
Abstract
Scientific progress requires the relentless correction of errors and refinement of hypotheses. Clarity of terminology is essential for clarity of thought and proper experimental interrogation of nature. Therefore, the application of the same scientific term to different and even conflicting phenomena and concepts is not useful and must be corrected. Such abuse of terminology has happened and is still increasing in the case of “neuroinflammation,” a term that until the 1990s meant classical inflammation affecting the central nervous system (CNS) and thereon was progressively used to mostly denote microglia activation. The resulting confusion is very wasteful and detrimental not only for scientists but also for patients, given the numerous failed clinical trials in acute and chronic CNS diseases over the last decade with “anti-inflammatory” drugs. Despite this failure, reassessments of the “neuroinflammation” concept are rare, especially considering the number of articles still using the term. This undesirable situation motivates this article. We review the origins and evolution of the term “neuroinflammation,” discuss the unique tissue defense and repair strategies in the CNS, define CNS immunity, and emphasize the notion of gliopathies to help readdress, if not bury, the term “neuroinflammation” as it stands in the way of scientific progress.
The Problem
In order to frame a discussion about the (mis)use of the term “neuroinflammation,” a few words are necessary to describe the field of inflammation initiated by Virchow and Metchnikoff in the early years of the 20th century (CitationTauber, 2003). In a leading immunology textbook (CitationBaker, 2022), “inflammation” is defined “as the process by which circulating leukocytes and plasma proteins are brought to sites of infection in tissues and are activated to destroy and eliminate the infection. Inflammation is also the main reaction to damaged or dead cells and to the accumulation of abnormal substances in cells and tissues.” Inflammation is thus a manifestation of the interaction between systemic and local innate immunities encompassing epithelial barriers, tissue-resident phagocytes (macrophages and neutrophils), circulating dendritic cells, cytolytic lymphoid cells, and a plethora of chemokines, cytokines, and interferons (typically TNF-α, IL-1β, IL-6, IFN-γ). Tissue destruction and repair are also hallmarks of inflammation. These classic concepts were recently extended to inflammation being “a response to deviations from homeostasis that cannot be reversed by homeostatic mechanisms alone” (CitationMeizlish et al., 2021). This definition rests on several principles. First, homeostatic adaptations, stress responses, inflammation, and adaptive immunity represent a continuum of responses to tissue perturbations. Extreme deviations of biological variables upon failure of homeostasis and stress responses trigger inflammation, eventually implicating adaptive immunity at the very end of the continuum. Second, homeostatic signals can act as inflammatory mediators and vice versa, depending on context, further supporting the gradual character of tissue protection strategies. Third, in restoring local and systemic homeostasis, inflammatory reactions act on key cellular functions and may override incompatible homeostatic signals, for tissue defense is prioritized over homeostasis. If the temporary disappearance of homeostasis has irreversible consequences, becomes chronic, or affects the wrong cells as in autoimmune diseases, inflammation causes disease. Fourth, and highly relevant to the present article, inflammatory reactions are organ and disease specific to a certain extent, although common principles such as tissue infiltration by cells supplied via the bloodstream and cytokine production exist. It is worth noting that cytokine expression by itself is not sufficient to indicate inflammation, for cytokines are pleiotropic, their actions context-dependent, and protection strategies between organs may differ as does tissue organization.
Historically, the prefix “neuro” was added to CNS inflammation by some authors merely to denote the anatomic location of inflammatory processes, for example, autoimmune reactions in multiple sclerosis and its experimental models. However, in the 1990s the same term “neuroinflammation” and variations thereof such as “sterile neuroinflammation” or “nonclassic neuroinflammation” began to be used to describe responses of glial cells, essentially what had been known as gliosis until then, now combined with the detection in the CNS of cytokines and members of the complement family thanks to the availability of sensitive new antibodies for immunohistochemistry and other novel detection methods. In analogy to peripheral inflammation, this evidence was interpreted as a sign of microglia behaving as macrophages, including the release of toxic-free radicals upon respiratory bursts that would kill neurons by pyroptosis, a form of cellular lysis whereby macrophages kill microbes (CitationMcGeer et al., 1994). This idea gained significant traction and launched the field of “neuroinflammation” research dedicated to the study of the reactions of microglia and astrocytes as though they were CNS macrophages. This second, alternative usage of the term “neuroinflammation” has become dominant among scientists, particularly with the arrival of positron emission tomography (PET) and ligands for the (micro)glial receptor protein, TSPO (CitationFiliou et al., 2017), and the astrocytic mitochondrial enzyme, MAOB (CitationHarada et al., 2022). “Neuroinflammation” has also attracted the attention of popular media. In January 2022, the U.S. National Public Radio dedicated a piece to microglia as a “hyperactive cell in the brain that might trigger Alzheimer’s disease” (*). The piece contends that amyloid-β and tau send “microglia cells into overdrive,” engaging in an “inflammatory immune response” that eventually kills neurons as if they were microbes. The fact that the main feature of Alzheimer’s disease is slow neuronal degeneration happening over decades was ignored.
Currently, the term “neuroinflammation” is highly ambiguous and de facto unusable as it is applied in completely opposing ways by the scientific community: to classical inflammation characterized by the infiltration of blood-derived immune cells, on the one hand, and to microglial reactions and variations thereof occurring in the absence of mononuclear perivascular infiltrates on the other. In fact, the presence of the latter constitutes a neuropathological diagnostic exclusion criterion for many diseases. A formal definition of “neuroinflammation” and consensus criteria accepted by the entire scientific community are consequently lacking. This was also the striking conclusion of a recent survey among microglia experts, who presented different views about the meaning of the term, causes of the phenomenon, mediators and cellular targets involved, anatomical localization, and consequences of “neuroinflammation” (CitationPaolicelli et al., 2022). The lack of a practical definition of “neuroinflammation” and the resulting confusion cause many misunderstandings among researchers, while fostering platitudes and ignorance about the complexity of mechanisms aimed to maintain and restore homeostasis in the CNS. This confusion also obscures important features of the spectrum of physiological and pathological interactions between the CNS and the systemic immune system, as well as of the basic biology of microglia and astrocytes. We and others have expressed these concerns earlier (CitationGraeber, 2010; CitationGraeber, 2014; CitationMasgrau et al., 2017; CitationWoodburn et al., 2021).
