Abstract
Copy‐number variations (CNVs) have been found in association with various types of diseases, including hematological malignancies. A recent array‐based study implicated the presence of CNVs of ZMAT4 in the genome of acute myelogenous leukemia. In our study, we collected 617 bone marrow samples from multitypes of hematological malignancies as well as healthy controls. We found significant association between the CNVs of ZMAT4 and these hematological malignancies, including acute lymphoblastic leukemia, acute myelogenous leukemia, chronic lymphocytic leukemia, chronic myelogenous leukemia, multiple myeloma, and myelodysplastic syndrome. We also examined the expression of ZMAT4 mRNA in the samples with 1 or 2 copies of DNA, and observed a weak yet positive correlation between the relative expression level and gene dosage. In conclusion, the CNVs of ZMAT4 have the potential to serve as a diagnostic indicator, alone or in combination with other markers, for hematological malignancies.
Introduction
Copy‐number variations (CNVs) were originally defined by the presence of variable numbers of copies of large, multikilobase genomic regions in the genomes of different individuals.Citation1–Citation9 However, recent high‐resolution genome maps have revealed smaller CNVs among healthy humans,Citation4,Citation10 thus extending the definition of CNVs to the length of regions being as short as several hundred bases. CNVs are present in the general population at varying degrees, and their role in disease is poorly defined so far. Most CNVs have either no phenotypic consequences or only subtle or benign ones. Several methodologies, such as the most commonly used array‐based comparative genomic hybridization (aCGH), were utilized for genome‐wide CNV detection and genotyping. CNVs have been discovered to have phenotypic consequences and associate with various types of diseases including mental disorders, rheumatoid arthritis, diabetes and so on.Citation11–Citation14 The association of CNVs in cancers has been increasingly topical over the past few years.Citation15–Citation17 Studies using single nucleotide polymorphism arrays and aCGH have suggested that CNVs are common in genomes of hematological malignancies.Citation18–Citation22 A recent array‐based study screened paired tumor and normal samples to identify genes that are somatically altered in leukemia genomes, and implicated the presence of ZMAT4 CNVs in tumor samples.Citation23 This was the first report suggesting the potential function of ZMAT4.
However, most of the aCGH experiments focused on genome‐wide screening of CNVs, and the data obtained are generally informative but not definitive. Thus, they require further molecular genetic experiments for validation. The functional impact of such CNV regions, especially the particular genes, warrants extensive studies and the significance of these identified CNVs needs to be confirmed in large numbers of clinical samples. Therefore, in our study, we collected 617 bone marrow samples from multitypes of hematological malignancies including acute lymphoblastic leukemia (ALL), acute myelogenous leukemia (AML), chronic lymphocytic leukemia (CLL), chronic myelogenous leukemia (CML), multiple myeloma (MM), myelodysplastic syndrome (MDS), and healthy controls. Our aim was to extensively examine the significance of CNVs of ZMAT4 in these hematological malignancies.
Materials and Methods
Controls and patient samples
Bone marrow samples from 114 AML, 86 ALL, 78 CML, 72 CLL, 67 MM, 98 MDS, and 102 healthy controls were collected at Peking University People’s Hospital. Definition and classification of hematological malignancies were based on International Classification of Diseases by WHO. The study was approved by the ethical committee of Peking University Shenzhen Hospital. The individuals gave their written informed consent. The investigations were conducted according to the Declaration of Helsinki principles.
DNA extraction and quantification of copy numbers
Genomic DNA was isolated from the tissues using the Genomic DNA Extraction Kit (Innogent, Shenzhen, China) according to the manufacturer’s instruction. Quantitative polymerase chain reaction (PCR) was performed through BioRad Chromo4 real‐time PCR system. Average copy numbers of RNAse P in normal candidates (copy numbers = 2) were used as control.Citation7 The copy numbers of ZMAT4 was calculated by using the comparative C(T) method. Cut‐off values of 0·25, 0·75, 1·25, and 1·75 were used to define the copy numbers as 0, 1, 2, and 3, respectively. The primers for RNAse P are: forward, 5′ AGACTAGGGTCAGAAGCAA and reverse, 5′ CATTTCACTGAATCCGTTC. The primers for ZMAT4 are: forward, 5′ TAACGACCATAGGCAATC and reverse, 5′ AGGCTCCTTCATAACAACAC. Statistical analysis was performed using chi‐square test or Fisher exact test. P values less than 0·05 were considered statistically significant.
