteins) (Citation53, Citation61, Citation64, Citation65). Some examples of autophagic remodeling in the establishment of a senescent proteome are found in experiments identifying large shifts in ubiquitin-modified proteins in senescent fibroblasts (Citation66) and in observations that the levels of GATA4, an upstream effector of the SASP, rises after genotoxic stress due to the relief of selective autophagy (Citation67).
The increase in mTORC1 activity that occurs at the onset of senescence has been shown to be a critical part of the induction and maintenance of senescent phenotypes. Conversely, inhibition of the mTORC1 complex has been shown to delay or suppress senescent phenotypes in response to the overexpression of stress factors (Citation68, Citation69), genotoxic stress (Citation70), OIS (Citation71), and RS (Citation71Citation–Citation73). Since the inhibition of mTORC1 is known to cause a global decrease in translation initiation, which is apparent from decreases in both protein synthesis and polysome content (Citation74), the necessity of mTORC1 in senescence suggests that there is substantial translation occurring at least during the onset of senescence.
The inhibition of mTORC1 primarily reduces the translation of a subset of mRNAs containing a 5′ oligopyrimidine tract (TOP) or similar motif in their 5′ UTRs (Citation74, Citation75). These mRNAs experience the most substantial decline in translation upon mTOR inhibition, and many of them encode components of the translation machinery, including all of the ribosomal proteins (Citation74Citation–Citation76). It has also been shown that while mTORC1 phosphorylates both S6K1 and 4E-BP to positively regulate translation initiation, the phosphorylation of 4E-BP is most responsible for the increase in protein output since a knockout of 4E-BP abrogates the effects of an mTOR inhibitor (Citation74) and a dominant negative mutant of 4E-BP reduces translation of mTOR-sensitive transcripts (Citation75).
The vast array of senescent phenotypes shown to be blunted by mTORC1 inhibition indicates that translation is important for the onset and maintenance of senescence (). In strong support of the direct involvement of mTORC1 in senescence, mTOR inhibitors suppress the SASP (Citation77Citation–Citation79). Surprisingly, mTORC1 inhibition primarily reduces the levels of mRNAs encoding SASP factors and not their translation, as would have been expected (Citation77, Citation78). Two separate models have been proposed to explain this observation. In one model, Laberge et al. showed that inhibiting mTORC1 decreases the translation of interleukin 1α (IL1A), a signaling cytokine that in turn promotes the transcription of SASP factors, suggesting that the reduced levels of SASP mRNAs is a consequence of the decrease in IL1A levels (Citation77). Supporting this possibility is evidence that the mTORC1 inhibitor rapamycin decreases IL1A protein expression while leaving IL1A mRNA levels mostly unchanged and that NF-κB activity, which is normally elevated by IL1A expression, is greatly reduced upon rapamycin treatment (Citation77).
In a second model, Herranz et al. demonstrated that mTORC1 inhibition reduces expression of the kinase mitogen-activated protein kinase-activated protein kinase 2 (MAPKAPK2) that would otherwise indirectly inhibit the degradation of mRNAs encoding SASP factors by the RNA-binding protein ZFP36L1 (Citation78). The authors show that MAPKAPK2 expression is substantially downregulated upon treatment with the mTORC1 inhibitor Torin-1 and that expression of nonphosphorylatable mutants of ZFP36L1 in cells reduced the levels of mRNAs encoding SASP factors (Citation78). Interestingly, MAPKAPK2 is an essential element of the p38 MAPK pathway that is closely related to most cellular senescence traits. This level of cross talk between both pathways suggests that MAPKs might coordinate with mTOR for the enhanced translation of certain transcripts necessary for cellular senescence (Citation80). Since the models presented by both authors convincingly explain the same phenomenon of reduced SASP transcripts after treatment with mTORC1 inhibitors, it is likely that there is synergy between the transcriptional increase induced by IL1A signaling and the stability increase from MAPKAPK2 activity (Citation81).
SENESCENCE INDUCERS TRIGGER eIF2α PHOSPHORYLATION
The phosphorylation of eIF2α is generally considered in the context of the integrated stress response (ISR) whereby cells responding to injury and metabolic stress reduce global protein synthesis (Citation58). The ISR is activated through the stress-sensing kinases heme-regulated inhibitor (HRI), double-stranded RNA-dependent protein kinase (PKR), PKR-like ER kinase (PERK), and general control nonderepressible 2 (GCN2) (Citation82). The HRI pathway can be activated by heme depletion (Citation83), intercellular infection (Citation84), and mitochondrial stress (Citation85). PERK is activated through binding immunoglobulin protein (BiP) by protein misfolding, and PKR appears to be activated by inflammation (Citation82). Finally, GCN2 is activated by diverse stressors, such as amino acid starvation, UV light, and oxidative stress (Citation82).
Senescence is triggered in skin keratinocytes by UV irradiation (Citation86, Citation87), which causes eIF2α phosphorylation by GCN2, resulting in a dramatic inhibition of protein synthesis that can be rescued by deletion of GCN2 (Citation88). The phosphorylation of eIF2α triggered by most ISR stressors enhances translation of the ATF4 transcription factor that then promotes transcription of downstream ISR genes. However, UV irradiation represses the transcription of ATF4 in addition to general protein synthesis and thus prevents the rise in the expression of ATF4 (Citation89). Instead, UV irradiation triggers a selective translational increase in DNA damage response proteins (Citation90).
UV irradiation has also been implicated in the translational upregulation of the cell cycle inhibitor p21, specifically of a splice variant containing an upstream open reading frame (uORF) (Citation91), a feature that is common to transcripts showing increased translation after eIF2α phosphorylation (Citation92). Furthermore, phosphorylation of eIF2α was shown to activate the NF-κB pathway, which promotes the SASP trait (Citation93) by reducing the translation of the NF-κB inhibitory protein IκBα (Citation94). The relationship between UV-mediated activation of the ISR and translation was recently highlighted in a report from the Green lab, demonstrating that ribosome collisions caused by UV-induced mRNA damage led to phosphorylation of eIF2α by GCN2 (Citation95).