Sizing the Problem
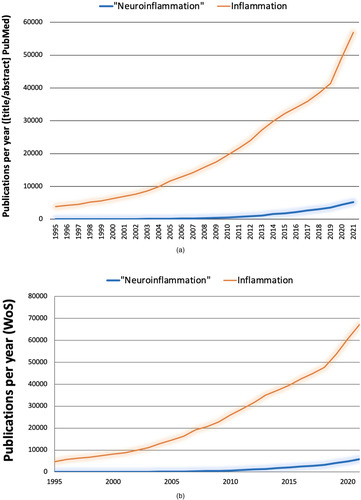
The extent of (mis)use of “neuroinflammation” is confirmed by numbers. A literature search was conducted in June 2022 using the PubMed database, as it is publicly accessible, to get a sense about the popularity of the term “neuroinflammation,” how researchers understand it, and how its usage compares to that of “inflammation.” A search for “inflammation” limited to title and abstract occurrences yielded more than half a million results starting in 1791. The same search for “neuroinflammation” yielded less than 30,000 hits in PubMed starting in 1995, the approximate time when the term was coined according to our earlier research (CitationGraeber, 2014). When using the same time window, that is, 1995 to 2021, “inflammation” still showed more than half a million results. That is, “inflammation” has been cited more than 15 times more frequently than “neuroinflammation.” When comparing the same stretches of time in PubMed for word occurrences in title and abstract of all published articles, “inflammation” appears 74,456 times between 1995 and 2005; 214,542 times between 2006 and 2015; and 255,899 between 2016 and 2021. In the same periods, “neuroinflammation” is cited 427 times between 1995 and 2005; 7,659 times between 2006 and 2015; and 21,033 between 2016 and 2021. In conclusion, the field of “inflammation” has grown very rapidly and may have influenced the growth of the field of “neuroinflammation” ( and (b)). It is worth noting that the usage of “neuroinflammation” has continued to increase in recent years.
Figure 1 Evolution of the inflammation and “neuroinflammation” literature. Comparable search strategies using different databases (a, b) yield consistent results.
What are the ideas associated with “neuroinflammation”? A search for the topic “neuroinflammation” in the Web of Science Core Collection yields around 36,000 publications, the category of Neurosciences accounting for more than 50% of them (). A few themes predominate: “neuroinflammation” is largely used as a synonym of microglia and astrocyte reactivity, particularly microglia activation/reaction—any reaction; the highly specialized nature of microglia in the CNS and microglia molecular and phenotypic heterogeneity are ignored; the detection of “inflammatory cytokines” in CNS tissues or fluids is considered a sign of “neuroinflammation”; and “neuroinflammation” is essentially always considered pathological and frequently labeled a cornerstone of neurological disorders. There is either no explanation of what “neuroinflammation” exactly is and why is detrimental, or it is assumed that “neuroinflammation” causes neuronal demise by a variety of mechanisms including various forms of cell death. A case in point is “neuroinflammation” in Alzheimer’s disease. Here, “neuroinflammation” has been linked to several aspects of microglia including the microglia-associated inflammasome and amyloid-β plaque seeding (CitationVenegas et al., 2017) and propagation (d’CitationErrico et al., 2022), phagocytosis and secretion of amyloid-β fibers (CitationHuang et al., 2021), defective phagocytosis of amyloid-β via TREM2 (CitationUlland & Colonna, 2018), excessive phagocytosis as in synaptic pruning (CitationHong et al., 2016), protection of neurons from amyloid-β plaques (CitationCondello et al., 2015), or none of the above because microglia are dystrophic (CitationStreit et al., 2009) or diseased (CitationSanchez-Mejias et al., 2016). In good logic, this evidence cannot be summarized as “Current evidence suggests that neuroinflammation has a vital role in the pathogenesis and progression of AD” without acknowledging that data are conflictive (CitationLeng & Edison, 2021). In our view, reviews in high-profile journals (CitationBright et al., 2019; CitationHeneka et al., 2015; CitationLeng & Edison, 2021) do not convincingly integrate the existing evidence in a coherent model and fail to address these questions: is the problem of “neuroinflammation” a deficit in CNS innate immunity or that CNS innate immunity is aberrant or loss of homeostatic functions unrelated to CNS immunity or a mix of the above? Is there such thing as “CNS immunity”? What CNS cells participate in “innate immunity”? Can microglia adopt opposite phenotypes at the same time? Which phenotype(s) do imaging markers detect? Are CNS diseases and reactive microglia and astrocytes appropriately modeled in mice? Are microglia and astrocyte reactions disease specific? Are the types of “inflammatory” neuronal death consistent with the mechanisms of neuronal degeneration and demise in human brains? The answers to these questions are crucial for the development of effective therapies and the correct interpretation of neuroimaging data.
Table 1 Web of Science Categories Associated With “Neuroinflammation” (Thomson Reuters).
Proof of abuse of the term “neuroinflammation” is nowhere more apparent than in the sheer number of conditions that are now referred to as “neuroinflammatory,” including obesity (CitationLorena et al., 2021; CitationMiller & Spencer, 2014), pain (CitationDe Logu et al., 2021; CitationLi et al., 2022a), epilepsy (CitationParsons et al., 2022; CitationPracucci et al., 2021), periodontitis (CitationLi et al., 2022b; CitationTeixeira et al., 2017), sleep loss (CitationKou et al., 2022; CitationWisor et al., 2011), autism (CitationMatta et al., 2019; CitationVargas et al., 2005), depression (CitationFurtado & Katzman, 2015; CitationSorensen et al., 2022), schizophrenia (CitationMonji & Mizoguchi, 2022; CitationVallee, 2022), Alzheimer’s disease (CitationLeng & Edison, 2021; CitationMcGeer et al., 2000), Parkinson’s disease (CitationPfeiffer, 2009; CitationYang et al., 2020), amyotrophic lateral sclerosis (CitationArnoux & Dupuis, 2021; CitationLewis et al., 2012), and the brain’s response to air pollution (CitationBlock & Calderon-Garciduenas, 2009; CitationBrockmeyer & D’Angiulli, 2016), to name a few. Arguably, this quite indiscriminate spread is facilitated by the fact that few research scientists are trained in neuropathology, a medical specialty that exists in only a handful of countries, making systematic and direct comparative studies between nervous system diseases very difficult.
In summary, the term “neuroinflammation” had its historical justification in the sudden need to name what had not been seen before, triggered by new technologies that have allowed the visualization of glial reactiveness with unprecedented sensitivity and cellular resolution. The need for new terms to grasp new concepts is inherent to discovery. However, gliosis has always been a microscopic hallmark of neuropathology, not infrequently the only one, especially under subtle subclinical conditions. It is important to point out that Alois Alzheimer was correct when reporting on the original case of the disease that was later named after him that there is no inflammation—that is, no immune cell infiltration. Gliosis yes, but no inflammation. This is also evident from the original case material (CitationGraeber et al., 1998).
We argue that the extension of the term and concepts of “inflammation” to neuro-“inflammation” of CNS parenchyma has lacked rigor, resulting in the dismal failure of clinical trials with drugs based on the “neuroinflammation” framework (reviewed in CitationMasgrau et al., 2017). Up to this day, no therapies exist targeting neuroinflammatory ailments of CNS parenchyma. Surprisingly, the principle of science being error-correcting has not worked here, for the failed clinical trials did not prompt criticism and pleas to urgently revise the tenets of “neuroinflammation.” We argue that progress of knowledge has suffered on this occasion from several traits of contemporary science: media hype, fads followed without reflection by scientists, funding agencies and publishers, and an excessive number of publications that cite others without looking at the original data, thereby perpetuating platitudes and falsehoods. More scientists should have noticed by now, at least in our view, that the emperor has no clothes.