RNA extraction and quantitative reverse transcription‐PCR
Total RNA was isolated from tissues by using AxyPrepTM Blood Total RNA MiniPrep Kit (Axygen, Union City, CA, USA) according to the manufacturer’s instruction. First strand cDNA was synthesized with RevertAidTM First Stand cDNA Synthesis Kit (Fermentas, Shenzhen, China). The relative expression level of ZMAT4 mRNA was calculated by using the comparative C(T) method with RNAse P as the internal control.Citation7 Then the average expression level of ZMAT4 mRNA in the group with 2 copies of DNA was calculated. Fold change of each sample was presented as follows: fold change = relative expression level/average expression level in the group with 2 copies of DNA. Statistical analysis was performed using chi‐square test or Fisher exact test. P values less than 0·05 were considered statistically significant.
Results
shows CNVs of ZMAT4 in hematological malignancies and healthy controls. A total of 617 samples were examined. Statistical differences were observed in all six types of hematological malignancies as compared with the controls (P<0·05). Interestingly, the distribution of the copy numbers was not consistent among different types of malignancies. ZMAT4 was deleted in most of the samples from CML, CLL, MM, and MDS, and was amplified in rare cases (a maximum of 7 of 78 for CML). However, a relatively high percentage of samples from AML and ALL showed amplification of ZMAT4 (15 of 114 for AML and 21 of 86 for ALL). We then compared the CNVs among different types of malignancies, and found that much higher frequencies of ZMAT4 deletion (a minimum of 43·1%, 31 of 72 for CLL) existed in CML, CLL, and MDS than that in AML, ALL, and MM.
Table 1. CNVs of ZMAT4 in controls and hematological malignancies
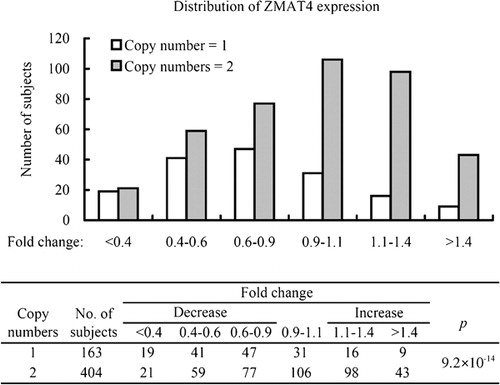
As gene CNVs can contribute to qualitative and quantitative diversities to their gene products, we selected the samples with 1, 2 or 3 copies of DNA, and tested whether the expression levels of ZMAT4 mRNA were correlated with the copy numbers. A total of 163, 404, and 38 samples were in the group with 1, 2, and 3 copies of DNA, respectively. A large variation of the expression levels was observed in each group although statistical difference among the three groups was obtained (data now shown). To further analyze the distribution of the expression, we calculated the fold change of expression in each sample as follows: fold change = relative expression level/average expression level in the group with 2 copies of DNA (as shown in the ‘Materials and methods’ section). Thus, the average fold change in the samples with 2 copies of DNA would be 1. Results of the fold change in each sample were shown in . The 38 samples with 3 copies of DNA were not included due to the small sample size. Again, a wide distribution of the fold changes was observed.
Figure 1. Expression of ZMAT4 mRNA in samples with 1 or 2 copies of DNA. The relative expression level of ZMAT4 mRNA was calculated by using the comparative C(T) method with RNAse P as the internal control. Fold change of each sample was calculated as follows: fold change = relative expression level/average expression level in the group with 2 copies of DNA. Y axis indicates the number of samples in each level of fold change. P value was calculated by chi‐square test.