TRANSLATION ELONGATION IN SENESCENT CELLS
Translation elongation in eukaryotes begins after the aminoacylated loaded into the P-site of the ribosome is positioned over a start codon, and a cognate aminoacyl-tRNA is transferred by eukaryotic elongation factor 1A (eEF1A) to the open A-site positioned over the codon immediately downstream of the start codon (Citation96). Upon codon-anticodon recognition, GTP hydrolysis releases eEF1A, and the peptide bond is formed as the tRNAs begin a hybrid transition from A and P sites into P and E sites on the ribosome, respectively, followed by translocation to the canonical P and E sites by eEF2-GTP hydrolysis (Citation96). The crucial involvement of eEF1A and eEF2 in translation explains both their natural abundance and their frequent dysregulation in hyperproliferative states, such as cancer (Citation97, Citation98). While no studies have systematically measured the abundance of translation elongation factors in senescence, individual studies have identified decreases of some elongation factors.
Cells subjected to OIS have decreased eEF1A expression compared with proliferating control cells (Citation99), and reduced eEF1A expression was an indicator of cancer cells entering senescence after exposure to gamma irradiation (Citation100). Genotoxic stress has also been shown to inhibit the activity of eEF2 through AMPK activation of the eEF2 regulatory kinase eEF2K (Citation101), although there is little information on eEF2 expression levels during senescence. Another elongation factor that appears to decline during senescence is eIF5A (Citation102), a translation factor that contains the unique hypusine modification that is implicated in the translation of polyproline stretches and in the resolution of ribosome stalling (Citation103, Citation104). The capacity of cells to generate the hypusine modification decreases during senescence, as do the levels of eIF5A mRNA (Citation102). Consistent with those observations, eIF5A hypusination in B cells was shown to decrease with age, and this decrease was found to reduce the translation of transcription factor EB (TFEB), an autophagy regulator (Citation105).
Similar to the measurement of translation factors, there are only a few reports on the actual translation efficiency and protein output of senescent cells. Earlier reports utilizing in vivo systems generally showed that protein synthesis decreased with organism age, although the effect was tissue specific and some results indicated either no change in translation or even an increase; cell-free experiments were generally more consistent, and ribosomes and mitochondria isolated from older rats exhibited reduced protein synthesis (Citation25). The majority of the earlier studies in cultured cells examining RS in fibroblasts, which best resemble contemporary senescence experiments, indicated that global protein synthesis was decreased (Citation25).
More recently, RS in fibroblasts was shown to cause reduced global protein synthesis, as measured by pulse labeling with 35S-labeled amino acids, while transcriptomic data appeared largely unchanged, suggesting that overall translation was suppressed (Citation23). Similarly, another study of RS in fibroblasts also indicated that senescent cells had reduced protein synthesis by using a puromycin incorporation assay and further showed that senescent cells had both reduced total polysomes and a reduced presence of mRNAs encoding ribosomal proteins within the polysome fraction (Citation24). The translation efficiency in senescent cells has also been examined using ribosome sequencing, a technique that infers translation efficiency based on ribosome occupancy on the mRNA. In response to OIS triggered by the expression of RASG12V, cells reduced the translation of ribosome biogenesis proteins and ribosomal proteins, which is anticipated to have the collective effect of reducing protein synthesis; however, when p53 and p16INK4A were stably knocked down, the translational repression caused by RASG12V was eliminated (Citation106).
PERSPECTIVES
Dichotomy of senescent cell translation.
The complex translational environment of senescent cells begins right at the onset of senescence. The sublethal genomic damage that triggers SIS, RS, and OIS includes nucleolar stress that leads to inhibition of ribosome biogenesis through the IRBC or the Rb protein (Citation30, Citation31). At the same time, the onset of senescence is marked by the activation of the mTORC1 pathway that phosphorylates 4E-BP and S6K1 to promote translation (Citation61). Furthermore, while inhibition of the mTORC1 pathway prevents the onset of senescence (Citation70), inhibition of ribosome biogenesis is sufficient to induce senescence (Citation30). These observations suggest that there are two essential and opposite forces acting upon translation in senescent cells, namely, a force of growth suppression that shuts down ribosome biogenesis and disengages the translation machinery and a force of translation activation through mTORC1.
A recent study suggested that the choice between apoptosis and senescence upon telomere shortening in zebrafish is dependent on the activation of the mTORC1 pathway, with activation being associated with senescence (Citation107). Translation in senescent cells may also be compartmentalized into regions of high and low protein synthesis, as has been suggested to explain the cooccurrence of mTORC1 activation and autophagy (Citation16, Citation61), wherein a burst of translation remodels the senescent cell proteome and translation-independent processes enforce those changes.