Liberating CNS Tissue Defense and Repair From “Neuroinflammation”
We recommend avoiding the term ‘neuroinflammation’ and to use ‘inflammation’ instead—since there is no hepato-inflammation, pulmo-inflammation or dermato‐inflammation used in a comparable way—and exclusively to indicate CNS diseases featuring infiltration of circulating cells from the systemic immune system. Precise description of phenomena (e.g., types of cells, mechanisms, and molecules involved) is also encouraged. In presenting this recommendation, we do not contend that the CNS lacks strategies to cope with homeostatic changes. The case we aim to build in the following is that the CNS has its own rules and that focus should be placed on understanding CNS defensive strategies in the homeostasis-to-immunity continuum rather than loosely applying peripheral organ inflammation stereotypes to CNS phenomena. Below, we describe CNS defense and repair and pinpoint popular misunderstandings and inappropriate generalizations.
Unique Barriers Afford Limited Inflammation in CNS Parenchyma
The CNS has physical, cellular, and chemical barriers that isolate neural-circuit signaling from external influence and provide protection from traumatic injuries and infections. The skull and meninges act as physical barriers against mechanical injuries. Cellular barriers are the blood-brain barrier (CitationAbbott et al., 2010), the blood-CSF barrier (CitationZamanian et al., 2012), and the meninges, which are surveyed by circulating immune cells and include a diversity of immune cell populations that withstand and resolve pathogen infections before they reach the brain parenchyma (CitationRua & McGavern, 2018). Furthermore, a blood-brain barrier in immunological terms exists at the level of MHC class II expressing perivascular cells which reside in the perivascular spaces (CitationGraeber et al., 1992) that immune cells have to traverse before they can enter the CNS parenchyma. Astrocytes perform CNS barrier functions in three ways. First, a lamina limitans gliae superficialis et perivascularis made mainly of astrocyte processes separates the brain and spinal cord parenchyma from the CNS surface and perivascular spaces, respectively; second, the lamina limitans induces via sonic hedgehog signaling the specialization of the endothelium that makes up the classical blood-brain barrier for molecules which resides at the level of endothelial tight junctions (CitationAlvarez et al., 2011); and third, subsets of astrocytes prevent the infiltration of T-cells by secreting proapoptotic factors such as TRAIL (CitationSanmarco et al., 2021) and FAS (CitationWang et al., 2013). Finally, neural-circuit activity itself may be immunosuppressive (CitationGalea et al., 2003) and contribute to blood-brain barrier maintenance (CitationKalinin et al., 2006). Thus, the CNS parenchyma is normally protected from systemic influence and free of inflammation. This fact has been long recognized and defined as the CNS immune privilege, a term initially coined upon the observation of absence of allograft rejection in CNS parenchyma (CitationMedawar, 1948). Although the choice of the word “privilege” is perhaps not fortunate, the idea of immune tolerance in the parenchyma to preserve the structural and functional integrity of neural circuits with limited regenerative capacity is still valid (CitationEngelhardt et al., 2017; CitationLiu et al., 2021). By contrast, CNS compartments accessible to the innate and adaptive immune system such as the ventricles, meninges, and perivascular spaces at postcapillary venules have been aptly described as dedicated to CNS immunity (CitationBuckley and McGavern, 2022; CitationEngelhardt et al., 2017; CitationRansohoff & Engelhardt, 2012). Thus, anatomical precision is crucial when referring to CNS immunity.
The Most Frequent Tasks of Microglia are not Related to Immunity
In healthy CNS, as well as in many CNS diseases, microglia do not present as macrophages indicating that their most common functions are different (CitationGraeber, 2010). This simple realization has fundamental implications: normal microglia have also nonphagocytic roles in the CNS, and some may be unknown, as suggested by the existence of recently discovered highly specific microglia-neuron contacts (CitationCserep et al., 2021); microglia turning into macrophages signifies extreme conditions, and microglia expressing a macrophage-like phenotype are truly exceptional. Nevertheless, microglia are commonly introduced in scientific literature as the resident immune cells of the CNS and the CNS macrophages. This description is not completely unjustified as microglia derive from yolk-sac erythromyeloid progenitors, present pattern recognition receptors, and are phagocytes that, in certain conditions, produce a plethora of cytokines and promote CNS repair—all staples of innate immunity. However, there has not been enough debate as to whether these traits are vestigial or functional and whether microglia-based immunity operates and is as effective as macrophage-based immunity. This debate is in order because molecular profiling of myeloid cells has proven that microglia are distinct from meningeal and circulating macrophages (CitationBorst & Prinz, 2020). Further, the characterization of microglia as CNS immune cells is at odds with the normally immunologically “silent” CNS parenchyma and overlooks the fact that microglia are specialized to perform day-to-day homeostatic functions, including correction of neuronal hyperactivity (CitationUmpierre & Wu, 2021), participating in neural-circuit remodeling (CitationTremblay et al., 2010) and neurogenesis (CitationSellner et al., 2016), and controlling CNS milieu composition (CitationNimmerjahn et al., 2005). Molecular profiling of microglia subtypes with single-cell RNA-seq confirms the predominance of transcriptional states lacking immunity-related signatures (CitationOlah et al., 2020). Thus, nine microglial clusters have been identified in human brain cortex samples from surgeries and autopsies, of which two clusters, lacking distinct transcription factor signatures or cell surface markers, account for over 50% of microglia. Clusters enriched in genes related to antigen presentation or interferon responses each represent less than 5% of total microglia. It might be premature to label the former as “homeostatic” and the latter as “pathophysiologic,” as proposed in (CitationGoldmann et al., 2015), because whether the identified microglia subtypes are fixed and universal across conditions, and what their functions are in the normal and diseased CNS, is not yet known. For instance, tonic IFN-dependent signaling exists in the brain in physiological conditions (CitationGoldmann et al., 2015). Single-cell RNA-seq also shows that genes associated with different diseases are distributed among microglia transcriptional clusters from healthy human brains, and only one cluster is less abundant in Alzheimer’s disease (CitationOlah et al., 2020). All in all, the molecular characterization of microglia at a high molecular resolution supports two notions: microglia exert a wide range of roles in the healthy CNS that challenge classic classifications of homeostatic versus immunologic, and microglia reactions in diseases are subtle and multifaceted and do not follow a single pattern. This phenotypic complexity demands an evolved view of microglia and a more precise terminology beyond “innate immunity” and its manifestation as “neuroinflammation.”
