
Abstract
The Fbw7 ubiquitin ligase critically regulates hematopoietic stem cell (HSC) function, though the precise contribution of individual substrate ubiquitination pathways to HSC homeostasis is unknown. In the work reported here, we used a mouse model in which we introduced two knock-in mutations (T74A and T393A [changes of T to A at positions 74 and 393]) to disrupt Fbw7-dependent regulation of cyclin E, its prototypic substrate, and to examine the consequences of cyclin E dysregulation for HSC function. Serial transplantation revealed that cyclin ET74A T393A HSCs self-renewed normally; however, we identified defects in their multilineage reconstituting capacity. By inducing hematologic stress, we exposed an impaired self-renewal phenotype in cyclin E knock-in HSCs that was associated with defective cell cycle exit and the emergence of chromosome instability (CIN). Importantly, p53 deletion induced both defects in self-renewal and multilineage reconstitution in cyclin E knock-in HSCs with serial transplantation and CIN in hematopoietic stem and progenitor cells. Moreover, CIN was a feature of fatal T-cell malignancies that ultimately developed in recipients of cyclin ET74A T393A; p53-null HSCs. Together, our findings demonstrate the importance of Fbw7-dependent cyclin E control to the hematopoietic system and highlight CIN as a characteristic feature of HSC dysfunction and malignancy induced by deregulated cyclin E.
INTRODUCTION
The hematopoietic system is maintained by stem cells with self-renewal and multilineage reconstituting potential. Hematopoietic stem cells (HSCs) are functionally heterogeneous with regard to their intrinsic self-renewal capacity, with a distinct subpopulation possessing long-term self-renewal potential (Citation1). Under steady-state conditions, HSCs are predominantly quiescent (Citation2, Citation3). Upon hematologic injury, dormant HSCs reenter the cell cycle to generate progenitor cells, which ultimately give rise to mature cells that occupy the bone marrow and peripheral blood. After reestablishment of hematopoietic homeostasis, injury-activated HSCs return to dormancy (Citation1).
Cell cycle regulators play key roles in establishing bistability in the switch between quiescence and cell cycle reentry (Citation4). The cyclin-dependent kinase inhibitors p21Cip1, p27Kip1, and p57Kip2 maintain hematopoietic stem and progenitor cell (HSPC) quiescence by restraining cyclin-dependent kinase (Cdk) activity, and their inactivation in hematopoietic cells can alter the HSPC pool size and repopulating capacity (Citation5Citation–Citation8). Additionally, the Retinoblastoma family proteins pRb, p107, and p130 cooperatively maintain HSC quiescence, and their loss of function results in increased proliferation and impaired self-renewal (Citation9). Taken together, these studies strongly support the conclusion that hyperproliferation is intrinsically linked to stem cell exhaustion. Other reports, however, challenge the notion that increased proliferation of HSCs necessarily opposes their self-renewal capacity. Notably, the loss of p18Ink4c, p53, and Junb all lead to increased HSC proliferation without apparent induction of stem cell exhaustion (Citation10Citation–Citation15). Thus, under certain conditions, HSC proliferation may be uncoupled from self-renewal.
Fbw7 is a substrate-binding module for an SCF (Skp1/Cul1/F-box protein)-type E3 ubiquitin ligase, and SCFFbw7 regulates the abundance of a number of oncoprotein substrates, including cyclin E, Notch, c-Jun, and c-Myc, by ubiquitin-mediated proteolysis (Citation16). Fbw7 controls HSC quiescence, and inactivation of Fbxw7 causes premature loss of HSCs due to excessive cycling and p53-dependent apoptosis (Citation17Citation–Citation19). Both c-Myc and Notch are deregulated in Fbw7-deficient hematopoietic cells, and yet the role of other substrates, including cyclin E, in phenotypes associated with Fbw7 loss or Fbxw7 mutations remains unclear.
Cyclin E is a critical mediator of S-phase reentry following quiescence exit (Citation20) and has diverse functions supporting cell cycle progression and the regulation of gene expression and epigenetic state (Citation21). We previously reported the generation of a cyclin ET74A T393A (knock-in mutations of T to A at positions 74 and 393) mouse model (Citation22), in which mutations were introduced into two conserved domains termed Cdc4 phosphodegrons (CPDs) that regulate the interaction between SCFFbw7 and its substrates, including cyclin E (Citation23, Citation24). In the cyclin ET74A T393A strain, anemia with defective erythropoiesis is associated with impaired control of oxidative metabolism and reactive oxygen species during terminal erythroid maturation (Citation25). In this study, we have examined the physiologic consequences of disrupting Fbw7-dependent cyclin E controls upon HSC function. We find that deregulation of cyclin E during steady-state hematopoiesis induces a cell-autonomous defect in multilineage reconstitution but not self-renewal with serial transplantation. Hematologic injury exposes deficient self-renewal in cyclin ET74A T393A HSCs, which we find is associated with the emergence of chromosome instability (CIN). Importantly, the loss of both p53- and Fbw7-dependent pathways for regulating cyclin E activity induces defective self-renewal and CIN in HSCs. Moreover, deregulated cyclin E and p53 mutations cooperatively promote genomically unstable, fatal T-cell neoplasms in recipients of cyclin E knock-in; p53-null HSCs. Thus, ubiquitin-mediated destruction of cyclin E is a critical component of SCFFbw7 function in both HSCs and cells of distinct hematopoietic lineages in vivo.
MATERIALS AND METHODS
Mice.
The generation of the cyclin ET74A T393A knock-in strain was previously described (Citation22). C57BL/6 CD45.1 congenic mice, Trp53 knockout (p53−/−) mice, Cdkn1a knockout (p21−/−) mice, Fbxw7flox/flox mice (here called Fbxw7fl/fl mice), and Mx1-cre+ mice were obtained from Jackson Laboratory. Cyclin ET74A T393A knock-in; p53 and p21 knockout mice were generated by crossing cyclin ET74A T393A mice with p53−/− or p21−/− mice, all in C57BL/6 backgrounds. 5-Fluorouracil (5FU) was obtained from APP Pharmaceuticals, LLC, and it was administered at a dose of 150 mg/kg of body weight via intraperitoneal injection. Mx1-cre+ Fbxw7fl/fl mice were generated by crossing Mx1-cre+ mice with Fbxw7fl/fl mice. To induce Cre expression in vivo, mice were injected with poly(I) · poly(C) (20 μg/g of body weight; GE Healthcare, Little Chalfont, United Kingdom) every other day for a total of three doses. Mice were analyzed 2 weeks after the end of the poly(I) · poly(C) treatment. All mice were genotyped by PCR using published protocols. The Northwestern University IACUC approved all mouse studies. In accordance with IACUC guidelines, in tumor induction studies, mice were euthanized when they appeared moribund, as defined by at least two of the following criteria: weight loss of more than 20% of the baseline weight, decreased movement and grooming, and hunched posture.
HSC transplantation.
Serial transplantation assays were performed as described in the legend to . For transplants of purified HSCs, CD45.1 recipients were placed on water containing trimethoprim-sulfamethoxazole (Hi-Tech Pharmacal) for 1 week before irradiation with a total of 11 Gy divided over two equal fractions (separated by 3 h) the day before transplantation. Recipient mice were then administered, via retroorbital sinus injection, 50 HSCs (CD45.2) of one of the immunophenotypes described in . In all, 3 × 105 Lin+ Sca1− bone marrow cells (CD45.1) were used as support cells. Peripheral blood was obtained monthly and stained with CD45.1 and CD45.2 antibodies to verify donor cell engraftment. For other transplantation experiments, recipient mice (CD45.1) were irradiated as previously described. One million total nucleated bone marrow cells (CD45.2+) from donor mice were injected retroorbitally into CD45.1 recipients. Twelve weeks after transplantation, recipient engraftment was monitored along with complete blood counts.
FIG 1 Cyclin ET74A T393A HSCs are defective for multilineage reconstitution but not long-term self-renewal. (A) Left, bone marrow cells from wild-type or cyclin ET74A T393A mice were enriched for HSPCs by immunomagnetic separation based on lineage-negative CD150+ surface marker expression. Western blot analyses of cyclin E abundance are shown. Right, transcript abundances of Ccne1 in primary bone marrow HSPCs were compared using quantitative RT-PCR. Error bars indicate standard deviations with two biological replicates. (B) Left, bone marrow cells from poly(I) · poly(C)-treated Mx1-cre− Fbxw7fl/fl and Mx1-cre+ Fbxw7fl/fl mice were harvested for Western blot analyses. Cyclin E protein abundances in bone marrow extracts were analyzed using affinity-purified anti-cyclin E antibody, with anti-Grb2 antibody as the loading control. Loss of the floxed band in bone marrow and excision of the deleted allele were confirmed by PCR. Right, E14.5 fetal liver cells from wild-type, Fbxw7fl/+, or Fbxw7fl/fl mice were lineage depleted, followed by transduction with Cre recombinase. Immunoblot analyses of cyclin E abundance in Cre-expressing fetal liver extracts are shown. (C) Schematic of serial adoptive transfer assays is shown. Donor (CD45.2) HSCs of the indicated genotypes were isolated based on criteria listed in and transplanted into irradiated (11 Gy) CD45.1 recipients, along with Lin+ Sca1− CD45.1 cells for short-term hematopoietic support. Recipient mice were monitored for 3 months, after which unfractionated bone marrow cells were prepared from primary recipients and injected into irradiated wild-type mice. This process was reiterated after 3 months. Symbols (1°, 2°, 3°) denote primary, secondary, and tertiary transplants, respectively. (D and E) Peripheral blood and total bone marrow cell donor chimerisms were calculated as shown in y axes at 3 months after each round of transplantation (n = 9 to 14 recipients per group). (F and G) Lineage-specific donor chimerism and bone marrow composition of donor-derived cells following secondary transplantation are shown. (H) Absolute numbers of donor-derived HSCs were enumerated in individual recipient mice 3 months after each round of transplantation. (D to G) Error bars display standard errors. *, P < 0.05; **, P < 0.005; ***, P < 0.0001, using Student's t test.