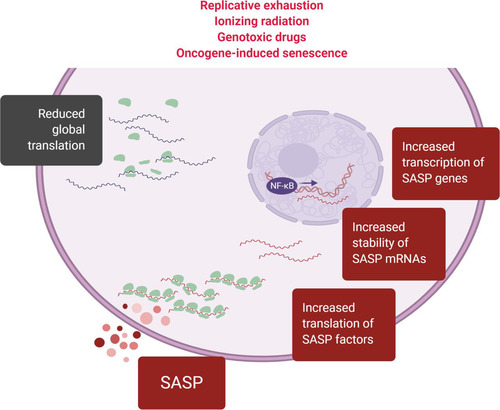
A major consequence of senescent translation is the SASP, a trait shared across senescence types and believed to be a key phenotype that makes senescent cells detrimental to the health of older organisms. The SASP requires an orchestrated and large-scale effort by the cellular machinery to transcribe, stabilize, and translate mRNAs encoding SASP factors, as well as to generate the proteins necessary for the processing and secretion of those SASP factors (). This translational effort is in direct opposition to the growth-suppressive functions of the p53 stress response pathway and is further unexpected given the observed reductions in components of the translation machinery. Instead, the levels of mRNAs encoding SASP factors increase during senescence (Citation18) and inhibition of the mTORC1 pathway suppresses the SASP (Citation77, Citation78), even though the translation of SASP factors was not affected by mTORC1 inhibition and SASP factors do not appear to contain mTORC1-sensitive 5′ TOP motifs. Thus, while the increase in mRNAs encoding SASP factors and the mTORC1 activation of senescence are undoubtedly essential to implementing the SASP, neither observation explains why the synthesis of SASP factors is bolstered amidst the large reductions in translation machinery components.
FIG 4 The SASP requires coordinated transcriptional and posttranscriptional regulation, including translational control. The SASP is induced in virtually all types of senescence. The production of SASP factors requires increased transcription, stabilization, and translation of mRNAs encoding SASP factors (“SASP mRNAs”). In light of the overall reduction of global translation in senescent cells, selective mechanisms that are as yet poorly understood specifically promote the biosynthesis of SASP factors during senescence.
Selective translation of subsets of mRNAs during senescence.
One possible explanation for the increased production of SASP factors during a time of translational repression is that SASP factors might be selectively translated. Such selective translation of mRNA subsets has been observed in response to other cellular stressors that similarly repress the translation machinery on a global scale. For example, the ISR selectively upregulates the translation of certain mRNAs containing upstream open reading frames (uORFs) (Citation92), while downregulating most protein translation. Similarly, the nucleolar stress that triggers the IRBC and reduces the synthesis of most ribosomal proteins selectively increases the translational efficiency of the 5S RNP component RPL11 (Citation108, Citation109). Other interesting cases of selective translation are the ribosomopathies 5q syndrome and Diamond-Blackfan anemia (DBA), both characterized by haploinsufficiency of several ribosomal proteins which can lead to activation of the IRBC (Citation110). A recent study revealed that a reduction in ribosome content due to the haploinsufficiency of ribosomal proteins characteristic of DBA specifically impaired the translation of well-translated transcripts that have short and unstructured 5′ UTRs (Citation111).
Translation in senescent cells could also be modulated by changes in the populations of available tRNAs, which could further alter the optimality of codons. It was previously observed that tRNA populations change between states of cancer, proliferation, quiescence, and senescence (Citation112) and that changes in tRNA expression are important for some cancers by changing the translation efficiency of certain codons (Citation113). Similarly, codon choice may offer another mechanism of selective translation, as codon optimality and codon pairs are recognized as important factors in translation efficiency as well as mRNA decay (Citation114, Citation115). It is possible that synonymous codons are treated differently by the cell during senescence or that mRNAs encoding members of the senescent proteome are enriched in codons that are refractory to changes in the translation machinery.
Goals for the future study of translation in senescent cells.
Upon genomic injury, a cell typically undergoes transformation, apoptosis, or senescence. As a means to prevent tumorigenesis, particularly in young organisms, understanding the molecular underpinnings of senescence has become an area of intense interest. Gaining a mechanistic understanding of senescence-associated translation is particularly important, given its direct role in the establishment and maintenance of the SASP.
An important future goal in the study of senescent cell translation is a comprehensive measurement of translation efficiency across the major senescent models to systematically determine global protein synthesis patterns, just as has been undertaken for the senescent transcriptome (Citation13, Citation14). Such global measurements have been performed in other systems by collectively quantifying protein levels and their rate of decay through stable isotopic labeling of amino acids in culture (SILAC) experiments (Citation116Citation–Citation118) and by integrating SILAC and ribosome sequencing analysis into one estimation of translation efficiency (Citation119). Another remaining gap in knowledge in senescent cell translation is quantifying the inhibition of rRNA processing and ribosome biogenesis that occur during senescence. Given the scale of these reductions, there should be a substantial impact on the ribosomes available for translation, of which the possible effects are not always obvious, as is evident from research on ribosomopathies.
There is a growing appreciation that suppressing senescence and the SASP can have a positive impact on age-associated diseases. In order to design therapeutic strategies that target senescent cells, a deeper understanding is needed of the basic mechanisms governing the transcription, turnover, and translation of mRNAs encoding senescence-relevant proteins. Given that the study of senescent cells has long been hampered by a lack of exclusive senescence markers, an improved knowledge of translation programs in senescence can further identify novel targets in elimination strategies. In closing, the delicate balance of progrowth and proarrest translation programs that coexist in senescent cells may offer unique venues for effective and specific therapies to treat age-associated diseases exacerbated by senescence.
ACKNOWLEDGMENTS
This work was supported in its entirety by the NIA IRP, NIH.
We thank Rachel Munk for critical reading of the manuscript.
REFERENCES
- Narita M, Lowe SW. 2005. Senescence comes of age. Nat Med 11:920–922. https://doi.org/10.1038/nm0905-920.
- Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, Lowe SW. 2013. Non-cell-autonomous tumor suppression by p53. Cell 153:449–460. https://doi.org/10.1016/j.cell.2013.03.020.
- Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, Iken M, Vucur M, Weiss S, Heikenwalder M, Khan S, Gil J, Bruder D, Manns M, Schirmacher P, Tacke F, Ott M, Luedde T, Longerich T, Kubicka S, Zender L. 2011. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479:547–551. https://doi.org/10.1038/nature10599.
- Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, Van Steeg H, Dolle ME, Hoeijmakers JH, de Bruin A, Hara E, Campisi J. 2014. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 31:722–733. https://doi.org/10.1016/j.devcel.2014.11.012.
- Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, Sansom OJ, Zender L, Keyes WM. 2017. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev 31:172–183. https://doi.org/10.1101/gad.290635.116.
- Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, Rodriguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, Serrano M. 2013. Programmed cell senescence during mammalian embryonic development. Cell 155:1104–1118. https://doi.org/10.1016/j.cell.2013.10.019.
- Franceschi C, Campisi J. 2014. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci 69:S4–S9. https://doi.org/10.1093/gerona/glu057.
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, Jin H, Dharmalingam G, Snijders AP, Carroll T, Capper D, Pritchard C, Inman GJ, Longerich T, Sansom OJ, Benitah SA, Zender L, Gil J. 2013. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15:978–990. https://doi.org/10.1038/ncb2784.
- Coppe JP, Desprez PY, Krtolica A, Campisi J. 2010. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol Mech Dis 5:99–118. https://doi.org/10.1146/annurev-pathol-121808-102144.
- Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, Luo Y, Wang X, Aykin-Burns N, Krager K, Ponnappan U, Hauer-Jensen M, Meng A, Zhou D. 2016. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 22:78–83. https://doi.org/10.1038/nm.4010.
- Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG, Onken JL, Johnson KO, Verzosa GC, Langhi LGP, Weigl M, Giorgadze N, LeBrasseur NK, Miller JD, Jurk D, Singh RJ, Allison DB, Ejima K, Hubbard GB, Ikeno Y, Cubro H, Garovic VD, Hou X, Weroha SJ, Robbins PD, Niedernhofer LJ, Khosla S, Tchkonia T, Kirkland JL. 2018. Senolytics improve physical function and increase lifespan in old age. Nat Med 24:1246–1256. https://doi.org/10.1038/s41591-018-0092-9.
- Cai Y, Zhou H, Zhu Y, Sun Q, Ji Y, Xue A, Wang Y, Chen W, Yu X, Wang L, Chen H, Li C, Luo T, Deng H. 2020. Elimination of senescent cells by beta-galactosidase-targeted prodrug attenuates inflammation and restores physical function in aged mice. Cell Res 30:574–589. https://doi.org/10.1038/s41422-020-0314-9.
- Hernandez-Segura A, de Jong TV, Melov S, Guryev V, Campisi J, Demaria M. 2017. Unmasking transcriptional heterogeneity in senescent cells. Curr Biol 27:2652–2660.e4. https://doi.org/10.1016/j.cub.2017.07.033.
- Casella G, Munk R, Kim KM, Piao Y, De S, Abdelmohsen K, Gorospe M. 2019. Transcriptome signature of cellular senescence. Nucleic Acids Res 47:7294–7305. https://doi.org/10.1093/nar/gkz555.
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. 2010. The essence of senescence. Genes Dev 24:2463–2479. https://doi.org/10.1101/gad.1971610.
- Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G, Gil J, Hara E, Krizhanovsky V, Jurk D, Maier AB, Narita M, Niedernhofer L, Passos JF, Robbins PD, Schmitt CA, Sedivy J, Vougas K, von Zglinicki T, Zhou D, Serrano M, Demaria M. 2019. Cellular senescence: defining a path forward. Cell 179:813–827. https://doi.org/10.1016/j.cell.2019.10.005.
- Rodier F, Campisi J. 2011. Four faces of cellular senescence. J Cell Biol 192:547–556. https://doi.org/10.1083/jcb.201009094.
- Coppé JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. 2008. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6:e301. https://doi.org/10.1371/journal.pbio.0060301.
- Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. 2008. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133:1019–1031. https://doi.org/10.1016/j.cell.2008.03.039.
- Childs BG, Durik M, Baker DJ, van Deursen JM. 2015. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 21:1424–1435. https://doi.org/10.1038/nm.4000.
- Correia-Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A, Rushton MD, Charles M, Jurk D, Tait SW, Czapiewski R, Greaves L, Nelson G, Bohlooly YM, Rodriguez-Cuenca S, Vidal-Puig A, Mann D, Saretzki G, Quarato G, Green DR, Adams PD, von Zglinicki T, Korolchuk VI, Passos JF. 2016. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J 35:724–742. https://doi.org/10.15252/embj.201592862.
- Li M, Durbin KR, Sweet SM, Tipton JD, Zheng Y, Kelleher NL. 2013. Oncogene-induced cellular senescence elicits an anti-Warburg effect. Proteomics 13:2585–2596. https://doi.org/10.1002/pmic.201200298.
- Srikantan S, Marasa BS, Becker KG, Gorospe M, Abdelmohsen K. 2011. Paradoxical microRNAs: individual gene repressors, global translation enhancers. Cell Cycle 10:751–759. https://doi.org/10.4161/cc.10.5.14825.
- Wu S, Xu S, Li R, Li K, Zhong X, Li Y, Zhou Z, Liu Y, Feng R, Zheng J, Songyang Z, Liu F. 2019. mTORC1-Rps15 axis contributes to the mechanisms underlying global translation reduction during senescence of mouse embryonic fibroblasts. Front Cell Dev Biol 7:337. https://doi.org/10.3389/fcell.2019.00337.
- Makrides SC. 1983. Protein synthesis and degradation during aging and senescence. Biol Rev Camb Philos Soc 58:343–422. https://doi.org/10.1111/j.1469-185X.1983.tb00394.x.
- Boisvert FM, van Koningsbruggen S, Navascues J, Lamond AI. 2007. The multifunctional nucleolus. Nat Rev Mol Cell Biol 8:574–585. https://doi.org/10.1038/nrm2184.
- Khatter H, Myasnikov AG, Natchiar SK, Klaholz BP. 2015. Structure of the human 80S ribosome. Nature 520:640–645. https://doi.org/10.1038/nature14427.