Immune Signaling in CNS Circuits is not “Neuroinflammation”
The discovery that selective ablation of meningeal lymphocyte populations results in cognitive deficits in healthy mice (CitationDerecki et al., 2010; CitationFiliano et al., 2016; CitationRibeiro et al., 2019) has led to the notion of neuromodulation by immune factors originating in the meninges, including IL-4, IL-17, and INF-γ (CitationAlves de Lima et al., 2020). This link between the immune and nervous systems does not render the principle of immune privilege of the CNS parenchyma obsolete, as repeatedly argued, because neuromodulation is a prime strategy of homeostatic adjustment of anatomically fixed neural circuits to rapidly changing brain states. Thus, should meningeal IL-4, IL-17, and INF-γ act as neuromodulators, it is in fact more appropriate to think of them as homeostatic rather than immunity signals activated in response to extreme deviations of homeostasis. Immune factors doubling as homeostatic regulators in the CNS abound, including neuron-derived IL-33, which modulates synaptic plasticity and memory consolidation (CitationNguyen et al., 2020); TNF-α, IL-1β, and chemokines (reviewed in CitationWoodburn et al., 2021); and several others (CitationBoulanger, 2009; CitationMirabella et al., 2021). Likewise, the capacity of brain cells to detect immune cues in the context of systemic infections must not be labeled as “neuroinflammation”; rather, what happens is that peripheral nerves sense cytokines and transmit this information to the brain to induce specific changes in behavior (CitationBonaz et al., 2017)—whether these are adaptive or maladaptive is discussed elsewhere (CitationAlves de Lima et al., 2020).
When “Neuroinflammation” Masks Gliopathies
A widespread belief about CNS parenchymal “neuroinflammation” during CNS diseases is that it is initially protective, but it turns out to be detrimental when it becomes chronic. Like many other tenets in neuroinflammation, this one is also borrowed from systemic immunity. Recall that a key principle of inflammation is that it takes temporary control of core cell functions and coordinates systemic responses; if cellular, tissue, or systemic homeostasis is overridden beyond recovery, inflammation becomes pathological (CitationMeizlish et al., 2021). For example, in rheumatoid arthritis TNF-α, IL-1, and IL6 disrupt the tight feedback control that exists between osteoclast-dependent bone resorption and osteoblast-dependent bone formation for proper bone remodeling, resulting in progressive erosion of bone tissue in joints (CitationHirsch & Hunot, 2009; CitationShaw & Gravallese, 2016). In the case of “neuroinflammation,” as the term is used rather generically, there is no such cellular and mechanistic understanding as to why “sustained parenchymal neuroinflammation” is deleterious. For example, “neuroinflammation” is, for some authors, the reactive astrocytosis detected by immunohistochemical and imaging approaches in Alzheimer’s disease (CitationHarada et al., 2022). What a more detailed analysis of the existing data shows is that amyloid-β damages astrocytes, particularly their endolysosomal system and mitochondria, resulting in the disruption of energy metabolism and ROS homeostasis in astrocyte-neuronal circuits, among other alterations (reviewed in CitationGalea et al., 2022). There is no evidence in human brains that reactive astrocytes are first “neuroprotective” and then “neurotoxic”. These are simplistic classifications that do not reflect the complex mix of adaptive and maladaptive changes occurring in any phenotypical transformation (CitationEscartin et al., 2021). Rather, based on correlations between gene expression and scoring of disease stages in Alzheimer’s disease, astrocytes seem to evolve from doing badly to doing worse (CitationGalea et al., 2022). This is a case of astrocytopathy and not innate immunity going rogue. Whether cytokines released by stressed astrocytes (CitationOrre et al., 2014) counteract or exacerbate the toxic actions of amyloid-β by acting on core astrocytic functions has not been determined because, beyond the detrimental actions attributed to the most highly cited cytokines (e.g., IL-1β, TNF-α), there is no analysis of the complex roles of secretomes of brain cells during stress. A case in point is the phenomenon of resilience in Alzheimer’s disease, that is, subjects harboring pathological hallmarks in the absence of cognitive decline. It has been proposed that reduced “neuroinflammation” accounts for resilience (CitationViviano et al., 2022). However, the data do not show reduced expression of “inflammatory” cytokines in resilient subjects, but a distinct cytokine profile as compared to patients with Alzheimer’s disease (CitationBarroeta-Espar et al., 2019), pinpointing the existence of a very successful protective response in the homeostasis-to-immunity continuum in resilient subjects, arguably mediated by adaptive changes in glia subsets. Interestingly, IL-1β is equally upregulated in both the brains of resilient subjects and patients (CitationBarroeta-Espar et al., 2019), indicating that context determines the roles of this cytokine. Yet another example of the “neuroinflammation” (mis)construction, in our view, is the microglial inflammasome in Alzheimer’s disease. The evidence causally linking the inflammasome with pathogenesis in mouse models (CitationIsing et al., 2019) is at odds with ample evidence supporting defective TREM2-mediated phagocytosis of amyloid-β by microglia (CitationGratuze et al., 2021; CitationUlland et al., 2017). Therapies in development to potentiate TREM2 (CitationWang et al., 2020) function or inhibit the inflammasome (CitationRavichandran & Heneka, 2021) will settle which scenario is correct or if both occur. We predict that the problem of microglia in Alzheimer’s disease is more of a loss than gain-of-toxic functions and that inflammasome-associated microglia phenotypes are malfunctional microglia, that is, a case of microgliopathy, perhaps due to extreme microglia de-differentiation upon loss of neural-circuit control (CitationHeneka et al., 2010; CitationIaccarino et al., 2016). As with astrocytes, there is no evidence of microglia transitions from protective to detrimental phenotypes in Alzheimer’s disease. Rather, patient heterogeneity with regard to disease onset and progression might originate in the proportion of resilient versus malfunctional microglia phenotypes determined by genetic backgrounds (e.g., TREM2 polymorphisms and APOE alleles).
What is and is not CNS Innate Immunity
In conclusion, there is an abuse of the term “neuroinflammation” when used to depict any morphological and functional change in resident brain cells (usually microglia but also astrocytes and vascular cells) accompanied by upregulation of cytokines, chemokines, and interferons or the PET ligands TSPO and deprenyl for MAOB, with a pathological connotation, with utter disregard for mechanistic detail, fact-checking, and contradictory evidence. We wish to end with a list of phenomena we think represent CNS immunity protecting the CNS from external influence and coming into play in extreme deviations of homeostasis. Not surprisingly, CNS immunity is specialized in maintaining and rebuilding CNS barriers and in interacting with the systemic immune system. Examples are (i) meningeal immunity (CitationRua & McGavern, 2018); (ii) formation of scar borders by astrocytes activated upon acute injury to restore the astrocyte limitans thereby preventing immune-cell infiltration (CitationWahane & Sofroniew, 2022)—here it is appropriate to talk about “neuroprotective” astrocytes; (iii) astrocytes inducing the apoptosis of T-lymphocytes through TRAIL (CitationSanmarco et al., 2021) and FAS (CitationWang et al., 2013); (iv) newly discovered subsets of LPS-sensitive astrocytes located near ventricles, blood vessels, and sub-pial zones (CitationHasel et al., 2021); and (v) the interplay of microglia with macrophages to restore CNS vasculature after injuries (CitationHalder and Milner, 2019; CitationYao et al., 2022).