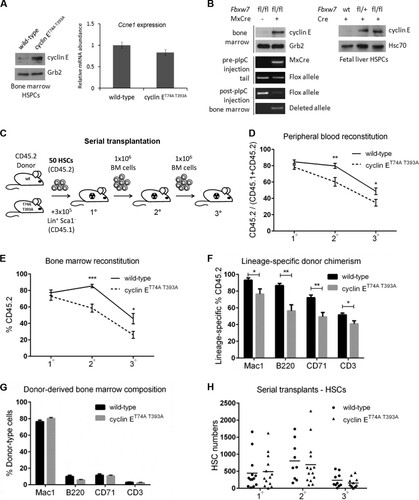
TABLE 1 Characterization of hematopoietic stem and progenitor cellsTable Footnotea
Hematopoietic analyses.
Thymuses, bilateral femurs, and tibiae were obtained from age- and sex-matched mice. Bone marrow cells were harvested from bilateral femurs and tibiae and resuspended in phosphate-buffered saline (PBS)–2% fetal bovine serum (FBS). For transplantation studies, highly purified HSCs and other bone marrow cells were collected using a MoFlo (Dako) or FACSAria (BD Biosciences) cell sorter. For bone marrow and thymus immunophenotyping studies, cells were antibody stained and analyzed using an LSRII flow cytometer (BD Biosciences). For HSC cell cycle studies, bone marrow cells were fixed in 2% paraformaldehyde (PFA)–PBS, permeabilized in 0.5% saponin–PBS–2% FBS, and labeled with fluorescein isothiocyanate (FITC)-conjugated Ki-67 (BD Pharmingen) and Hoechst 33342 (Sigma-Aldrich) for fluorescence-activated cell sorting (FACS) analysis. For B-lymphocyte apoptosis studies, bone marrow cells were stained with anti-B220 antibodies and allophycocyanin (APC)-conjugated annexin V and 7-aminoactinomycin D (7-AAD), following the manufacturer's instructions (BD Pharmingen). For thymus cell cycle studies, thymocytes were fixed in 1% PFA, washed, and stained in propidium iodide mix containing PBS with 0.1% bovine serum albumin (Sigma-Aldrich), 0.1% saponin (Sigma-Aldrich), 10 μg/ml propidium iodide, and 10 U/ml RNase A (Qiagen). FACS data were analyzed using FACSDiva (BD Biosciences) and FlowJo (Tree Star). Morphological analyses were performed on bone marrow and peripheral blood using Wright-Giemsa stain solution (Sigma-Aldrich).
Antibodies.
An EasySep mouse hematopoietic progenitor enrichment kit (Stemcell Technologies) was used to isolate lineage-negative bone marrow cells. For other bone marrow cells and thymocytes, we used the following antibodies: APC-conjugated anti-Mac1 (M1/70), anti-CD45.1 (A20), anti-Gr1 (RB6-8C5), and anti-CD4 (RM4-5) antibodies and anti-IgM (II/41); APC-Cy7-conjugated anti-c-kit antibody (2B8); phycoerythrin (PE)-conjugated anti-CD8a (53-6.7) and anti-FcγR (2.4G2) antibodies; PE-Cy7-conjugated anti-Sca1 antibody (D7); peridinin chlorophyll protein (PerCP)-Cy5.5-conjugated streptavidin antibody; FITC-conjugated anti-CD41 antibody (MW Reg30); and FITC- or PE-conjugated anti-CD34 (RAM34), anti-B220 (RA3-6B2), anti-Mac1 (M1/70), anti-CD3 (17A2), anti-CD71 (C1), and anti-CD45.2 (104) antibodies, all purchased from BD Pharmingen. APC-conjugated anti-CD150 antibody (TC15-12F12.2) was purchased from BioLegend. The following antibodies were purchased from eBioscience: APC-conjugated anti-Flt3 antibody (A2F10); efluor 450-conjugated anti-B220 (RA3-6B2) and antistreptavidin antibodies; Pacific blue-conjugated anti-CD34 antibody (RAM34); PE-conjugated anti-CD48 antibody (HM48-1); and PerCP-Cy5.5-conjugated anti-interleukin-7 receptor α (anti-IL-7Rα) antibody (A7R34). Antibodies used in immunoblots and immunoprecipitations were obtained from the following sources: affinity-purified polyclonal anti-cyclin E antibody (Citation26); anti-Grb2 monoclonal antibody (BD Biosciences); anti-Hsc70 (B-6) and anti-p21 (C-19) antibodies (Santa Cruz Biotechnology); and anti-p53 (1C12) and anti-phospho-p53 (Ser15) (D4S1H) antibodies (Cell Signaling Technology). Immunoblot analyses and kinase assays were performed as previously described (Citation27).
Quantitative real-time PCR.
HSC RNA was extracted from FACS-sorted bone marrow cells using TRIzol (Life Technologies) and amplified using the Ovation PicoSL WTA system V2 (NuGen Technologies, Inc.). RNA was isolated from thymocytes using the RNeasy minikit (Qiagen) and reverse transcribed using the AffinityScript quantitative PCR (qPCR) cDNA synthesis kit (Agilent Technologies). All quantitative real-time PCRs were performed using SYBR green mix (Roche) on a LightCycler 480 machine. The expression levels of target mRNA transcripts were normalized to the level of Gapdh. Primer sequences are available upon request.
BrdU incorporation.
Mice were treated with a single dose (180 μg) of 5-bromo-2′-deoxyuridine (BrdU) (Sigma-Aldrich) via intraperitoneal injection and fed BrdU (0.8 mg/ml) in drinking water for 7 days. BrdU detection in HSCs was performed using the FITC BrdU flow kit, following the manufacturer's instructions (BD Biosciences).
Cell culture.
Embryonic day 14.5 (E14.5) fetal liver cells from wild-type, Fbxw7fl/+ or Fbxw7fl/fl mice were enriched for hematopoietic stem and progenitor cells using a customized EasySep mouse hematopoietic progenitor enrichment kit (Stemcell Technologies). These cells were cultured in Iscove's modified Dulbecco's medium (Life Technologies) supplemented with 15% FBS, 10−4 M beta-mercaptoethanol, 100 U/ml penicillin-streptomycin, 2 mM l-glutamine, 50 ng/ml murine recombinant SCF, and 10 ng/ml murine recombinant IL-3 (Peprotech) at 37°C for 6 h. To induce Fbxw7 deletion, cells were then transduced via spinoculation with retrovirus expressing Cre recombinase (murine stem cell virus containing the internal ribosome entry site and expressing green fluorescent protein and Cre [MSCV-IRES-GFP-Cre]; gift from Weiguo Cui at BloodCenter of Wisconsin) on two consecutive days, followed by FACS. Primary thymocytes from wild-type, Fbxw7fl/+, or Fbxw7fl/fl mice were cocultured with OP9/DL stromal cells (gift from Barbara Kee and Renee De Pooter at the University of Chicago) in Opti-MEM medium (Life Technologies) supplemented with 10% FBS, 100 U/ml penicillin-streptomycin, 2 mM l-glutamine, 5 ng/ml human recombinant Flt3, 5 ng/ml murine recombinant IL-7, and 0.0004% beta-mercaptoethanol. The cells were then transduced with MSCV-IRES-GFP-Cre retrovirus, followed by FACS. For retroviral transduction, cells were spinoculated with retroviral supernatants (prepared by transfecting Phoenix cells [G. Nolan, Stanford] using the calcium phosphate precipitation method) and 4 μg/ml Polybrene for 90 min at room temperature. For protein stability assays, primary thymocytes from either wild-type cyclin E (cyclin Ewt) or cyclin ET74A T393A mice were cocultured with OP9/DL stromal cells as described above. The cells were treated with 5 μg/ml aphidicolin (Sigma-Aldrich) overnight. The cells were then washed and treated with 25 μg/ml cycloheximide (Sigma-Aldrich) for the times indicated (see ). Cell extracts were prepared using NP-40 lysis buffer. Thymocyte extracts for kinase assays were prepared by lysing snap-frozen thymuses in radioimmunoprecipitation assay (RIPA) buffer.
Chromosome analysis.
FACS-sorted Lin− Sca1+ c-kit+ (LSK) cells (hematopoietic stem and progenitor cells [HSPCs]) were cultured in RPMI-DMEM (1:1) medium (Life Technologies) supplemented with 100 U/ml penicillin-streptomycin, 2 mM l-glutamine, 0.075% sodium bicarbonate, 10% fetal bovine serum, 50 ng/ml murine recombinant SCF, 10−6 M cortisol, 10 ng/ml murine recombinant IL-3 (Peprotech), 20 ng/ml IL-6 (Peprotech), 2 U/ml erythropoietin, 20 ng/ml thrombopoietin (Peprotech), and 50 ng/ml Flt3 ligand (Peprotech) at 37°C for 57 h and then treated with 0.1 μg/ml Colcemid for 3 h. For metaphase chromosome counts, thymocytes were cocultured with OP9/DL stromal cells overnight and then treated with 0.1 μg/ml Colcemid for 3 h. Metaphase spreads were prepared as described previously (Citation28). Images of chromosomes were obtained using a Nikon C2+ confocal microscope.