- Henras AK, Plisson-Chastang C, O'Donohue M-F, Chakraborty A, Gleizes P-E. 2015. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip Rev RNA 6:225–242. https://doi.org/10.1002/wrna.1269.
- Ciganda M, Williams N. 2011. Eukaryotic 5S rRNA biogenesis. Wiley Interdiscip Rev RNA 2:523–533. https://doi.org/10.1002/wrna.74.
- Nishimura K, Kumazawa T, Kuroda T, Katagiri N, Tsuchiya M, Goto N, Furumai R, Murayama A, Yanagisawa J, Kimura K. 2015. Perturbation of ribosome biogenesis drives cells into senescence through 5S RNP-mediated p53 activation. Cell Rep 10:1310–1323. https://doi.org/10.1016/j.celrep.2015.01.055.
- Lessard F, Igelmann S, Trahan C, Huot G, Saint-Germain E, Mignacca L, Del Toro N, Lopes-Paciencia S, Le Calve B, Montero M, Deschenes-Simard X, Bury M, Moiseeva O, Rowell MC, Zorca CE, Zenklusen D, Brakier-Gingras L, Bourdeau V, Oeffinger M, Ferbeyre G. 2018. Senescence-associated ribosome biogenesis defects contributes to cell cycle arrest through the Rb pathway. Nat Cell Biol 20:789–799. https://doi.org/10.1038/s41556-018-0127-y.
- Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI. 2010. The nucleolus under stress. Mol Cell 40:216–227. https://doi.org/10.1016/j.molcel.2010.09.024.
- Ogawa LM, Baserga SJ. 2017. Crosstalk between the nucleolus and the DNA damage response. Mol Biosyst 13:443–455. https://doi.org/10.1039/C6MB00740F.
- Lindström MS, Jurada D, Bursac S, Orsolic I, Bartek J, Volarevic S. 2018. Nucleolus as an emerging hub in maintenance of genome stability and cancer pathogenesis. Oncogene 37:2351–2366. https://doi.org/10.1038/s41388-017-0121-z.
- Bieging KT, Mello SS, Attardi LD. 2014. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 14:359–370. https://doi.org/10.1038/nrc3711.
- Hu W, Feng Z, Levine AJ. 2012. The regulation of multiple p53 stress responses is mediated through MDM2. Genes Cancer 3:199–208. https://doi.org/10.1177/1947601912454734.
- Kruse JP, Gu W. 2009. Modes of p53 regulation. Cell 137:609–622. https://doi.org/10.1016/j.cell.2009.04.050.
- Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. 2003. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 3:577–587. https://doi.org/10.1016/S1535-6108(03)00134-X.
- Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA, Xiong Y. 2003. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol 23:8902–8912. https://doi.org/10.1128/MCB.23.23.8902-8912.2003.
- Dai MS, Lu H. 2004. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J Biol Chem 279:44475–44482. https://doi.org/10.1074/jbc.M403722200.
- Sun XX, Wang YG, Xirodimas DP, Dai MS. 2010. Perturbation of 60 S ribosomal biogenesis results in ribosomal protein L5- and L11-dependent p53 activation. J Biol Chem 285:25812–25821. https://doi.org/10.1074/jbc.M109.098442.
- Bursac S, Brdovcak MC, Pfannkuchen M, Orsolic I, Golomb L, Zhu Y, Katz C, Daftuar L, Grabusic K, Vukelic I, Filic V, Oren M, Prives C, Volarevic S. 2012. Mutual protection of ribosomal proteins L5 and L11 from degradation is essential for p53 activation upon ribosomal biogenesis stress. Proc Natl Acad Sci U S A 109:20467–20472. https://doi.org/10.1073/pnas.1218535109.
- Horn HF, Vousden KH. 2008. Cooperation between the ribosomal proteins L5 and L11 in the p53 pathway. Oncogene 27:5774–5784. https://doi.org/10.1038/onc.2008.189.
- Donati G, Peddigari S, Mercer CA, Thomas G. 2013. 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep 4:87–98. https://doi.org/10.1016/j.celrep.2013.05.045.
- Sloan KE, Bohnsack MT, Watkins NJ. 2013. The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep 5:237–247. https://doi.org/10.1016/j.celrep.2013.08.049.
- Gentilella A, Moron-Duran FD, Fuentes P, Zweig-Rocha G, Riano-Canalias F, Pelletier J, Ruiz M, Turon G, Castano J, Tauler A, Bueno C, Menendez P, Kozma SC, Thomas G. 2017. Autogenous control of 5′TOP mRNA stability by 40S ribosomes. Mol Cell 67:55–70.e4. https://doi.org/10.1016/j.molcel.2017.06.005.
- Bensaude O. 2011. Inhibiting eukaryotic transcription: which compound to choose? How to evaluate its activity? Transcription 2:103–108. https://doi.org/10.4161/trns.2.3.16172.
- Kruhlak M, Crouch EE, Orlov M, Montano C, Gorski SA, Nussenzweig A, Misteli T, Phair RD, Casellas R. 2007. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature 447:730–734. https://doi.org/10.1038/nature05842.
- Govoni M, Farabegoli F, Pession A, Novello F. 1994. Inhibition of topoisomerase II activity and its effect on nucleolar structure and function. Exp Cell Res 211:36–41. https://doi.org/10.1006/excr.1994.1055.
- Zhu Y, Poyurovsky MV, Li Y, Biderman L, Stahl J, Jacq X, Prives C. 2009. Ribosomal protein S7 is both a regulator and a substrate of MDM2. Mol Cell 35:316–326. https://doi.org/10.1016/j.molcel.2009.07.014.
- Rubbi CP, Milner J. 2003. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J 22:6068–6077. https://doi.org/10.1093/emboj/cdg579.
- Zhai W, Comai L. 2000. Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol Cell Biol 20:5930–5938. https://doi.org/10.1128/MCB.20.16.5930-5938.2000.
- Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavare S, Arakawa S, Shimizu S, Watt FM, Narita M. 2009. Autophagy mediates the mitotic senescence transition. Genes Dev 23:798–803. https://doi.org/10.1101/gad.519709.
- Jackson RJ, Hellen CU, Pestova TV. 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11:113–127. https://doi.org/10.1038/nrm2838.
- Saxton RA, Sabatini DM. 2017. mTOR signaling in growth, metabolism, and disease. Cell 168:960–976. https://doi.org/10.1016/j.cell.2017.02.004.
- Bernard M, Yang B, Migneault F, Turgeon J, Dieude M, Olivier MA, Cardin GB, El-Diwany M, Underwood K, Rodier F, Hebert MJ. 2020. Autophagy drives fibroblast senescence through MTORC2 regulation. Autophagy 16:2004–2016. https://doi.org/10.1080/15548627.2020.1713640.
- Jung SH, Hwang HJ, Kang D, Park HA, Lee HC, Jeong D, Lee K, Park HJ, Ko Y-G, Lee J-S. 2019. mTOR kinase leads to PTEN-loss-induced cellular senescence by phosphorylating p53. Oncogene 38:1639–1650. https://doi.org/10.1038/s41388-018-0521-8.
- Sonenberg N, Hinnebusch AG. 2009. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136:731–745. https://doi.org/10.1016/j.cell.2009.01.042.
- Viniegra JG, Martinez N, Modirassari P, Hernandez Losa J, Parada Cobo C, Sanchez-Arevalo Lobo VJ, Aceves Luquero CI, Alvarez-Vallina L, Ramon y, Cajal S, Rojas JM, Sanchez-Prieto R. 2005. Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM. J Biol Chem 280:4029–4036. https://doi.org/10.1074/jbc.M410344200.
- Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. 2004. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A 101:13489–13494. https://doi.org/10.1073/pnas.0405659101.
- Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, Hong S, Berry LS, Reichelt S, Ferreira M, Tavare S, Inoki K, Shimizu S, Narita M. 2011. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 332:966–970. https://doi.org/10.1126/science.1205407.
- Cho S, Hwang ES. 2012. Status of mTOR activity may phenotypically differentiate senescence and quiescence. Mol Cells 33:597–604. https://doi.org/10.1007/s10059-012-0042-1.
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 92:9363–9367. https://doi.org/10.1073/pnas.92.20.9363.
- Ma XM, Blenis J. 2009. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 10:307–318. https://doi.org/10.1038/nrm2672.
- Salama R, Sadaie M, Hoare M, Narita M. 2014. Cellular senescence and its effector programs. Genes Dev 28:99–114. https://doi.org/10.1101/gad.235184.113.
- Bengsch F, Tu Z, Tang HY, Zhu H, Speicher DW, Zhang R. 2015. Comprehensive analysis of the ubiquitinome during oncogene-induced senescence in human fibroblasts. Cell Cycle 14:1540–1547. https://doi.org/10.1080/15384101.2015.1026492.
- Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, Elledge SJ. 2015. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349:aaa5612. https://doi.org/10.1126/science.aaa5612.
- Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. 2009. Rapamycin decelerates cellular senescence. Cell Cycle 8:1888–1895. https://doi.org/10.4161/cc.8.12.8606.
- Leontieva OV, Blagosklonny MV. 2016. Gerosuppression by pan-mTOR inhibitors. Aging (Albany NY) 8:3535–3551. https://doi.org/10.18632/aging.101155.
- Leontieva OV, Blagosklonny MV. 2010. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY) 2:924–935. https://doi.org/10.18632/aging.100265.
- Kolesnichenko M, Hong L, Liao R, Vogt PK, Sun P. 2012. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle 11:2391–2401. https://doi.org/10.4161/cc.20683.
- Pospelova TV, Leontieva OV, Bykova TV, Zubova SG, Pospelov VA, Blagosklonny MV. 2012. Suppression of replicative senescence by rapamycin in rodent embryonic cells. Cell Cycle 11:2402–2407. https://doi.org/10.4161/cc.20882.
- Walters HE, Deneka-Hannemann S, Cox LS. 2016. Reversal of phenotypes of cellular senescence by pan-mTOR inhibition. Aging (Albany NY) 8:231–244. https://doi.org/10.18632/aging.100872.
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. 2012. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485:109–113. https://doi.org/10.1038/nature11083.
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, Wang S, Ren P, Martin M, Jessen K, Feldman ME, Weissman JS, Shokat KM, Rommel C, Ruggero D. 2012. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485:55–61. https://doi.org/10.1038/nature10912.
- Gentilella A, Thomas G. 2012. Cancer biology: the director’s cut. Nature 485:50–51. https://doi.org/10.1038/485050a.
- Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, Limbad C, Demaria M, Li P, Hubbard GB, Ikeno Y, Javors M, Desprez PY, Benz CC, Kapahi P, Nelson PS, Campisi J. 2015. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 17:1049–1061. https://doi.org/10.1038/ncb3195.
- Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito A, Georgilis A, Montoya A, Wolter K, Dharmalingam G, Faull P, Carroll T, Martinez-Barbera JP, Cutillas P, Reisinger F, Heikenwalder M, Miller RA, Withers D, Zender L, Thomas GJ, Gil J. 2015. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol 17:1205–1217. https://doi.org/10.1038/ncb3225.
- Aarts M, Georgilis A, Beniazza M, Beolchi P, Banito A, Carroll T, Kulisic M, Kaemena DF, Dharmalingam G, Martin N, Reik W, Zuber J, Kaji K, Chandra T, Gil J. 2017. Coupling shRNA screens with single-cell RNA-seq identifies a dual role for mTOR in reprogramming-induced senescence. Genes Dev 31:2085–2098. https://doi.org/10.1101/gad.297796.117.