Whether phagocytosis of misfolded proteins (CitationPlowey et al., 2022) and neuronal (CitationAndoh & Koyama, 2021) or oligodendrocyte (CitationAndoh & Koyama, 2021) materials by microglia belongs to homeostasis or immunity is debatable. Plausibly, different mechanisms and microglia populations come into play in the homeostasis-to-immunity continuum, although we argue that, given their highly specialized nature, microglia are at their fittest when dealing with small to moderate deviations from homeostasis.
Finally, we support the use of terms such as “neuroimmune” interactions between systemic or CNS meningeal immunity and CNS resident cells and “autoimmune diseases” or “inflammatory diseases of the CNS” when referring to CNS diseases caused or exacerbated by systemic immunity or aberrant meningeal immunity. The advent of molecular tools to characterize CNS and circulating cells with unprecedented molecular resolution will help refine the understanding of CNS defense and repair and neuroimmune interactions and possibly guide the generation of new terminology to describe novel phenomena.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Australian Research Council, Spanish Ministerio de Investigación e Innovación (grant number DP150104472, Project PID2019-107633rb-I00).
References
- https://www.npr.org/sections/health-shots/2022/01/30/1076166807/how-a-hyperactive-cell-in-the-brain-might-trigger-alzheimers-disease?t=1656320845618
- Abbott N. J., Patabendige A. A., Dolman D. E., Yusof S. R., Begley D. J. (2010). Structure and function of the blood-brain barrier. Neurobiology of Disease, 37(1), 13–25. https://doi.org/10.1016/j.nbd.2009.07.030
- Alvarez J. I., Dodelet-Devillers A., Kebir H., Ifergan I., Fabre P. J., Terouz S., Sabbagh M., Wosik K., Bourbonnière L., Bernard M., van Horssen J., de Vries H. E., Charron F., Prat A. (2011). The hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science, 334(6063), 1727–1731. https://doi.org/10.1126/science.1206936
- Alves de Lima K., Rustenhoven J., Kipnis J. (2020). Meningeal immunity and its function in maintenance of the central nervous system in health and disease. Annual Review of Immunology, 38, 597–620. https://doi.org/10.1146/annurev-immunol-102319-103410
- Andoh M., Koyama R. (2021). Comparative review of microglia and monocytes in CNS phagocytosis. Cells, 10(10), 1–27. https://doi.org/10.3390/cells10102555
- Arnoux A., Dupuis L. (2021). Linking neuroinflammation to motor neuron degeneration in ALS: The critical role of CXCL13/CXCR5. EBioMedicine, 63, 103149. https://doi.org/10.1016/j.ebiom.2020.103149
- Baker D. L (2022) Editorial in Abbas, Abul K., Lichtman, Andrew H. and Pillai, Shiv, Cellular and molecular immunology. Elsevier.
- Barroeta-Espar I,et al.. (2019). Distinct cytokine profiles in human brains resilient to Alzheimer’s pathology. Neurobiology of Disease, 121, 327–337. https://doi.org/10.1016/j.nbd.2018.10.009
- Block M. L., Calderon-Garciduenas L. (2009). Air pollution: Mechanisms of neuroinflammation and CNS disease. Trends in Neurosciences, 32(9), 506–516. https://doi.org/10.1016/j.tins.2009.05.009
- Bonaz B., Sinniger V., Pellissier S. (2017). The Vagus nerve in the neuro-immune axis: Implications in the pathology of the gastrointestinal tract. Frontiers in Immunology, 8, 1452. https://doi.org/10.3389/fimmu.2017.01452
- Borst K., Prinz M. (2020). Deciphering the heterogeneity of myeloid cells during neuroinflammation in the single-cell era. Brain Pathology, 30(6), 1192–1207. https://doi.org/10.1111/bpa.12910
- Boulanger L. M. (2009). Immune proteins in brain development and synaptic plasticity. Neuron, 64(1), 93–109. https://doi.org/10.1016/j.neuron.2009.09.001
- Bright F., Werry E. L., Dobson-Stone C., Piguet O., Ittner L. M., Halliday G. M., Hodges J. R., Kiernan M. C., Loy C. T., Kassiou M., Kril J. J. (2019). Neuroinflammation in frontotemporal dementia. Nature Reviews. Neurology, 15(9), 540–555. https://doi.org/10.1038/s41582-019-0231-z
- Brockmeyer S., D’Angiulli A (2016) How air pollution alters brain development: The role of neuroinflammation. Translational Neuroscience, 7(1), 24–30. https://doi.org/10.1515/tnsci-2016-0005
- Buckley M. W., McGavern D. B. (2022). Immune dynamics in the CNS and its barriers during homeostasis and disease. Immunological Reviews, 306(1), 58–75. https://doi.org/10.1111/imr.13066
- Condello C., Yuan P., Schain A., Grutzendler J. (2015). Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nature Communications, 6, 6176. https://doi.org/10.1038/ncomms7176
- Cserep C., Posfai B., Denes A. (2021). Shaping neuronal fate: Functional heterogeneity of direct microglia-neuron interactions. Neuron, 109(2), 222–240. https://doi.org/10.1016/j.neuron.2020.11.007
- d’Errico P., Ziegler-Waldkirch S., Aires V., Hoffmann P., Mezo C., Erny D., Monasor L. S., Liebscher S., Ravi V. M., Joseph K., Schnell O., Kierdorf K., Staszewski O., Tahirovic S., Prinz M., Meyer-Luehmann M. (2022). Microglia contribute to the propagation of Abeta into unaffected brain tissue. Nature Neuroscience, 25(1), 20–25. https://doi.org/10.1038/s41593-021-00951-0
- De Logu F., Boccella S., Guida F. (2021). Editorial: The role of neuroinflammation in chronic pain development and maintenance. Frontiers in Pharmacology, 12, 821534. https://doi.org/10.3389/fphar.2021.821534
- Derecki N. C., Cardani A. N., Yang C. H., Quinnies K. M., Crihfield A., Lynch K. R., Kipnis J. (2010). Regulation of learning and memory by meningeal immunity: A key role for IL-4. Journal of Experimental Medicine, 207(5), 1067–1080. https://doi.org/10.1084/jem.20091419
- Engelhardt B., Vajkoczy P., Weller R. O. (2017). The movers and shapers in immune privilege of the CNS. Nature Immunology, 18(2), 123–131. https://doi.org/10.1038/ni.3666
- Escartin C., et al. (2021). Reactive astrocyte nomenclature, definitions, and future directions. Nature Neuroscience, 24(3), 312–325. https://doi.org/10.1038/s41593-020-00783-4
- Filiano A. J., Xu Y., Tustison N. J., Marsh R. L., Baker W., Smirnov I., Overall C. C., Gadani S. P., Turner S. D., Weng Z., Peerzade S. N., Chen H., Lee K. S., Scott M. M., Beenhakker M. P., Litvak V., Kipnis J. (2016). Unexpected role of interferon-γ in regulating neuronal connectivity and social behaviour. Nature, 535(7612), 425–429. https://doi.org/10.1038/nature18626
- Filiou M. D., Banati R. B., Graeber M. B. (2017). The 18-kDa translocator protein as a CNS drug target: Finding our way through the neuroinflammation fog. CNS & Neurological Disorders Drug Targets, 16(9), 990–999. https://doi.org/10.2174/1871527316666171004125107