Immunofluorescence.
FACS-sorted LSK cells were cultured in RPMI-DMEM with the appropriate cytokines as described above. Cells were harvested, cytospun onto microscope slides, and fixed in prechilled methanol at −20°C for 15 min. The cells were then rehydrated in PBS for 30 min and blocked in 5% BSA–PBS at 4°C overnight. The slides were air dried and stained with a primary antibody against γ-tubulin (AK-15; Sigma-Aldrich) and an Alexa Fluor 488-conjugated anti-rabbit secondary antibody (Life Technologies). Cells were washed with PBS and counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich). Images of centrosomes were obtained using a Nikon A1R confocal microscope.
Array comparative genomic hybridization analysis.
Genomic DNA was prepared from mouse thymocytes using the DNeasy blood & tissue kit (Qiagen). Tumor and reference DNA (thymocytes from wild-type C57BL/6 mice) were labeled with Cy3 or Cy5 and then hybridized to oligonucleotide microarrays (G4425B; Agilent Technologies) for scanning. The labeling, hybridization, and scanning steps were performed by the University of Chicago Functional Genomics Facility. Data analysis was performed as described previously (Citation29).
RESULTS
Impaired Fbw7-mediated degradation leads to cyclin E protein stabilization in hematopoietic tissues.
We previously demonstrated increased cyclin E protein stability with insensitivity to Fbw7 expression in primary fibroblasts from cyclin ET74A T393A knock-in embryos. Our previous studies of cyclin ET74A T393A adult mice revealed increased cyclin E protein abundance in total bone marrow and bone marrow erythroid progenitor cells (Citation22, Citation25). Here, we examined cyclin E protein levels in cyclin ET74A T393A hematopoietic stem and progenitor cells (HSPCs) and confirmed increased protein expression without a corresponding increase in Ccne1 mRNA (). To confirm directly that the Fbw7 pathway regulates cyclin E in hematopoietic cells in vivo, we measured cyclin E protein levels in adult bone marrow cells after Fbxw7 deletion in vivo. We also isolated hematopoietic progenitor cells from conditional Fbxw7 knockout embryos (Citation17, Citation18), and after transduction with Cre recombinase, we documented increased cyclin E protein abundance (). These results demonstrate that Fbw7 loss or CPD mutations lead to cyclin E protein stabilization in hematopoietic progenitor cells.
Cyclin ET74A T393A HSCs are defective for multilineage reconstitution but not long-term self-renewal.
The long-term repopulating potential of hematopoietic cells resides in the quiescent HSC pool (Citation1Citation–Citation3). To evaluate the functional consequences for HSC function of ablating the Fbw7-dependent cyclin E ubiquitination pathway, we first performed serial transplantation experiments (). Fifty HSCs, defined using stringent criteria, including signaling lymphocyte activation molecule (SLAM) family markers () (Citation30), were isolated from the bone marrows of adult, wild-type, or cyclin ET74A T393A mice and transplanted into syngeneic recipient animals conditioned with 11 Gy radiation from a cesium source. We coadministered Lin+ Sca1− cells for short-term hematopoietic support. Peripheral blood and bone marrow donor chimerism were examined 3 months after each round of transplantation. We found that cyclin ET74A T393A HSCs are defective for repopulating the hematopoietic system of irradiated recipients, as shown by reduced donor contribution to both peripheral blood and bone marrow compared to that of wild-type counterparts, following secondary transplantation ( and ). Recipients of cyclin ET74A T393A HSCs showed defects in multilineage reconstitution, with reduced donor contribution to lymphoid (B220+ and CD3+), myeloid (Mac1+), and erythroid (CD71+) lineages (). The overall bone marrow composition of donor-derived cells in cyclin ET74A T393A reconstituted animals was comparable to that in wild-type controls following engraftment (). To assess donor HSC long-term self-renewal capacity, we enumerated HSCs at the end of each adoptive transfer experiment. Contrary to our original expectations, the cyclin ET74A T393A HSC numbers were similar to the wild-type HSC numbers after three iterations (). Together, these experiments demonstrated that cyclin E phosphorylations controlling its ubiquitination by the Fbw7 pathway are required for multilineage reconstitution but not for HSC self-renewal during steady-state hematopoiesis.
Cyclin ET74A T393A hematopoietic stem and progenitor cells show proliferative defects in response to hematologic stress.
To maintain hematopoietic homeostasis, HSCs switch reversibly between the quiescent and proliferative states in response to physiologic stimuli (Citation1). Cyclin E is an essential regulator of the G0- to S-phase transition, and its activity as a kinase increases during early cell cycle progression (Citation20). Thus, we hypothesized that induction of hematologic stress would induce HSC exit from quiescence and unmask defects in HSC function due to loss of Fbw7-dependent cyclin E regulation. First, we enumerated HSCs in cyclin ET74A T393A mice at steady state and observed fewer than in wild-type counterparts (). We used a combination of intracellular Ki67 and Hoechst 33342 labeling to study the cell cycle distributions of wild-type and cyclin ET74A T393A HSCs and found that at steady state, both populations were highly quiescent, with over 90% residing in G0 (). We also labeled adult mice with 5-bromo-2′-deoxyuridine (BrdU) for 10 days, followed by a BrdU-free chase for 70 days. After 70 days, we observed approximately 2-fold fewer BrdU label-retaining HSCs in cyclin ET74A T393A mice than in wild-type mice (). These data indicate that the cyclin ET74A T393A mutation only modestly altered the proliferation status of HSCs at steady state. Moreover, the finding of absolute reduction of HSCs in cyclin ET74A T393A mice compared to the numbers in controls (), interpreted with our aforementioned serial transplantation data (), suggests that HSC self-renewal within the knock-in strain was compromised by a cell-nonautonomous mechanism. We then studied cyclin ET74A T393A HSCs after exposing mice to the myelosuppressive agent 5-fluorouracil (5FU) to induce injury to mature hematopoietic elements and HSC cycling (Citation31). Cyclin ET74A T393A and wild-type mice were injected with a single dose (150 mg/kg) of 5FU. We found that a significant amount (29 to 33%) of both cyclin ET74A T393A and wild-type HSCs exited quiescence (G0) and entered the mitotic cell cycle 10 days after 5FU exposure. At this time, the absolute numbers of HSCs were equivalent in cyclin ET74A T393A and wild-type mice (). Both cyclin ET74A T393A and wild-type mice recovered from 5FU-induced stress 21 days posttreatment, confirmed by complete blood count analysis. Enumerating HSCs at this time point revealed that the wild-type HSC compartment regenerated significantly, while cyclin ET74A T393A HSCs showed diminished expansion following 5FU challenge. Interestingly, cell cycle analysis showed that cyclin ET74A T393A HSCs failed to efficiently undergo quiescence arrest after 21 days, with ∼37% of the cells remaining in active cycle ().
FIG 2 Cyclin ET74A T393A hematopoietic stem and progenitor cells show proliferative defects in response to hematologic stress. (A) Absolute numbers of HSCs in wild-type and cyclin ET74A T393A mice were enumerated at steady state. Inset, steady-state HSC cell cycle distributions were determined using intracellular Ki67 and Hoechst 33342 staining. (B) Assay of BrdU label-retaining cells was performed as previously described (Citation1). Percentage of BrdU+ HSCs after 10 days of labeling in vivo and percentage of BrdU label-retaining HSCs after 70 days of chase are shown. Shown are means ± standard errors of the means of results for three mice per time point. (C and D) HSC numbers and cell cycle distributions (insets) were determined 10 days and 21 days after 5FU treatment. (E and F) Absolute numbers of the indicated multipotent progenitor cell populations (MPP1- to -3) in wild-type and cyclin ET74A T393A mice were enumerated at steady state and 21 days following 5FU treatment. *, P < 0.05; **, P < 0.02, using Student's t test.
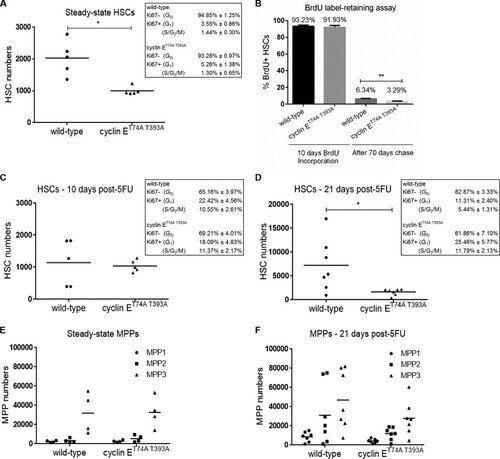
We speculated that cyclin E deregulation at the HSC-multipotent progenitor cell population (MPP) transition may result in production of more MPPs at the expense of HSCs during injury-induced proliferation. We examined the various MPP subsets (MPP1 to -3) () (Citation1) in wild-type and cyclin ET74A T393A mice at steady state and 21 days after 5FU treatment. We found that at 21 days posttreatment, the total number of MPPs in cyclin ET74A T393A mice remained unchanged compared to the number at steady state, whereas wild-type mice showed marked expansion of MPPs following 5FU challenge ( and ). Thus, the overall pool size of wild-type HSPCs increased upon hematologic injury. In contrast, cyclin ET74A T393A HSPCs failed to proliferate normally following hematologic challenge. Collectively, these results suggest that Fbw7-mediated cyclin E regulation is required for HSCs to exit the cell cycle efficiently following hematologic stress and that failure to resolve the transition between the mitotic cell cycle and reestablishment of quiescence impairs HSC self-renewal and the generation of MPP reserves.