- Anerillas C, Abdelmohsen K, Gorospe M. 2020. Regulation of senescence traits by MAPKs. Geroscience 42:397–408. https://doi.org/10.1007/s11357-020-00183-3.
- Tomimatsu K, Narita M. 2015. Translating the effects of mTOR on secretory senescence. Nat Cell Biol 17:1230–1232. https://doi.org/10.1038/ncb3244.
- Costa-Mattioli M, Walter P. 2020. The integrated stress response: from mechanism to disease. Science 368:eaat5314. https://doi.org/10.1126/science.aat5314.
- Chen J-J, London IM. 1995. Regulation of protein synthesis by heme-regulated eIF-2 alpha kinase. Trends Biochem Sci 20:105–108. https://doi.org/10.1016/S0968-0004(00)88975-6.
- Abdel-Nour M, Carneiro LAM, Downey J, Tsalikis J, Outlioua A, Prescott D, Da Costa LS, Hovingh ES, Farahvash A, Gaudet RG, Molinaro R, van Dalen R, Lau CCY, Azimi FC, Escalante NK, Trotman-Grant A, Lee JE, Gray-Owen SD, Divangahi M, Chen JJ, Philpott DJ, Arnoult D, Girardin SE. 2019. The heme-regulated inhibitor is a cytosolic sensor of protein misfolding that controls innate immune signaling. Science 365:eaaw4144. https://doi.org/10.1126/science.aaw4144.
- Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin YT, Wiita AP, Xu K, Correia MA, Kampmann M. 2020. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 579:427–432. https://doi.org/10.1038/s41586-020-2078-2.
- Gu Y, Han J, Jiang C, Zhang Y. 2020. Biomarkers, oxidative stress and autophagy in skin aging. Ageing Res Rev 59:101036. https://doi.org/10.1016/j.arr.2020.101036.
- Wang Y, Wang L, Wen X, Hao D, Zhang N, He G, Jiang X. 2019. NF-kappaB signaling in skin aging. Mech Ageing Dev 184:111160. https://doi.org/10.1016/j.mad.2019.111160.
- Deng J, Harding HP, Raught B, Gingras AC, Berlanga JJ, Scheuner D, Kaufman RJ, Ron D, Sonenberg N. 2002. Activation of GCN2 in UV-irradiated cells inhibits translation. Curr Biol 12:1279–1286. https://doi.org/10.1016/S0960-9822(02)01037-0.
- Baird TD, Wek RC. 2012. Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv Nutr 3:307–321. https://doi.org/10.3945/an.112.002113.
- Powley IR, Kondrashov A, Young LA, Dobbyn HC, Hill K, Cannell IG, Stoneley M, Kong YW, Cotes JA, Smith GC, Wek R, Hayes C, Gant TW, Spriggs KA, Bushell M, Willis AE. 2009. Translational reprogramming following UVB irradiation is mediated by DNA-PKcs and allows selective recruitment to the polysomes of mRNAs encoding DNA repair enzymes. Genes Dev 23:1207–1220. https://doi.org/10.1101/gad.516509.
- Collier AE, Spandau DF, Wek RC. 2018. Translational control of a human CDKN1A mRNA splice variant regulates the fate of UVB-irradiated human keratinocytes. Mol Biol Cell 29:29–41. https://doi.org/10.1091/mbc.E17-06-0362.
- Young SK, Wek RC. 2016. Upstream open reading frames differentially regulate gene-specific translation in the integrated stress response. J Biol Chem 291:16927–16935. https://doi.org/10.1074/jbc.R116.733899.
- Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, Kogan SC, Lowe SW. 2011. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev 25:2125–2136. https://doi.org/10.1101/gad.17276711.
- Jiang HY, Wek RC. 2005. GCN2 phosphorylation of eIF2alpha activates NF-kappaB in response to UV irradiation. Biochem J 385:371–380. https://doi.org/10.1042/BJ20041164.
- Wu CC-C, Peterson A, Zinshteyn B, Regot S, Green R. 2020. Ribosome collisions trigger general stress responses to regulate cell fate. Cell 182:404–416.e14. https://doi.org/10.1016/j.cell.2020.06.006.
- Dever TE, Green R. 2012. The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harb Perspect Biol 4:a013706. https://doi.org/10.1101/cshperspect.a013706.
- Knight JRP, Garland G, Poyry T, Mead E, Vlahov N, Sfakianos A, Grosso S, De-Lima-Hedayioglu F, Mallucci GR, von der Haar T, Smales CM, Sansom OJ, Willis AE. 2020. Control of translation elongation in health and disease. Dis Model Mech 13:dmm043208. https://doi.org/10.1242/dmm.043208.
- Abbas W, Kumar A, Herbein G. 2015. The eEF1A proteins: at the crossroads of oncogenesis. Front Oncol 5:75. https://doi.org/10.3389/fonc.2015.00075.
- Byun H-O, Han N-K, Lee H-J, Kim K-B, Ko Y-G, Yoon G, Lee Y-S, Hong S-I, Lee J-S. 2009. Cathepsin D and eukaryotic translation elongation factor 1 as promising markers of cellular senescence. Cancer Res 69:4638–4647. https://doi.org/10.1158/0008-5472.CAN-08-4042.
- Kim BC, Yoo HJ, Lee HC, Kang KA, Jung SH, Lee H-J, Lee M, Park S, Ji Y-H, Lee Y-S, Ko Y-G, Lee J-S. 2014. Evaluation of premature senescence and senescence biomarkers in carcinoma cells and xenograft mice exposed to single or fractionated irradiation. Oncol Rep 31:2229–2235. https://doi.org/10.3892/or.2014.3069.