- Furtado M., Katzman M. A. (2015). Examining the role of neuroinflammation in major depression. Psychiatry Research, 229(1-2), 27–36. https://doi.org/10.1016/j.psychres.2015.06.009
- Galea E., Heneka M. T., Russo D., Feinstein C., L D. (2003). Intrinsic regulation of brain inflammatory responses. Cellular and Molecular Neurobiology, 23(4-5), 625–635. https://doi.org/10.1023/A:1025084415833
- Galea E., Weinstock L. D., Larramona-Arcas R., Pybus A. F., Giménez-Llort L., Escartin C., Wood L. B. (2022). Multi-transcriptomic analysis points to early organelle dysfunction in human astrocytes in Alzheimer’s disease. Neurobiology of Disease, 166, 105655. https://doi.org/10.1016/j.nbd.2022.105655
- Goldmann T., et al. (2015). USP18 Lack in microglia causes destructive interferonopathy of the mouse brain. EMBO Journal, 34(12), 1612–1629. https://doi.org/10.15252/embj.201490791
- Graeber M. B. (2010). Changing face of microglia. Science, 330(6005), 783–788. https://doi.org/10.1126/science.1190929
- Graeber M. B. (2014). Neuroinflammation: No rose by any other name. Brain Pathology, 24(6), 620–622. https://doi.org/10.1111/bpa.12192
- Graeber M. B., Streit W. J., Buringer D., Sparks D. L., Kreutzberg G. W. (1992) Ultrastructural location of major histocompatibility complex (MHC) class II positive perivascular cells in histologically normal human brain. Journal of Neuropathology & Experimental Neurology, 51(3), 303–311. https://doi.org/10.1097/00005072-199205000-00009
- Graeber M. B., Kosel S., Grasbon-Frodl E., Moeller H. J., Mehraein P. (1998). Histopathology and APOE genotype of the first Alzheimer disease patient, Auguste D. Neurogenetics, 1(3), 223–228. https://doi.org/10.1007/s100480050033
- Gratuze M., Chen Y., Parhizkar S., Jain N., Strickland M. R., Serrano J. R., Colonna M., Ulrich J. D., Holtzman D. M. (2021). Activated microglia mitigate Aβ-associated tau seeding and spreading. Journal of Experimental Medicine, 218(8), 1–11. https://doi.org/10.1084/jem.20210542
- Halder S. K., Milner R. (2019). A critical role for microglia in maintaining vascular integrity in the hypoxic spinal cord. Proceedings of the National Academy of Sciences of the United States of America, 116, 26029–26037. https://doi.org/10.1073/pnas.1912178116
- Harada R., Furumoto S., Kudo Y., Yanai K., Villemagne V. L., Okamura N. (2022). Imaging of reactive astrogliosis by positron emission tomography. Frontiers in Neuroscience, 16, 807435. https://doi.org/10.3389/fnins.2022.807435
- Hasel P., Rose I. V. L., Sadick J. S., Kim R. D., Liddelow S. A. (2021). Neuroinflammatory astrocyte subtypes in the mouse brain. Nature Neuroscience, 24(10), 1475–1487. https://doi.org/10.1038/s41593-021-00905-6
- Heneka M. T., Nadrigny F., Regen T., Martinez-Hernandez A., Dumitrescu-Ozimek L., Terwel D., Jardanhazi-Kurutz D., Walter J., Kirchhoff F., Hanisch U. K., Kummer M. P. (2010). Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proceedings of the National Academy of Sciences of the United States of America, 107(13), 6058–6063. https://doi.org/10.1073/pnas.0909586107
- Heneka M. T., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurology, 14(4), 388–405. https://doi.org/10.1016/S1474-4422(15)70016-5
- Hirsch E. C., Hunot S. (2009). Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurology, 8(4), 382–397. https://doi.org/10.1016/S1474-4422(09)70062-6
- Hong S., Beja-Glasser V. F., Nfonoyim B. M., Frouin A., Li S. M., Ramakrishnan S., Merry K. M., Shi Q. Q., Rosenthal A., Barres B. A., Lemere C. A., Selkoe D. J., Stevens B. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science, 352(6286), 712–716. https://doi.org/10.1126/science.aad8373
- Huang Y., Happonen K. E., Burrola P. G., O’Connor C., Hah N., Huang L., Nimmerjahn A., Lemke G. (2021) Microglia use TAM receptors to detect and engulf amyloid beta plaques. Nature Immunology, 22(5), 586–594. https://doi.org/10.1038/s41590-021-00913-5
- Iaccarino H. F., Singer A. C., Martorell A. J., Rudenko A., Gao F., Gillingham T. Z., Mathys H., Seo J., Kritskiy O., Abdurrob F., Adaikkan C., Canter R. G., Rueda R., Brown E. N., Boyden E. S., Tsai L. H. (2016). Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature, 540(7632), 230–235. https://doi.org/10.1038/nature20587
- Ising C., et al. (2019). NLRP3 Inflammasome activation drives tau pathology. Nature, 575(7784), 669–673. https://doi.org/10.1038/s41586-019-1769-z
- Kalinin S., Feinstein D. L., Xu H. L., Huesa G., Pelligrino D. A., Galea E. (2006). Degeneration of noradrenergic fibres from the locus coeruleus causes tight-junction disorganisation in the rat brain. European Journal of Neuroscience, 24(12), 3393–3400. https://doi.org/10.1111/j.1460-9568.2006.05223.x
- Kou L., Chi X., Sun Y., Han C., Wan F., Hu J., Yin S., Wu J., Li Y., Zhou Q., Zou W., Xiong N., Huang J., Xia Y., Wang T. (2022) The circadian clock protein Rev-erbalpha provides neuroprotection and attenuates neuroinflammation against Parkinson’s disease via the microglial NLRP3 inflammasome. Journal of Neuroinflammation, 19(1), 133. https://doi.org/10.1186/s12974-022-02494-y
- Leng F. D., Edison P. (2021). Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nature Reviews Neurology, 17(3), 157–172. https://doi.org/10.1038/s41582-020-00435-y
- Lewis C. A., Manning J., Rossi F., Krieger C. (2012). The neuroinflammatory response in ALS: The roles of microglia and T cells. Neurology Research international, 2012: 1–8. https://doi.org/10.1155/2012/803701
- Li Q. Y., Chen S. X., Liu J. Y., Yao P. W., Duan Y. W., Li Y. Y., Zang Y. ( 2022a). Neuroinflammation in the anterior cingulate cortex: The potential supraspinal mechanism underlying the mirror-image pain following motor fiber injury. Journal of Neuroinflammation, 19(1), 162. https://doi.org/10.1186/s12974-022-02525-8
- Li X., Kiprowska M., Kansara T., Kansara P., Li P. ( 2022b) Neuroinflammation: A distal consequence of periodontitis. Journal of Dental Research, 101(12), 1441–1449. https://doi.org/10.1177/00220345221102084
- Liu X. L., Zhang M. M., Liu H. N., Zhu R., He H., Zhou Y. Q., Zhang Y. L., Li C., Liang D. H., Zeng Q., Huang G. Z. (2021). Bone marrow mesenchymal stem cell-derived exosomes attenuate cerebral ischemia-reperfusion injury-induced neuroinflammation and pyroptosis by modulating microglia M1/M2 phenotypes. Experimental Neurology, 341, 1–16.