Cyclin E deregulation induces chromosomal instability in HSPCs after hematologic stress.
To determine whether the observed defects in cyclin ET74A T393A HSC homeostasis after 5FU-mediated injury are cell autonomous, we established a cohort of wild-type recipient mice reconstituted with either cyclin ET74A T393A or wild-type bone marrows. Following engraftment, we enumerated fewer HSCs in cyclin ET74A T393A recipients than in wild-type controls (). At 10 days after 5FU administration, we noted increased cyclin ET74A T393A HSC numbers compared to the numbers of wild-type cells, which was not observed in our experiments with cyclin ET74A T393A mice (), suggesting that the early response of cyclin E knock-in HSCs to injury is modified by a cell-nonautonomous mechanism. At 21 days, however, wild-type HSCs had increased in number, while the cyclin E knock-in cells were significantly reduced compared to their numbers at the earlier time point (), as we observed in the earlier experiment without transplantation (). To evaluate whether this aberrant response to hematologic stress in the cyclin ET74A T393A HSC pool was physiologically significant, we challenged wild-type recipients of either wild-type or cyclin ET74A T393A bone marrow cells with repeated exposures to 5FU dosed every 10 days. We observed significantly shorter survival of recipients of cyclin ET74A T393A cells than of wild-type recipients (). Although we cannot rule out the possibility that cyclin ET74A T393A HSCs have increased sensitivity to potential DNA-damaging effects of 5FU, our results demonstrate that cyclin ET74A T393A HSCs exhaust more rapidly than wild-type cells under conditions of repeated hematologic injury.
FIG 3 Cyclin E deregulation induces chromosomal instability in HSPCs after hematologic stress. (A) Wild-type (CD45.1) recipients were transplanted with 1 × 106 (CD45.2) bone marrow cells isolated from wild-type or cyclin ET74A T393A mice. Three months after engraftment, recipients of bone marrow cells of the indicated genotypes were either injected with a single dose of 5FU or remained untreated, and absolute numbers of donor-derived HSCs were enumerated at 10 days or 21 days later. *, P < 0.05; **, P < 0.0005, using Student's t test. (B) Cohorts of mice transplanted with bone marrow cells of the indicated genotypes were established, and 3 months after engraftment, recipient mice with equivalent peripheral blood donor chimerism (>85%) were treated with 5FU (150 mg/kg) every 10 days (stars). The survival of the groups was analyzed using the Gehan-Breslow-Wilcoxon test (P = 0.0172) and expressed as Kaplan-Meier survival curves (n = 13). (C) Three months after engraftment, recipients of bone marrow cells of the indicated genotypes were either injected with a single dose of 5FU or left untreated. The cohort was then labeled with bromodeoxyuridine (BrdU) for 7 days. At the end of the in vivo labeling period, HSCs were analyzed for BrdU incorporation. Error bars display standard errors. (D) Top, HSPCs of the indicated genotypes were isolated from wild-type recipients 17 days after 5FU exposure, briefly cultured, and arrested in metaphase with Colcemid. Chromosomes were examined using a confocal microscope, and the results of enumerating chromosomes for 50 metaphase cells are shown. The chi-square test was used to determine statistical significance for the difference in metaphase chromosome count distribution (<40, 40, or >40) between wild-type and cyclin ET74A T393A HSPCs. Fisher's exact test was used to determine the statistical significance for the difference in normal and abnormal chromosome counts between wild-type and cyclin ET74A T393A cells. *, P < 0.02. Bottom, representative confocal images of metaphase chromosomes are shown (100× magnification). (E) Enumeration of metaphase chromosomes from untreated, wild-type, and cyclin ET74A T393A HSPCs is shown. n.s., not significant, using Fisher's exact test.
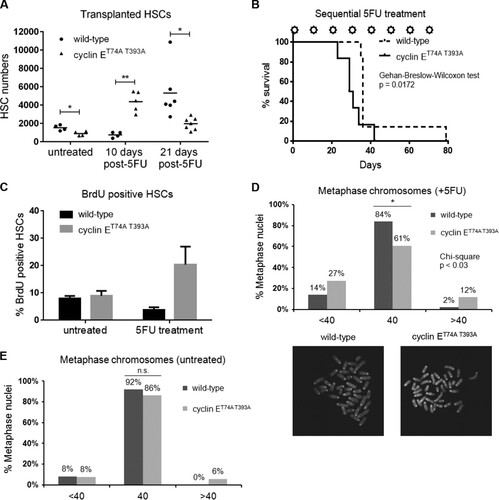
We examined DNA synthesis in cyclin ET74A T393A and wild-type HSCs following recovery from hematologic stress using in vivo BrdU labeling. Although BrdU incorporation in untreated cyclin ET74A T393A HSCs was comparable to that in wild-type HSCs, we observed a significant increase in the percentage of BrdU-positive cyclin ET74A T393A HSCs after 5FU treatment (). One possible explanation for this finding and the observation of reduced cyclin ET74A T393A HSCs following recovery from 5FU injury () is that cyclin ET74A T393A cells are both proliferating faster and being eliminated from the HSC pool with increased frequency. Alternatively, our results are consistent with delayed S-phase transit in cyclin ET74A T393A HSCs, resulting in overall lowering of the rate at which the population in aggregate expands after 5FU challenge. Indeed, we previously described delayed S-phase transit in primary fibroblasts expressing Fbw7-resistant cyclin E (Citation32).
Deregulated cyclin E activity has been shown to induce chromosomal instability (CIN) by interfering with prereplication complex assembly, perturbing S-phase progression, and impairing chromosome alignment on the metaphase plate (Citation32Citation–Citation34). To investigate the possibility that the observed proliferative defect in cyclin ET74A T393A HSCs is associated with genomic instability, leading to reduced HSC fitness, we examined metaphase-arrested chromosomes isolated from Lin− Sca1+ c-kit+ (LSK) cells of 5FU-treated animals. Strikingly, we found that cyclin ET74A T393A hematopoietic stem and progenitor cells (HSPCs) demonstrate significant chromosomal instability (39%), signified by gain or loss of chromosomes per metaphase (). In comparison, at steady state, the majority of cyclin ET74A T393A metaphases (86%) appear normal, similar to wild-type HSPCs (). Collectively, these results show that hematologic stress unmasks CIN in cooperation with defective-Fbw7-mediated cyclin E regulation and that CIN is associated with impaired S-phase completion and self-renewal of HSCs.
Loss of both p53- and Fbw7-dependent cyclin E regulatory pathways impairs self-renewal and induces chromosome instability in HSCs.
Excess cyclin E activity is known to activate p53 in primary human and murine fibroblasts (Citation32), and in cyclin ET74A T393A bone marrow erythroid cells, an induced p53-dependent DNA damage pathway promotes compensated erythropoiesis (Citation25). In cyclin ET74A T393A HSCs, we similarly observed induced expression of canonical p53 target genes. Notably, Cdkn1a and Gadd45A are uniformly upregulated in response to high cyclin E activity, with dramatic induction of the latter (). Cdkn1a encodes p21, which inhibits cyclin E-Cdk2 kinase activity (Citation35) and protects cells from cyclin E-induced apoptosis (Citation36), whereas Gadd45 proteins act to restrain genomic instability and inhibit apoptosis triggered by genotoxic stress (Citation37, Citation38). Thus, we reasoned that p53 might play a role in preserving HSC function when the Fbw7 pathway controlling cyclin E expression is impaired.
FIG 4 Loss of the p53 pathway reveals self-renewal defects in cyclin ET74A T393A HSCs. (A) Quantitative RT-PCR analyses of the indicated p53 gene targets in wild-type and cyclin ET74A T393A HSCs are shown. Each bar represents the average value from triplicate quantitative RT-PCR assays from one cyclin ET74A T393A mouse, expressed as log(fold difference) compared to the results for an age- and sex-matched wild-type control. (B) Peripheral blood cell donor chimerism was calculated as shown in the y axis at 3 months after each round of transplantation (n = 7 or 8 wild-type recipients per group) for recipients of p53-null HSCs of the indicated cyclin E genotypes. (C and D) Total bone marrow donor chimerism and lineage-specific donor chimerism were determined 3 months after engraftment. (E) Bone marrow compositions of donor-derived cells of the indicated genotypes following secondary transplantation are shown. (F) Absolute numbers of donor-derived HSCs were enumerated in individual recipient mice 3 months after each round of transplantation. (G) Serial adoptive transfer assays were performed using cyclin Ewt; p21−/− or cyclin ET74A T393A; p21−/− HSCs as donor cells. Absolute numbers of donor-derived HSCs in individual recipient mice following primary and secondary transplantation are shown. Error bars display standard errors. *, P < 0.05; **, P < 0.005; ***, P < 0.001; ****, P < 0.0001, using Student's t test.