Discussion
CNVs have been clearly shown to have the potential to directly or indirectly influence a healthy individual’s susceptibility to cancer, for example by varying the gene dosage of tumor suppressors or oncogenes.Citation24,Citation25 However, there are many discrepancies among previous studies which used high‐resolution approaches to screen CNVs.Citation18–Citation22 Thus, validation of such CNVs by studying larger numbers is required. It is suggested that the genes present in very small regions of CNVs are excellent candidates for evaluation in cancer pathogenesis. Examination of the CNVs for such genes helps to understand the functional consequences of these CNVs. A previous study implied that the region encompassing ZMAT4 was amplified in AML samples.Citation23 However, in our study, we found that ZMAT4 was only amplified in a small percentage of AML samples (13·2%, 15 of 114), but was deleted with a rather high percentage (28·1%, 32 of 114). This discrepancy might be due to the different races and populations used in the studies. The different methodologies may also account for the differences. The previous study used array‐based screening to target relatively long genomic regions (e.g., ∼800 kb encompassing ZMAT4), and may be unable to effectively detect the CNVs of short fragments.Citation23 In contrast, we used gene‐specific strategy to target short fragments (several hundred basepairs), and the sensitivity was increased. We also found that CNVs of ZMAT4 were present in other hematological malignancies including ALL, CML, CLL, MM, and MDS. This suggests that the association with CNVs of ZMAT4 might be a common feature of hematological malignancies. The unique feature (the presence of 13·2 and 24·4% of amplification in AML and ALL, respectively) of acute leukemia indicates that the etiology of acute leukemia might be different from the other hematological malignancies: deletion of ZMAT4 may generally contribute to the development of hematological malignancies, while amplification of ZMAT4 may specifically associate to the outbreak of acute leukemia. More likely, however, other factors such as epigenetic alterations may contribute to the development and progression of acute leukemia. The frequencies of ZMAT4 deletion were relatively lower in acute leukemia than in other malignancies (), further suggesting the presence of some unknown influences in the development of acute leukemia, which warrants extensive study in the future.
It is expected that the CNVs will indeed have phenotypic consequences. Phenotypic effects of genetic differences, such as CNVs, are supposedly brought about by changes in expression levels.Citation26,Citation27 We investigated the correlation between the expression of ZMAT4 mRNA and the copy numbers of its DNA. Surprisingly, the correlation was not as strong as expected, although a statistical difference was obtained (). This is consistent with two recent reports which assessed an over‐representation of differentially expressed genes among CNV‐mapping transcripts, and observed a weak yet significant positive correlation between relative expression level and gene dosage.Citation28,Citation29 In our study, only 26·2% (106 of 404) of samples with 2 copies of DNA had the fold change of 0·9–1·1, indicating that the expression levels were widely distributed. As mentioned above, the tremendous heterogeneity of expression may be caused by other contributing factors such as epigenetic alterations. On the other hand, the highest percentage (28·8%, 47 of 163) of samples with 1 copy of DNA had the fold change of 0·6–0·9 rather than 0·4–0·6, and in some cases (31 of 163), the number of copies had no effect on relative expression levels, suggesting either dosage compensation mechanisms or the incomplete inclusion of regulatory elements in the deletion/duplication event.Citation28,Citation29 Furthermore, in a small percentage of samples (25 of 163), the recorded relative expression levels were inversely correlated with copy numbers. Such kind of inverse correlation was also observed by other previous studies.Citation28,Citation29 The mechanism of this phenomenon is still poorly understood, but may be explained by two models: in the first model, a negative correlation between number of copies and relative expression is explained by immediate early genes which induce directly or indirectly the expression of a repressor which, by a negative feedback loop, reduces or even abolishes the expression of the CNV gene; in the second model, the extra copies of a gene impair through steric hindrance their access to a specific transcription factory, where this particular locus should be transcribed.Citation30
Conclusion
In general, the CNVs of ZMAT4 have the potential to serve as a diagnostic indicator, alone or in combination with other markers, for hematological malignancies. However, the functional consequences of CNVs, the different feature of CNVs between acute leukemia and other malignancies and the underlying mechanisms of the heterogeneous expression levels need to be extensively investigated in the future.
References
- Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK. A high-resolution survey of deletion polymorphism in the human genome. Nat Genet 2006;38:75–81.
- Hinds DA, Kloek AP, Jen M, Chen X, Frazer KA. Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat Genet 2006;38:82–5.
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, et al.. Detection of large-scale variation in the human genome. Nat Genet 2004;36:949–51.
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, et al.. Mapping and sequencing of structural variation from eight human genomes. Nature 2008;453:56–64.
- Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, et al.. Paired-end mapping reveals extensive structural variation in the human genome. Science 2007;318:420–6.
- McCarroll SA, Hadnott TN, Perry GH, Sabeti PC, Zody MC, Barrett JC, et al.. Common deletion polymorphisms in the human genome. Nat Genet 2006;38:86–92.
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al.. Global variation in copy number in the human genome. Nature 2006;444:444–54.
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, et al.. Large-scale copy number polymorphism in the human genome. Science 2004;305:525–8.
- Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, et al.. Fine-scale structural variation of the human genome. Nat Genet 2005;37:727–32.