- Lorena F. B., do Nascimento B. P. P., Camargo E., Bernardi M. M., Fukushima A. R., do N. P. J., de B. N. P., Brandao M. E. S., Ribeiro M. O. (2021). Long-term obesity is associated with depression and neuroinflammation. Archives of Endocrinology and Metabolism, 65(5), 537–548. https://doi.org/10.20945/2359-3997000000400
- Masgrau R., Guaza C., Ransohoff R. M., Galea E. (2017). Should we stop saying ‘glia’ and ‘neuroinflammation’? Trends in Molecular Medicine, 23(6), 486–500. https://doi.org/10.1016/j.molmed.2017.04.005
- Matta S. M., Hill-Yardin E. L., Crack P. J. (2019). The influence of neuroinflammation in autism Spectrum disorder. Brain Behavior and Immunity, 79, 75–90. https://doi.org/10.1016/j.bbi.2019.04.037
- McGeer P. L., McGeer E. G., Yasojima K. (2000). Alzheimer disease and neuroinflammation. Journal of Neural Transmission Supplement, 59, 53–57. https://doi.org/10.1007/978-3-7091-6781-6_8
- McGeer P. L., Klegeris A., Walker D. G., Yasuhara O., McGeer E. G. (1994). Pathological proteins in senile plaques. Tohoku Journal of Experimental Medicine, 174(3), 269–277. https://doi.org/10.1620/tjem.174.269
- Medawar P. B. (1948). Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. British Journal of Experimental Pathology, 29(1), 58–69.
- Meizlish M. L., Franklin R. A., Zhou X., Medzhitov R. (2021). Tissue homeostasis and inflammation. Annual Review of Immunology, 39, 557–581. https://doi.org/10.1146/annurev-immunol-061020-053734
- Miller A. A., Spencer S. J. (2014). Obesity and neuroinflammation: A pathway to cognitive impairment. Brain Behavior and Immunity, 42, 10–21. https://doi.org/10.1016/j.bbi.2014.04.001
- Mirabella F., Desiato G., Mancinelli S., Fossati G., Rasile M., Morini R., Markicevic M., Grimm C., Amegandjin C., Termanini A. (2021). Prenatal interleukin 6 elevation increases glutamatergic synapse density and disrupts hippocampal connectivity in offspring. Immunity, 54(11), 2611–2631. https://doi.org/10.1016/j.immuni.2021.10.006
- Monji A., Mizoguchi Y. (2022). Neuroinflammation in late-onset schizophrenia: Viewing from the standpoint of the microglia hypothesis. Neuropsychobiology, 81(2), 98–103. https://doi.org/10.1159/000517861
- Nguyen P. T., Dorman L. C., Pan S., Vainchtein I. D., Han R. T., Nakao-Inoue H., Taloma S. E., Barron J. J., Molofsky A. B., Kheirbek M. A., Molofsky A. V. (2020) Microglial remodeling of the extracellular matrix promotes synapse plasticity. Cell, 182(2), 388–403. e315. https://doi.org/10.1016/j.cell.2020.05.050
- Nimmerjahn A., Kirchhoff F., Helmchen F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science, 308(5726), 1314–1318. https://doi.org/10.1126/science.1110647
- Olah M., et al. (2020). Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nature Communications, 11(1), 6129. https://doi.org/10.1038/s41467-020-19737-2
- Orre M., Kamphuis W., Osborn L. M., Jansen A. H. P., Kooijman L., Bossers K., Hol E. M. (2014). Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiology of Aging, 35(12), 2746–2760. https://doi.org/10.1016/j.neurobiolaging.2014.06.004
- Paolicelli R. C., et al. (2022). Microglia states and nomenclature: A field at its crossroads. Neuron, 110(21), 3458–3483. https://doi.org/10.1016/j.neuron.2022.10.020
- Parsons A. L. M., Bucknor E. M. V., Castroflorio, E., Soares, T. R., Oliver P. L., Rial D. (2022) The interconnected mechanisms of oxidative stress and neuroinflammation in epilepsy. Antioxidants (Basel), 11(1), 1–17. https://doi.org/10.3390/antiox11010157
- Pfeiffer R. F. (2009). Neuroinflammation and Parkinson disease: The silent battleground. Neurology, 73(18), 1434–1435. https://doi.org/10.1212/WNL.0b013e3181c2f07d
- Plowey E. D., Bussiere T., Rajagovindan R., Sebalusky J., Hamann S., von Hehn C., Castrillo-Viguera C., Sandrock A., Budd Haeberlein S., van Dyck C. H., Huttner A. (2022). Alzheimer disease neuropathology in a patient previously treated with aducanumab. Acta Neuropathologica, 144(1), 143–153. https://doi.org/10.1007/s00401-022-02433-4
- Pracucci E., Pillai V., Lamers D., Parra R., Landi S. (2021). Neuroinflammation: A signature or a cause of epilepsy? International Journal of Molecular Sciences, 22(13), 1–18. https://doi.org/10.3390/ijms22136981
- Ransohoff R. M., Engelhardt B. (2012). The anatomical and cellular basis of immune surveillance in the central nervous system. Nature Reviews Immunology, 12(9), 623–635. https://doi.org/10.1038/nri3265
- Ravichandran K. A., Heneka M. T. (2021). Inflammasome activation in neurodegenerative diseases. Essays in Biochemistry, 65(7), 885–904. https://doi.org/10.1042/EBC20210021
- Ribeiro M., Brigas H. C., Temido-Ferreira M., Pousinha P. A., Regen T., Santa C., Coelho J. E., Marques-Morgado I., Valente C. A., Omenetti S., Stockinger B., Waisman A., Manadas B., Lopes L. V., Silva-Santos B., Ribot J. C. (2019) Meningeal γδ T cell-derived IL-17 controls synaptic plasticity and short-term memory. Sci Immunol, 4(40), 1–13. https://doi.org/10.1126/sciimmunol.aay5199
- Rua R., McGavern D. B. (2018). Advances in meningeal immunity. Trends in Molecular Medicine, 24(6), 542–559. https://doi.org/10.1016/j.molmed.2018.04.003
- Sanchez-Mejias E., Navarro V., Jimenez S., Sanchez-Mico M., Sanchez-Varo R., Nuñez-Diaz C., Trujillo-Estrada L., Davila J. C., Vizuete M., Gutierrez A., Vitorica J. (2016). Soluble phospho-tau from Alzheimer’s disease hippocampus drives microglial degeneration. Acta Neuropathologica, 132(6), 897–916. https://doi.org/10.1007/s00401-016-1630-5
- Sanmarco L. M., Wheeler M. A., Gutiérrez-Vázquez C., Polonio C. M., Linnerbauer M., Pinho-Ribeiro F. A., Li Z., Giovannoni F., Batterman K. V., Scalisi G., Zandee S. E. J., Heck E. S., Alsuwailm M., Rosene D. L., Becher B., Chiu I. M., Prat A., Quintana F. J. (2021). Gut-licensed IFNγ(+) NK cells drive LAMP1(+)TRAIL(+) anti-inflammatory astrocytes. Nature, 590(7846), 473–479. https://doi.org/10.1038/s41586-020-03116-4
- Sellner S., Paricio-Montesinos R., Spieß A., Masuch A., Erny D., Harsan L. A., Elverfeldt D. V., Schwabenland M., Biber K., Staszewski O., Lira S., Jung S., Prinz M., Blank T. (2016). Microglial CX3CR1 promotes adult neurogenesis by inhibiting Sirt 1/p65 signaling independent of CX3CL1. Acta Neuropathologica Communications, 4(1), 102. https://doi.org/10.1186/s40478-016-0374-8
- Shaw A. T., Gravallese E. M. (2016). Mediators of inflammation and bone remodeling in rheumatic disease. Seminars in Cell & Developmental Biology, 49, 2–10. https://doi.org/10.1016/j.semcdb.2015.10.013
- Sorensen N. V., Orlovska-Waast S., Jeppesen R., Klein-Petersen A. W., Christensen R. H. B., Benros M. E. (2022). Neuroinflammatory Biomarkers in Cerebrospinal Fluid From 106 Patients With Recent-Onset Depression Compared With 106 Individually Matched Healthy Control Subjects. Biol Psychiatry.