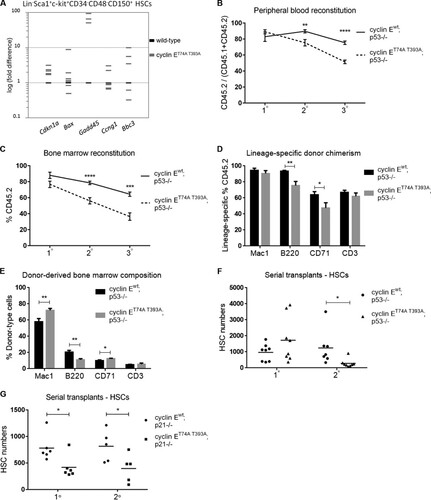
To assess the role of p53 in HSC function in the setting of deregulated cyclin E, we performed serial transplantation experiments as shown in , using cyclin ET74A T393A; p53−/− and cyclin Ewt; p53−/− HSCs as donor cells. As with recipients of cyclin ET74A T393A HSCs with wild-type p53, the peripheral blood and bone marrow donor-type chimerism was significantly reduced in recipients of cyclin ET74A T393A; p53−/− HSCs compared to the level in cyclin Ewt; p53−/− controls following secondary transplantation ( and ). Further investigation into specific bone marrow lineages revealed that cyclin ET74A T393A; p53−/− HSCs are deficient for reconstituting B cells (B220+) and erythroid cells (CD71+) compared to cyclin Ewt; p53−/− HSCs (). With analysis of the overall bone marrow composition, we found that donor-derived myeloid (Mac1+) and CD71-positive erythroid populations were expanded, with concomitant reduction of B cells in recipients of cyclin ET74A T393A; p53−/− HSCs compared to the numbers of B cells in their counterparts that received cyclin Ewt; p53−/− HSCs (). Previous studies of p53-null mice suggested that loss of p53 enhances HSC proliferation (Citation10, Citation11). In contrast, we found that cyclin ET74A T393A; p53−/− HSCs were substantially depleted after two rounds of transplantation (). Thus, the self-renewal capacity of cyclin ET74A T393A HSCs is impaired by the loss of p53. The Cdk inhibitor p21, a downstream target of p53, is upregulated in cyclin ET74A T393A HSCs (). We therefore determined whether p53 restrains cyclin E-induced HSC defects in part through induction of p21. When we performed serial adoptive transfer of cyclin E wild-type versus T74A T393A HSCs in the p21-null background, we observed self-renewal defects in recipients of cyclin ET74A T393A; p21−/− HSCs after one round of transplantation that persisted through the end of secondary transplantation (). These data suggest that a functional p53/p21 pathway preserves HSC self-renewal in the setting of defective Fbw7-mediated cyclin E control.
We next enumerated chromosomes in metaphase spreads prepared from cyclin ET74A T393A; p53−/− and cyclin Ewt; p53−/− HSPCs. We found that cyclin ET74A T393A; p53−/− cells demonstrated CIN, signified by gain or loss of chromosomes per metaphase. Importantly, the percentage of abnormal metaphases was significantly higher in cyclin ET74A T393A; p53−/− HSPCs (57%) than in cyclin Ewt; p53−/− controls (29%) (). Similar abnormalities were observed in cyclin ET74A T393A; p21−/− HSPCs (). These results suggest that the loss of both p53- and Fbw7-dependent cyclin E controls unmasks genomic instability in HSPCs.
FIG 5 Ablation of the p53 pathway unmasks chromosome instability in cyclin ET74A T393A HSPCs. (A and B) Enumeration of metaphase chromosomes from HSPCs of the indicated genotypes was performed as described in the legend to . *, P < 0.04; **, P < 0.01, using Fisher's exact test. (C) Results of centrosome enumeration in cyclin Ewt; p53−/− and cyclin ET74A T393A; p53−/− HSPCs using γ-tubulin and DAPI costaining are shown. (D) BrdU incorporation of cyclin Ewt; p53−/− and cyclin ET74A T393A; p53−/− HSCs was determined as described in the legend to . Error bars indicate standard deviation with biological replicates (n = 3 per genotype).
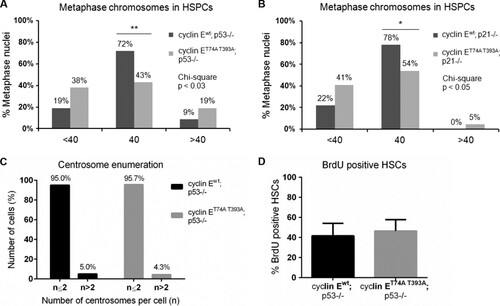
Missegregation of whole chromosomes in the setting of cyclin E deregulation and the loss of p53 has been associated with centrosome hyperamplification (Citation39). Supernumerary centrosomes increase the frequency of merotelic kinetochore attachments, thereby promoting CIN (Citation40). We thus examined centrosome numbers in wild-type and cyclin ET74A T393A HSPCs with p53 deletion and found no signs of increased amplification in the latter, suggesting that deregulated cyclin E promotes CIN in HSPCs via a centrosome-independent mechanism ().
Uncontrolled proliferation can lead to HSC exhaustion (Citation41). We therefore examined the possibility that cyclin ET74A T393A; p53−/− HSPC depletion and CIN is associated with hyperproliferation and measured BrdU incorporation in cyclin ET74A T393A; p53−/− and cyclin Ewt; p53−/− HSCs established in wild-type recipient animals by adoptive transfer. We found that BrdU incorporation in cyclin ET74A T393A; p53−/− HSCs is comparable to that in cyclin Ewt; p53−/− HSCs (). Thus, we cannot attribute the observed depletion of cyclin ET74A T393A; p53−/− HSCs to excessive proliferation. Rather, the defective self-renewal in these cells may be directly related to loss of genomic integrity controls and CIN.
Recipients of cyclin ET74A T393A; p53−/− HSCs develop T-cell malignancies in a background of abnormal hematopoiesis.
We previously reported defective maturation of cyclin ET74A T393A erythroid cells with morphological dysplasia and an accumulation of erythroid precursors. Additionally, p53 loss worsens maturation defects induced by deregulated cyclin E expression within the erythroid lineage (Citation22, Citation25). To determine whether the modest increase in the donor-type, CD71-positive subset underestimates the erythroid cell population size of cyclin ET74A T393A; p53−/− HSC recipients (), we compared cyclin ET74A T393A; p53−/− and cyclin Ewt; p53−/− bone marrow recipient morphologies and observed a significant expansion of erythroid cells in the former (). Thus, erythroid hyperplasia induced by deregulated cyclin E persisted in a cell-autonomous manner even in the background of p53 loss. Besides this abnormality in the erythroid lineage, we also observed a B-cell maturation defect in recipients of cyclin ET74A T393A; p53−/− bone marrow cells. Although pro- and pre-B-cell numbers were equivalent (data not shown), both recirculating/mature (B220high IgM+) and immature (B220low IgM+) bone marrow B220-positive cells were significantly reduced in cyclin ET74A T393A; p53−/− bone marrows compared to their levels in cyclin Ewt; p53−/− controls. These findings likely result from increased apoptosis within the B-lymphocyte lineage of cyclin ET74A T393A; p53−/− recipients (). We examined cyclin E abundance in primary bone marrow B cells and confirmed increased cyclin E protein levels in cyclin ET74A T393A B cells ().
FIG 6 Recipients of cyclin ET74A T393A; p53−/− HSCs develop T-cell malignancies in a background of erythroid hyperplasia and dysplasia. (A) Representative 40× micrographs of Wright-Giemsa-stained bone marrow cytospins from wild-type recipients of cyclin Ewt; p53−/− and cyclin ET74A T393A; p53−/− bone marrow cells are shown. Myeloid-to-erythroid ratios were calculated by scoring 400 to 700 nucleated cells per genotype based on morphology. (B) Left, absolute numbers of bone marrow B-lymphocytes from recipients of cyclin Ewt; p53−/− and cyclin ET74A T393A; p53−/− HSCs (n = 14) are shown, demonstrating reduced donor-type cyclin ET74A T393A immature B cells (B220low IgM+) and recirculating/mature B cells (B220high IgM+). Right, donor-type B220+ cells from recipient mice (n = 9) were assessed for apoptosis using annexin V and 7-AAD staining. Error bars display standard errors. *, P < 0.05; **, P < 0.00001, using Student's t test. (C) Bone marrow cells from wild-type or cyclin ET74A T393A mice were enriched for B220+ B cells by immunomagnetic separation. Western blot analyses of cyclin E abundance are shown. (D) Survival curves of wild-type recipients of cyclin Ewt; p53−/− and cyclin ET74A T393A; p53−/− HSCs after tertiary transplantation are shown for the cohort subset (n = 9 out of 14 recipients) with comparable donor-type bone marrow chimerism (>60%). Survival data were analyzed using the Gehan-Breslow-Wilcoxon test (P = 0.0150) and expressed as Kaplan-Meier survival curves. (E) Representative donor-type thymocyte immunophenotyping from cyclin Ewt; p53−/− and cyclin ET74A T393A; p53−/− tertiary transplant recipients. (F) Representative flow cytometry plots displaying donor-type bone marrow CD3 expression versus side scatter (SSC) are shown for tertiary transplant recipients (top). Representative hematoxylin and eosin stains of formalin-fixed, paraffin-embedded bone marrows (middle; 40× magnification) and Wright-Giemsa-stained peripheral blood smears (bottom; 100× magnification) from recipients of cyclin Ewt; p53−/− and cyclin ET74A T393A; p53−/− HSCs analyzed as described for panel D are shown.