- Kruiswijk F, Yuniati L, Magliozzi R, Low TY, Lim R, Bolder R, Mohammed S, Proud CG, Heck AJ, Pagano M, Guardavaccaro D. 2012. Coupled activation and degradation of eEF2K regulates protein synthesis in response to genotoxic stress. Sci Signal 5:ra40. https://doi.org/10.1126/scisignal.2002718.
- Chen ZP, Chen KY. 1997. Dramatic attenuation of hypusine formation on eukaryotic initiation factor 5A during senescence of IMR-90 human diploid fibroblasts. J Cell Physiol 170:248–254. https://doi.org/10.1002/(SICI)1097-4652(199703)170:3<248::AID-JCP5>3.0.CO;2-O.
- Dever TE, Gutierrez E, Shin B-S. 2014. The hypusine-containing translation factor eIF5A. Crit Rev Biochem Mol Biol 49:413–425. https://doi.org/10.3109/10409238.2014.939608.
- Schuller AP, Green R. 2018. Roadblocks and resolutions in eukaryotic translation. Nat Rev Mol Cell Biol 19:526–541. https://doi.org/10.1038/s41580-018-0011-4.
- Zhang H, Simon AK. 2020. Polyamines reverse immune senescence via the translational control of autophagy. Autophagy 16:181–182. https://doi.org/10.1080/15548627.2019.1687967.
- Loayza-Puch F, Drost J, Rooijers K, Lopes R, Elkon R, Agami R. 2013. p53 induces transcriptional and translational programs to suppress cell proliferation and growth. Genome Biol 14:R32. https://doi.org/10.1186/gb-2013-14-4-r32.
- El Mai M, Marzullo M, de Castro IP, Ferreira MG. 2020. Opposing p53 and mTOR/AKT promote an in vivo switch from apoptosis to senescence upon telomere shortening in zebrafish. Elife 9:e54935. https://doi.org/10.7554/eLife.54935.
- Fumagalli S, Di Cara A, Neb-Gulati A, Natt F, Schwemberger S, Hall J, Babcock GF, Bernardi R, Pandolfi PP, Thomas G. 2009. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat Cell Biol 11:501–508. https://doi.org/10.1038/ncb1858.
- Fumagalli S, Ivanenkov VV, Teng T, Thomas G. 2012. Suprainduction of p53 by disruption of 40S and 60S ribosome biogenesis leads to the activation of a novel G2/M checkpoint. Genes Dev 26:1028–1040. https://doi.org/10.1101/gad.189951.112.
- Teng T, Thomas G, Mercer CA. 2013. Growth control and ribosomopathies. Curr Opin Genet Dev 23:63–71. https://doi.org/10.1016/j.gde.2013.02.001.
- Khajuria RK, Munschauer M, Ulirsch JC, Fiorini C, Ludwig LS, McFarland SK, Abdulhay NJ, Specht H, Keshishian H, Mani DR, Jovanovic M, Ellis SR, Fulco CP, Engreitz JM, Schutz S, Lian J, Gripp KW, Weinberg OK, Pinkus GS, Gehrke L, Regev A, Lander ES, Gazda HT, Lee WY, Panse VG, Carr SA, Sankaran VG. 2018. Ribosome levels selectively regulate translation and lineage commitment in human hematopoiesis. Cell 173:90–103.e19. https://doi.org/10.1016/j.cell.2018.02.036.
- Gingold H, Tehler D, Christoffersen NR, Nielsen MM, Asmar F, Kooistra SM, Christophersen NS, Christensen LL, Borre M, Sørensen KD, Andersen LD, Andersen CL, Hulleman E, Wurdinger T, Ralfkiær E, Helin K, Grønbæk K, Ørntoft T, Waszak SM, Dahan O, Pedersen JS, Lund AH, Pilpel Y. 2014. A dual program for translation regulation in cellular proliferation and differentiation. Cell 158:1281–1292. https://doi.org/10.1016/j.cell.2014.08.011.
- Goodarzi H, Nguyen HCB, Zhang S, Dill BD, Molina H, Tavazoie SF. 2016. Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell 165:1416–1427. https://doi.org/10.1016/j.cell.2016.05.046.
- Brule CE, Grayhack EJ. 2017. Synonymous codons: choose wisely for expression. Trends Genet 33:283–297. https://doi.org/10.1016/j.tig.2017.02.001.
- Presnyak V, Alhusaini N, Chen YH, Martin S, Morris N, Kline N, Olson S, Weinberg D, Baker KE, Graveley BR, Coller J. 2015. Codon optimality is a major determinant of mRNA stability. Cell 160:1111–1124. https://doi.org/10.1016/j.cell.2015.02.029.
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. 2011. Global quantification of mammalian gene expression control. Nature 473:337–342. https://doi.org/10.1038/nature10098.
- Jovanovic M, Rooney MS, Mertins P, Przybylski D, Chevrier N, Satija R, Rodriguez EH, Fields AP, Schwartz S, Raychowdhury R, Mumbach MR, Eisenhaure T, Rabani M, Gennert D, Lu D, Delorey T, Weissman JS, Carr SA, Hacohen N, Regev A. 2015. Dynamic profiling of the protein life cycle in response to pathogens. Science 347:1259038. https://doi.org/10.1126/science.1259038.
- Lahtvee PJ, Sánchez BJ, Smialowska A, Kasvandik S, Elsemman IE, Gatto F, Nielsen J. 2017. Absolute quantification of protein and mRNA abundances demonstrate variability in gene-specific translation efficiency in yeast. Cell Syst 4:495–504.e5. https://doi.org/10.1016/j.cels.2017.03.003.
- Liu T-Y, Huang HH, Wheeler D, Xu Y, Wells JA, Song YS, Wiita AP. 2017. Time-resolved proteomics extends ribosome profiling-based measurements of protein synthesis dynamics. Cell Syst 4:636–644.e9. https://doi.org/10.1016/j.cels.2017.05.001.