- Korbel JO, Urban AE, Grubert F, Du J, Royce TE, Starr P, et al.. Systematic prediction and validation of breakpoints associated with copy-number variants in the human genome. Proc Natl Acad Sci USA 2007;104:10110–5.
- Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, et al.. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science 2005;307:1434–40.
- McKinney C, Merriman ME, Chapman PT, Gow PJ, Harrison AA, Highton J, et al.. Evidence for an influence of chemokine ligand 3-like 1 (CCL3L1) gene copy number on susceptibility to rheumatoid arthritis. Ann Rheum Dis 2008;67:409–13.
- Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, et al.. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet 2006;38:24–6.
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al.. Alpha-synuclein locus triplication causes Parkinson’s disease. Science 2003;302:841.
- Andrews J, Kennette W, Pilon J, Hodgson A, Tuck AB, Chambers AF, et al.. Multi-platform whole-genome microarray analyses refine the epigenetic signature of breast cancer metastasis with gene expression and copy number. PLoS One 2010;5:e8665.
- Diskin SJ, Hou C, Glessner JT, Attiyeh EF, Laudenslager M, Bosse K, et al.. Copy number variation at 1q21·1 associated with neuroblastoma. Nature 2009;459:987–91.
- Liu W, Sun J, Li G, Zhu Y, Zhang S, Kim ST, et al.. Association of a germ-line copy number variation at 2p24·3 and risk for aggressive prostate cancer. Cancer Res 2009;69:2176–9.
- Grubor V, Krasnitz A, Troge JE, Meth JL, Lakshmi B, Kendall JT, et al.. Novel genomic alterations and clonal evolution in chronic lymphocytic leukemia revealed by representational oligonucleotide microarray analysis (ROMA). Blood 2009;113:1294–303.
- Gunnarsson R, Staaf J, Jansson M, Ottesen AM, Goransson H, Liljedahl U, et al.. Screening for copy-number alterations and loss of heterozygosity in chronic lymphocytic leukaemia – a comparative study of four differently designed, high resolution microarray platforms. Genes Chromosomes Cancer 2008;47:697–711.
- Schafer M, Schwender H, Merk S, Haferlach C, Ickstadt K, Dugas M. Integrated analysis of copy number alterations and gene expression: a bivariate assessment of equally directed abnormalities. Bioinformatics 2009;25:3228–35.
- Strefford JC, van Delft FW, Robinson HM, Worley H, Yiannikouris O, Selzer R, et al.. Complex genomic alterations and gene expression in acute lymphoblastic leukemia with intrachromosomal amplification of chromosome 21. Proc Natl Acad Sci USA 2006;103:8167–72.
- Sulong S, Moorman AV, Irving JA, Strefford JC, Konn ZJ, Case MC, et al.. A comprehensive analysis of the CDKN2A gene in childhood acute lymphoblastic leukemia reveals genomic deletion, copy number neutral loss of heterozygosity, and association with specific cytogenetic subgroups. Blood 2009;113:100–7.
- Walter MJ, Payton JE, Ries RE, Shannon WD, Deshmukh H, Zhao Y, et al.. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc Natl Acad Sci USA 2009;106:12950–5.
- Dear PH. Copy-number variation: the end of the human genome? Trends Biotechnol 2009;27:448–54.
- Shlien A, Tabori U, Marshall CR, Pienkowska M, Feuk L, Novokmet A, et al.. Excessive genomic DNA copy number variation in the Li–Fraumeni cancer predisposition syndrome. Proc Natl Acad Sci USA 2008;105:11264–9.
- Dermitzakis ET, Stranger BE. Genetic variation in human gene expression. Mamm Genome 2006;17:503–8.
- Reymond A, Henrichsen CN, Harewood L, Merla G. Side effects of genome structural changes. Curr Opin Genet Dev 2007;17:381–6.
- Guryev V, Saar K, Adamovic T, Verheul M, van Heesch SA, Cook S, et al.. Distribution and functional impact of DNA copy number variation in the rat. Nat Genet 2008;40:538–45.
- Henrichsen CN, Vinckenbosch N, Zollner S, Chaignat E, Pradervand S, Schutz F, et al.. Segmental copy number variation shapes tissue transcriptomes. Nat Genet 2009;41:424–9.
- Sexton T, Umlauf D, Kurukuti S, Fraser P. The role of transcription factories in large-scale structure and dynamics of interphase chromatin. Semin Cell Dev Biol 2007;18:691–7.