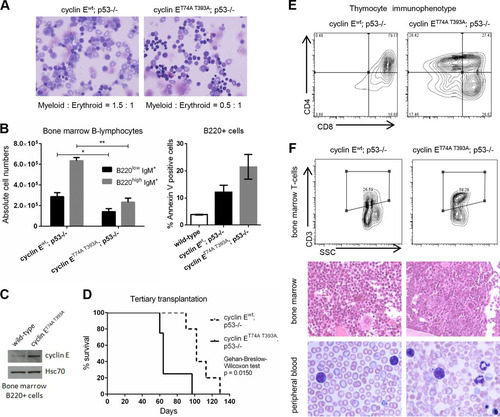
With serially transplanted p53-null HSCs, we ultimately observed the induction of fatal T-cell malignancies in all recipients. We transplanted a total of 14 wild-type mice with either cyclin Ewt; p53−/− or cyclin ET74A T393A; p53−/− bone marrow cells in the third iteration. Given the highly variable donor-type chimerism with tertiary transplantation of p53-knockout cells, we limited our comparisons of survival to recipients with comparable donor-type bone marrow reconstitution (>60% bone marrow donor chimerism; n = 9). Recipients of cyclin ET74A T393A; p53−/− HSCs succumbed to T-cell malignancies with significantly shorter latency than recipients of cyclin Ewt; p53−/− HSCs (). All recipient mice developed massive thymic enlargement, with accumulation of immature (CD4 and CD8 double-positive) T cells in thymi of cyclin Ewt; p53−/− recipients, whereas cyclin ET74A T393A; p53−/− recipients showed a broader representation of T-cell immunophenotypes (). Additionally, we observed leukemias characterized by >20% bone marrow CD3-positive lymphoblasts in all cyclin ET74A T393A; p53−/− recipients. In contrast, only 40% of cyclin Ewt; p53−/− recipients showed aberrant T-cell expansion in bone marrow (, top and middle). Analyses of peripheral blood of tumor-afflicted recipients of cyclin ET74A T393A; p53−/− HSCs showed lymphoblasts within a background of marked red cell morphological atypia, whereas lymphoblasts from cyclin Ewt; p53−/− HSCs occurred as the dominant peripheral blood finding (, bottom). In summary, deregulated cyclin E activity cooperates with p53 loss in a hematopoietic cell-autonomous manner to induce T-cell neoplasia that occurs in the context of multilineage hematologic disease.
Deregulated cyclin E activity cooperates with p53 loss to promote chromosomally unstable T-cell neoplasms.
We examined cyclin E protein levels in Fbxw7-deficient primary thymocytes and found stabilization of cyclin E after ex vivo deletion of Fbxw7 (). We next confirmed that cyclin E protein stability was increased in primary cyclin ET74A T393A thymocytes compared to its stability in wild-type cells. Furthermore, quantitative reverse transcription (RT)-PCR analysis showed that the Ccne1 transcript abundance in cyclin ET74A T393A thymocytes was equivalent to that in wild-type controls (). These data indicate that increased cyclin ET74A T393A protein expression results from enhanced stability due to loss of the phosphorylations that promote binding to SCFFbw7 (Citation23, Citation24). Next, we used quantitative RT-PCR to determine whether p53 is activated by deregulated cyclin E in primary thymocytes. We found induction of p53 target genes (Cdkn1a, Gadd45a, Bax, and Bbc3) in cyclin ET74A T393A thymocytes. The transcript abundance of the aforementioned genes was significantly reduced in p53-null thymocytes, suggesting that the expression of these genes is p53 dependent. Fbxw7 was not significantly induced in cyclin ET74A T393A thymocytes, though previous reports using different cell types demonstrate that Fbxw7 expression can be induced downstream from activated p53 (Citation42, Citation43). Immunoblot analyses further confirmed that both p53 serine 15 phosphorylation and total p53 abundance were elevated in cyclin ET74A T393A thymocytes ().
FIG 7 Deregulated cyclin E activity cooperates with p53 loss to promote chromosomally unstable T-cell neoplasms. (A) Primary thymocytes from wild-type, Fbxw7fl/+, or Fbxw7fl/fl mice were transduced with Cre recombinase to generate conditional Fbxw7 knockout allele. Western blot analyses of cyclin E abundance in Cre-expressing thymocyte extracts are shown. (B) Primary thymocytes from wild-type and cyclin ET74A T393A mice were cocultured with OP9/DL stromal cells overnight and then treated with 25 μg/ml of cycloheximide for the indicated times. Left, cyclin E immunoblots are shown with relative abundances, quantified using ImageJ. Right, abundances of Ccne1 mRNA in primary thymocytes were compared using quantitative RT-PCR. (C) Left, quantitative RT-PCR analysis of the indicated p53 target genes in primary thymocytes isolated from mice of the indicated genotypes is shown, with expression values normalized to relative expression in wild-type controls. Right, immunoblot analyses of p53 serine 15 phosphorylation, total p53, and cyclin E abundance in thymocyte extracts from wild-type and cyclin ET74A T393A mice are shown. Grb2 immunoblot is shown as loading control. (D) Top, total lysates were prepared from primary thymocytes of the indicated genotypes and immunoblotted to detect endogenous cyclin E and p21. Histone H1 and Rb were used to measure endogenous cyclin E kinase activities from immunoprecipitated (IP) cyclin E complexes. Grb2 immunoblot is shown as loading control. Bottom, kinase activities were quantified and normalized to activity measured in the wild-type samples. Student's t test was used to determine the statistical significance for the difference in cyclin E kinase activities. (E) Enumeration of metaphase chromosomes from wild-type and cyclin ET74A T393A thymocytes is shown. (F and G) Enumeration of metaphase chromosomes from thymocytes of the indicated genotypes is shown. (F) Bottom, representative confocal images of metaphase chromosomes from thymocytes of the indicated genotypes. (H) Array CGH results for representative thymic tumors obtained from recipients of cyclin Ewt; p53−/− and cyclin ET74A T393A; p53−/− HSCs are shown. Fluorescence intensities in thymic tumors were normalized to genomic DNA from wild-type thymocytes and are expressed as log2 ratios. Chromosome identification is shown on the x axis. Arrows indicate gains and arrowheads indicate losses of chromosomes. Error bars indicate standard deviations with two biological replicates. n.s., not significant; *, P < 0.04; **, P < 0.01, using Fisher's exact test.
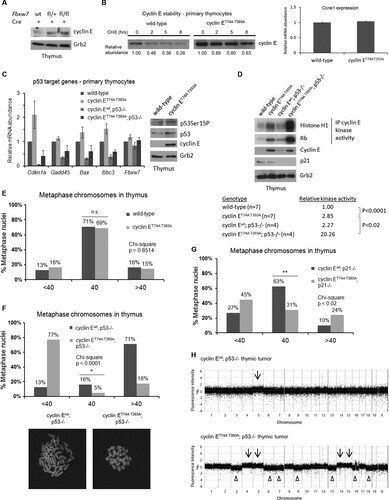
We next compared cyclin E kinase activity in wild-type, cyclin ET74A T393A, cyclin Ewt; p53−/−, and cyclin ET74A T393A; p53−/− thymocytes. As previously shown (Citation22), cyclin E activity is increased in cyclin ET74A T393A thymocytes compared to the level in wild-type cells. Here, we observed markedly increased kinase activity in cyclin E knock-in thymocytes with loss of p53 () (Citation32). In primary fibroblasts, genomic instability associated with deregulated cyclin E expression correlated with the relative increase in its kinase activity (Citation36). We examined whether the massive increase in cyclin ET74A T393A activity associated with deleting p53 in thymocytes is also associated with genome instability. To do this, we enumerated chromosomes in metaphase spreads prepared from thymi of transplanted recipients. We found that the majority of cyclin ET74A T393A metaphases appeared normal, similar to those of wild-type controls (). Both cyclin Ewt; p53−/− and cyclin ET74A T393A; p53−/− thymocytes exhibited CIN, with the majority of cyclin Ewt; p53−/− thymocytes showing gain of chromosomes and the majority of cyclin ET74A T393A; p53−/− thymocytes demonstrating chromosome loss. Importantly, cyclin ET74A T393A; p53−/− thymocytes possessed more abnormal chromosomes than p53-null thymocytes (P < 0.04) (). We next examined metaphase chromosomes prepared from thymi of cyclin Ewt; p21−/− and cyclin ET74A T393A; p21−/− recipient animals. We found that the percentage of abnormal metaphases was significantly higher in cyclin ET74A T393A; p21−/− thymocytes (69%) than in cyclin Ewt; p21−/− controls (37%) (). Since loss of p53 and p21 significantly increased the frequency of CIN in thymocytes that expressed cyclin ET74A T393A, our results indicate that the p53/p21 pathway is a barrier against cyclin E-induced genomic instability in vivo. We then examined the effect of cyclin ET74A T393A; p53−/− mutations on thymic tumor genome structure by using array-based comparative genomic hybridization (array CGH). Tumors in wild-type recipients of cyclin ET74A T393A; p53−/− HSCs showed significant gains and losses of whole chromosomes. In contrast, evidence of CIN was less prominent in cyclin Ewt; p53−/− thymic tumors (). Collectively, these results demonstrate that clonal CIN is associated with fully deregulated cyclin E, resulting from the loss of both Fbw7- and p53-dependent restraints on its activity in vivo.
DISCUSSION
Cyclin ET74A T393A HSCs phenocopy several features of Fbxw7-null HSCs, including p53 activation and impaired self-renewal and repopulating capacities (Citation17, Citation18). Importantly, multiple mechanisms can modulate Fbw7 expression, including microRNA-mediated repression of transcription/translation (Citation44, Citation45), transcriptional repression (Citation46), and gene hypermethylation (Citation47). Additionally, since phosphorylation of cyclin E CPDs by at least two kinases, Cdk2 itself and glycogen synthase kinase 3, is required for its interaction with SCFFbw7, molecular lesions that impair these phosphorylations (e.g., hyperactive phosphatidylinositol 3-kinase signaling) can also impair Fbw7-mediated cyclin E ubiquitination (Citation16).