- Streit W. J., Braak H., Xue Q. S., Bechmann I. (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathologica, 118(4), 475–485. https://doi.org/10.1007/s00401-009-0556-6
- Tauber A. I. (2003). Metchnikoff and the phagocytosis theory. Nature Reviews Molecular Cell Biology, 4(11), 897–901. https://doi.org/10.1038/nrm1244
- Teixeira F. B., Saito M. T., Matheus F. C., Prediger R. D., Yamada E. S., Maia C. S. F., Lima R. R. (2017). Periodontitis and Alzheimer’s disease: A possible comorbidity between oral chronic inflammatory condition and neuroinflammation. Frontiers in Aging Neuroscience, 9, 327. https://doi.org/10.3389/fnagi.2017.00327
- Tremblay M., Lowery R. L., Majewska A. K. (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biology, 8(11), e1000527. https://doi.org/10.1371/journal.pbio.1000527
- Ulland T. K., Colonna M. (2018). TREM2 - a key player in microglial biology and Alzheimer disease. Nature Reviews. Neurology, 14(11), 667–675. https://doi.org/10.1038/s41582-018-0072-1
- Ulland T. K., Song W. M., Huang S. C., Ulrich J. D., Sergushichev A., Beatty W. L., Loboda A. A., Zhou Y., Cairns N. J., Kambal A., Loginicheva E., Gilfillan S., Cella M., Virgin H. W., Unanue E. R., Wang Y., Artyomov M. N., Holtzman D. M., Colonna M. (2017). TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell, 170(4), 649–663.e613. https://doi.org/10.1016/j.cell.2017.07.023
- Umpierre A. D., Wu L. J. (2021). How microglia sense and regulate neuronal activity. Glia, 69(7), 1637–1653. https://doi.org/10.1002/glia.23961
- Vallee A. (2022). Neuroinflammation in schizophrenia: The key role of the WNT/beta-catenin pathway. International Journal of Molecular Sciences, 23(5), 1–15. https://doi.org/10.3390/ijms23052810
- Vargas D. L., Nascimbene C., Krishnan C., Zimmerman A. W., Pardo C. A. (2005). Neuroglial activation and neuroinflammation in the brain of patients with autism. Annals of Neurology, 57(1), 67–81. https://doi.org/10.1002/ana.20315
- Venegas C., Kumar S., Franklin B. S., Dierkes T., Brinkschulte R., Tejera D., Vieira-Saecker A., Schwartz S., Santarelli F., Kummer M. P., Griep A., Gelpi E., Beilharz M., Riedel D., Golenbock D. T., Geyer M., Walter J., Latz E., Heneka M. T. (2017). Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature, 552, 355–361. https://doi.org/10.1038/nature25158
- Viviano M., Barresi E., Simeon F. G., Costa B., Taliani S., Da Settimo F., Pike V. W., Castellano S. (2022) Essential principles and recent progress in the development of TSPO PET ligands for neuroinflammation imaging. Current Medicinal Chemistry, 29(28), 4862–4890. https://doi.org/10.2174/0929867329666220329204054
- Wahane S., Sofroniew M. V. (2022). Loss-of-function manipulations to identify roles of diverse glia and stromal cells during CNS scar formation. Cell and Tissue Research, 387(3), 337–350. https://doi.org/10.1007/s00441-021-03487-8
- Wang S., Mustafa M., Yuede C. M., Salazar S. V., Kong P., Long H., Ward M., Siddiqui O., Paul R., Gilfillan S., Ibrahim A., Rhinn H., Tassi I., Rosenthal A., Schwabe T., Colonna M. (2020). Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. Journal of Experimental Medicine, 217(9), 1–19. https://doi.org/10.1084/jem.20200785
- Wang X., Haroon F., Karray S., Martina D., Schlüter D. (2013). Astrocytic Fas ligand expression is required to induce T-cell apoptosis and recovery from experimental autoimmune encephalomyelitis. European Journal of Immunology, 43(1), 115–124. https://doi.org/10.1002/eji.201242679
- Wisor J. P., Schmidt M. A., Clegern W. C. (2011). Evidence for neuroinflammatory and microglial changes in the cerebral response to sleep loss. Sleep, 34(3), 261–272. https://doi.org/10.1093/sleep/34.3.261
- Woodburn S. C., Bollinger J. L., Wohleb E. S. (2021). The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. Journal of Neuroinflammation, 18(1), 258. https://doi.org/10.1186/s12974-021-02309-6
- Yang L., Mao K., Yu H., Chen J. (2020). Neuroinflammatory responses and Parkinson’ disease: Pathogenic mechanisms and therapeutic targets. Journal of Neuroimmune Pharmacology, 15(4), 830–837. https://doi.org/10.1007/s11481-020-09926-7
- Yao C., Cao Y., Wang D., Lv Y., Liu Y., Gu X., Wang Y., Wang X., Yu B. (2022). Single-cell sequencing reveals microglia induced angiogenesis by specific subsets of endothelial cells following spinal cord injury. FASEB Journal, 36(7), e22393. https://doi.org/10.1096/fj.202200337R
- Zamanian J. L., Xu L. J., Foo L. C., Nouri N., Zhou L., Giffard R. G., Barres B. A. (2012). Genomic analysis of reactive astrogliosis. Journal of Neuroscience, 32(18), 6391–6410. https://doi.org/10.1523/JNEUROSCI.6221-11.2012