In addition to its function as a kinase with its catalytic partner Cdk2, cyclin E has a kinase-independent role in supporting prereplication complex formation (Citation48). The association of CIN with impaired self-renewal of cyclin ET74A T393A HSCs suggests that deregulation of cyclin E-Cdk2 activity (and not of cyclin E expression alone) is a key driver of HSC defects in our experiments, since cyclin E-associated kinase activity is required for it to engender genomic instability (Citation36). Moreover, in this study, we directly measured cyclin E kinase activity in primary thymocytes and found that it was dramatically potentiated by p53 loss, leading to marked aneuploidy. Thus, deregulated cyclin E activity likely promotes genomically unstable tumors that later develop in these tissues. These results are consistent with observations made in a recently reported model of Fbw7-associated intestinal neoplasia, in which Fbxw7 deletion in mouse gut epithelial cells deregulates SCFFbw7 substrates, including cyclin E, and in combination with p53 loss drives the formation of aggressive, genomically unstable tumors (Citation49).
Previous results by Matsuoka et al. show that Fbxw7 deletion promotes leukemia-initiating cell development in part by exerting selective pressure on HSPCs to acquire secondary genetic lesions that lead to impaired p53 function (Citation18). The lack of spontaneous transformation of hematopoietic cells with cyclin ET74A T393A expression alone suggests that the contribution of other SCFFbw7 substrates, such as c-Myc, Notch, and others, is required to drive p53 loss-of-function in cooperation with deregulated cyclin E. Though p53 acts as a critical barrier protecting hematopoietic cells against cyclin E-induced genomic instability and neoplasia, our data nonetheless indicate that the ablation of a single ubiquitination pathway controlling active cyclin E protein abundance is sufficient to impair the multilineage reconstitution capacity of HSCs and, in the setting of stress hematopoiesis, to reduce their self-renewal capacity.
It is striking that cyclin E-induced CIN is associated with tumor induction within the T-cell lineage but a cell exhaustion phenotype in the HSPC pool. We investigated whether CIN was associated with increased apoptosis as a mechanism underpinning cyclin ET74A T393A; p53−/− stem and progenitor cell loss following transplantation. Using annexin V detection assays, we found no significant differences in the frequency of apoptotic cyclin ET74A T393A; p53−/− HSPCs compared to the rate of apoptosis of control HSPCs (data not shown). Thus, nonapoptotic mechanisms, including oncogene-induced senescence or mitotic catastrophe (Citation50, Citation51), likely account for the reduction in cyclin ET74A T393A HSPCs associated with p53 deletion and recovery from hematologic stress.
Other studies have linked genomic instability to HSC failure, including a report of MLH1 deficiency showing the accumulation of microsatellite instability in HSCs of older humans (Citation52) and a telomerase-deficient model showing that telomeric instability shortens HSC life span (Citation53). Deregulated cyclin E activity in HSCs may induce multiple molecular lesions that lead to CIN, including aberrant replication fork movement (Citation50), impairment of prereplication complex assembly (Citation33), and misalignment of chromosomes on the metaphase plates (Citation34). The varied mechanisms through which cyclin E can induce genomic instability when its activity is deregulated likely account for its potency as a cell-intrinsic mutagen. Our discovery that hematologic stress can promote CIN in HSPCs in cooperation with disabled Fbw7-mediated cyclin E control has clinical implications for understanding early steps in hematologic tumorigenesis, in that deregulated cyclin E could elicit a mutator phenotype in the setting of acute hematopoietic injuries, such as exposure to cytotoxic chemotherapy, even with intact p53 function. Additionally, our observation that CIN reduces HSC fitness suggests that, in some established cancers, synthetic lethal approaches could be evaluated in combination with standard cytotoxic therapies to exploit potential antagonism between CIN and self-renewal in tumor-initiating cells.
ACKNOWLEDGMENTS
We acknowledge technical support from the Robert H. Lurie Comprehensive Cancer Center Flow Cytometry and Cell Imaging core facilities and helpful advice from John Crispino, Kathleen Green, and Steven Kosak (all at Northwestern) and Ulrich Steidl (Albert Einstein College of Medicine).
K.T.S. was supported by the Malkin Scholars Program and the Chicago Biomedical Consortium, which is funded by the Searle Funds at The Chicago Community Trust. A.C.M. received funding for these studies from the American Society of Hematology (Scholar Award), American Cancer Society, Sidney Kimmel Foundation for Cancer Research, and the Leukemia Research Foundation. This work was supported by NIH/NHLBI grant R01HL098608.
We have no conflicting financial interests.
REFERENCES
- Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, Lio P, Macdonald HR, Trumpp A. 2008. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135:1118–1129. http://dx.doi.org/10.1016/j.cell.2008.10.048.
- Cheshier SH, Morrison SJ, Liao X, Weissman IL. 1999. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc. Natl. Acad. Sci. U. S. A. 96:3120–3125. http://dx.doi.org/10.1073/pnas.96.6.3120.
- Passegue E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. 2005. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J. Exp. Med. 202:1599–1611. http://dx.doi.org/10.1084/jem.20050967.
- Coller HA. 2011. Cell biology. The essence of quiescence. Science 334:1074–1075. http://dx.doi.org/10.1126/science.1216242.
- Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT. 2000. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 287:1804–1808. http://dx.doi.org/10.1126/science.287.5459.1804.
- Cheng T, Rodrigues N, Dombkowski D, Stier S, Scadden DT. 2000. Stem cell repopulation efficiency but not pool size is governed by p27(kip1). Nat. Med. 6:1235–1240. http://dx.doi.org/10.1038/81335.
- Matsumoto A, Takeishi S, Kanie T, Susaki E, Onoyama I, Tateishi Y, Nakayama K, Nakayama KI. 2011. p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell 9:262–271. http://dx.doi.org/10.1016/j.stem.2011.06.014.
- Zou P, Yoshihara H, Hosokawa K, Tai I, Shinmyozu K, Tsukahara F, Maru Y, Nakayama K, Nakayama KI, Suda T. 2011. p57(Kip2) and p27(Kip1) cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell 9:247–261. http://dx.doi.org/10.1016/j.stem.2011.07.003.
- Viatour P, Somervaille TC, Venkatasubrahmanyam S, Kogan S, McLaughlin ME, Weissman IL, Butte AJ, Passegue E, Sage J. 2008. Hematopoietic stem cell quiescence is maintained by compound contributions of the retinoblastoma gene family. Cell Stem Cell 3:416–428. http://dx.doi.org/10.1016/j.stem.2008.07.009.
- Chen J, Ellison FM, Keyvanfar K, Omokaro SO, Desierto MJ, Eckhaus MA, Young NS. 2008. Enrichment of hematopoietic stem cells with SLAM and LSK markers for the detection of hematopoietic stem cell function in normal and Trp53 null mice. Exp. Hematol. 36:1236–1243. http://dx.doi.org/10.1016/j.exphem.2008.04.012.
- Liu Y, Elf SE, Miyata Y, Sashida G, Huang G, Di Giandomenico S, Lee JM, Deblasio A, Menendez S, Antipin J, Reva B, Koff A, Nimer SD. 2009. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 4:37–48. http://dx.doi.org/10.1016/j.stem.2008.11.006.
- Milyavsky M, Gan OI, Trottier M, Komosa M, Tabach O, Notta F, Lechman E, Hermans KG, Eppert K, Konovalova Z, Ornatsky O, Domany E, Meyn MS, Dick JE. 2010. A distinctive DNA damage response in human hematopoietic stem cells reveals an apoptosis-independent role for p53 in self-renewal. Cell Stem Cell 7:186–197. http://dx.doi.org/10.1016/j.stem.2010.05.016.
- Santaguida M, Schepers K, King B, Sabnis AJ, Forsberg EC, Attema JL, Braun BS, Passegue E. 2009. JunB protects against myeloid malignancies by limiting hematopoietic stem cell proliferation and differentiation without affecting self-renewal. Cancer Cell 15:341–352. http://dx.doi.org/10.1016/j.ccr.2009.02.016.
- Yu H, Yuan Y, Shen H, Cheng T. 2006. Hematopoietic stem cell exhaustion impacted by p18 INK4C and p21 Cip1/Waf1 in opposite manners. Blood 107:1200–1206. http://dx.doi.org/10.1182/blood-2005-02-0685.
- Yuan Y, Shen H, Franklin DS, Scadden DT, Cheng T. 2004. In vivo self-renewing divisions of haematopoietic stem cells are increased in the absence of the early G1-phase inhibitor, p18INK4C. Nat. Cell Biol. 6:436–442. http://dx.doi.org/10.1038/ncb1126.
- Welcker M, Clurman BE. 2008. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 8:83–93. http://dx.doi.org/10.1038/nrc2290.
- Thompson BJ, Jankovic V, Gao J, Buonamici S, Vest A, Lee JM, Zavadil J, Nimer SD, Aifantis I. 2008. Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J. Exp. Med. 205:1395–1408. http://dx.doi.org/10.1084/jem.20080277.
- Matsuoka S, Oike Y, Onoyama I, Iwama A, Arai F, Takubo K, Mashimo Y, Oguro H, Nitta E, Ito K, Miyamoto K, Yoshiwara H, Hosokawa K, Nakamura Y, Gomei Y, Iwasaki H, Hayashi Y, Matsuzaki Y, Nakayama K, Ikeda Y, Hata A, Chiba S, Nakayama KI, Suda T. 2008. Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes Dev. 22:986–991. http://dx.doi.org/10.1101/gad.1621808.
- Onoyama I, Tsunematsu R, Matsumoto A, Kimura T, de Alboran IM, Nakayama K, Nakayama KI. 2007. Conditional inactivation of Fbxw7 impairs cell-cycle exit during T cell differentiation and results in lymphomatogenesis. J. Exp. Med. 204:2875–2888. http://dx.doi.org/10.1084/jem.20062299.
- Geng Y, Yu Q, Sicinska E, Das M, Schneider JE, Bhattacharya S, Rideout WM, Bronson RT, Gardner H, Sicinski P. 2003. Cyclin E ablation in the mouse. Cell 114:431–443. http://dx.doi.org/10.1016/S0092-8674(03)00645-7.
- Siu KT, Rosner MR, Minella AC. 2012. An integrated view of cyclin E function and regulation. Cell Cycle 11:57–64. http://dx.doi.org/10.4161/cc.11.1.18775.
- Minella AC, Loeb KR, Knecht A, Welcker M, Varnum-Finney BJ, Bernstein ID, Roberts JM, Clurman BE. 2008. Cyclin E phosphorylation regulates cell proliferation in hematopoietic and epithelial lineages in vivo. Genes Dev. 22:1677–1689. http://dx.doi.org/10.1101/gad.1650208.
- Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ. 2001. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 294:173–177. http://dx.doi.org/10.1126/science.1065203.
- Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. 2001. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature 413:316–322. http://dx.doi.org/10.1038/35095076.
- Xu Y, Swartz KL, Siu KT, Bhattacharyya M, Minella AC. 2014. Fbw7-dependent cyclin E regulation ensures terminal maturation of bone marrow erythroid cells by restraining oxidative metabolism. Oncogene 33:3161–3171. http://dx.doi.org/10.1038/onc.2013.289.
- Clurman BE, Sheaff RJ, Thress K, Groudine M, Roberts JM. 1996. Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev. 10:1979–1990. http://dx.doi.org/10.1101/gad.10.16.1979.
- Minella AC, Welcker M, Clurman BE. 2005. Ras activity regulates cyclin E degradation by the Fbw7 pathway. Proc. Natl. Acad. Sci. U. S. A. 102:9649–9654. http://dx.doi.org/10.1073/pnas.0503677102.
- Xu D, Liu X, Yu WM, Meyerson HJ, Guo C, Gerson SL, Qu CK. 2011. Non-lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. J. Exp. Med. 208:1977–1988. http://dx.doi.org/10.1084/jem.20110450.
- Loo LW, Grove DI, Williams EM, Neal CL, Cousens LA, Schubert EL, Holcomb IN, Massa HF, Glogovac J, Li CI, Malone KE, Daling JR, Delrow JJ, Trask BJ, Hsu L, Porter PL. 2004. Array comparative genomic hybridization analysis of genomic alterations in breast cancer subtypes. Cancer Res. 64:8541–8549. http://dx.doi.org/10.1158/0008-5472.CAN-04-1992.
- Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. 2005. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121:1109–1121. http://dx.doi.org/10.1016/j.cell.2005.05.026.
- Randall TD, Weissman IL. 1997. Phenotypic and functional changes induced at the clonal level in hematopoietic stem cells after 5-fluorouracil treatment. Blood 89:3596–3606.
- Minella AC, Swanger J, Bryant E, Welcker M, Hwang H, Clurman BE. 2002. p53 and p21 form an inducible barrier that protects cells against cyclin E-cdk2 deregulation. Curr. Biol. 12:1817–1827. http://dx.doi.org/10.1016/S0960-9822(02)01225-3.
- Ekholm-Reed S, Mendez J, Tedesco D, Zetterberg A, Stillman B, Reed SI. 2004. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J. Cell Biol. 165:789–800. http://dx.doi.org/10.1083/jcb.200404092.
- Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C. 2004. Inactivation of hCDC4 can cause chromosomal instability. Nature 428:77–81. http://dx.doi.org/10.1038/nature02313.
- Sherr CJ, Roberts JM. 1999. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 13:1501–1512. http://dx.doi.org/10.1101/gad.13.12.1501.
- Minella AC, Grim JE, Welcker M, Clurman BE. 2007. p53 and SCFFbw7 cooperatively restrain cyclin E-associated genome instability. Oncogene 26:6948–6953. http://dx.doi.org/10.1038/sj.onc.1210518.
- Gupta M, Gupta SK, Balliet AG, Hollander MC, Fornace AJ, Hoffman B, Liebermann DA. 2005. Hematopoietic cells from Gadd45a- and Gadd45b-deficient mice are sensitized to genotoxic-stress-induced apoptosis. Oncogene 24:7170–7179. http://dx.doi.org/10.1038/sj.onc.1208847.
- Hollander MC, Sheikh MS, Bulavin DV, Lundgren K, Augeri-Henmueller L, Shehee R, Molinaro TA, Kim KE, Tolosa E, Ashwell JD, Rosenberg MP, Zhan Q, Fernandez-Salguero PM, Morgan WF, Deng CX, Fornace AJJr. 1999. Genomic instability in Gadd45a-deficient mice. Nat. Genet. 23:176–184. http://dx.doi.org/10.1038/13802.
- Mussman JG, Horn HF, Carroll PE, Okuda M, Tarapore P, Donehower LA, Fukasawa K. 2000. Synergistic induction of centrosome hyperamplification by loss of p53 and cyclin E overexpression. Oncogene 19:1635–1646. http://dx.doi.org/10.1038/sj.onc.1203460.
- Thompson SL, Bakhoum SF, Compton DA. 2010. Mechanisms of chromosomal instability. Curr. Biol. 20:R285–R295. http://dx.doi.org/10.1016/j.cub.2010.01.034.
- Orford KW, Scadden DT. 2008. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat. Rev. Genetics 9:115–128. http://dx.doi.org/10.1038/nrg2269.
- Kimura T, Gotoh M, Nakamura Y, Arakawa H. 2003. hCDC4b, a regulator of cyclin E, as a direct transcriptional target of p53. Cancer Sci. 94:431–436. http://dx.doi.org/10.1111/j.1349-7006.2003.tb01460.x.
- Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, Brown K, Bryson S, Balmain A. 2004. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 432:775–779. http://dx.doi.org/10.1038/nature03155.
- Xu Y, Sengupta T, Kukreja L, Minella AC. 2010. MicroRNA-223 regulates cyclin E activity by modulating expression of F-box and WD-40 domain protein 7. J. Biol. Chem. 285:34439–34446. http://dx.doi.org/10.1074/jbc.M110.152306.
- Mavrakis KJ, Van Der Meulen J, Wolfe AL, Liu X, Mets E, Taghon T, Khan AA, Setty M, Rondou P, Vandenberghe P, Delabesse E, Benoit Y, Socci NB, Leslie CS, Van Vlierberghe P, Speleman F, Wendel HG. 2011. A cooperative microRNA-tumor suppressor gene network in acute T-cell lymphoblastic leukemia (T-ALL). Nat. Genet. 43:673–678. http://dx.doi.org/10.1038/ng.858.
- Balamurugan K, Wang JM, Tsai HH, Sharan S, Anver M, Leighty R, Sterneck E. 2010. The tumour suppressor C/EBPdelta inhibits FBXW7 expression and promotes mammary tumour metastasis. EMBO J. 29:4106–4117. http://dx.doi.org/10.1038/emboj.2010.280.
- Figueroa ME, Skrabanek L, Li Y, Jiemjit A, Fandy TE, Paietta E, Fernandez H, Tallman MS, Greally JM, Carraway H, Licht JD, Gore SD, Melnick A. 2009. MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood 114:3448–3458. http://dx.doi.org/10.1182/blood-2009-01-200519.
- Geng Y, Lee YM, Welcker M, Swanger J, Zagozdzon A, Winer JD, Roberts JM, Kaldis P, Clurman BE, Sicinski P. 2007. Kinase-independent function of cyclin E. Mol. Cell 25:127–139. http://dx.doi.org/10.1016/j.molcel.2006.11.029.
- Grim JE, Knoblaugh SE, Guthrie KA, Hagar A, Swanger J, Hespelt J, Delrow JJ, Small T, Grady WM, Nakayama KI, Clurman BE. 2012. Fbw7 and p53 cooperatively suppress advanced and chromosomally unstable intestinal cancer. Mol. Cell. Biol. 32:2160–2167. http://dx.doi.org/10.1128/MCB.00305-12.
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, Takaoka M, Nakagawa H, Tort F, Fugger K, Johansson F, Sehested M, Andersen CL, Dyrskjot L, Orntoft T, Lukas J, Kittas C, Helleday T, Halazonetis TD, Bartek J, Gorgoulis VG. 2006. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444:633–637. http://dx.doi.org/10.1038/nature05268.
- Fragkos M, Beard P. 2011. Mitotic catastrophe occurs in the absence of apoptosis in p53-null cells with a defective G1 checkpoint. PLoS One 6:e22946. http://dx.doi.org/10.1371/journal.pone.0022946.
- Kenyon J, Fu P, Lingas K, Thomas E, Saurastri A, Santos Guasch G, Wald D, Gerson SL. 2012. Humans accumulate microsatellite instability with acquired loss of MLH1 protein in hematopoietic stem and progenitor cells as a function of age. Blood 120:3229–3236. http://dx.doi.org/10.1182/blood-2011-12-401950.
- Allsopp RC, Morin GB, DePinho R, Harley CB, Weissman IL. 2003. Telomerase is required to slow telomere shortening and extend replicative lifespan of HSCs during serial transplantation. Blood 102:517–520. http://dx.doi.org/10.1182/blood-2002-07-2334.