Abstract
Previous work from our laboratory has confirmed that human ether-à-go-go-related gene 1 (hERG1) K+ channels are constitutively expressed in leukemia cells and enhanced cell proliferation. More importantly, it has shown that stromal cell-derived factor-1a (SDF-1a) significantly increases hERG1 K+ tail current and a specific hERG1 K+ channels inhibitor significantly blocks SDF-1a-induced migration of leukemic cells. In this study, we investigated a possible regulatory effect of hERG1 K+ channels upon SDF-1a-mediated cell proliferation as a mean to uncover new molecular events involved in bone marrow microenvironment and leukemogenesis. RT-PCR showed that SDF-1a enhanced hERG1 expression in a dose-dependent manner. Cell proliferation assay illustrated that SDF-1a promoted cell proliferation in a dose-dependent manner, whereas this effect was impaired by E-4031. In addition, E-4031 inhibited SDF-1a-stimulated leukemic cell proliferation by inducing G0/G1 arrest. Interestingly, E-4031 promoted SDF-1a-induced apoptosis in HL-60 and leukemic blasts, which markedly impaired the protection effect of SDF-1a in AML. Moreover, SDF-1a increased the expression of Wnt/beta-catenin target genes, including beta-catenin, cyclin-D1, and c-myc; however, this manner was abolished by blockage with the hERG1 K+ channels. Taken together, our results provide evidence of a novel mechanism involved in the proliferative effects of SDF-1a and highlight hERG1 K+ channels as a therapeutic target for leukemia treatment and prevention.
Introduction
Acute myeloid leukaemia (AML) has been extensively characterized as a stem cell disease with a hierarchy analogous to normal hematopoietic development. As normal hematopoietic cells, leukemic hematopoietic cells, and stem cells reside in the bone marrow in specialize areas (niches) that provide the structural and physiological conditions for their growth and survival.Citation1,Citation2 Subpopulations of leukemic cells can be sequestered in niches and thereby evade chemotherapy induced death.Citation3 Furthermore, stromal cells protect AML cells from the apoptosis induced by conventional chemotherapy.Citation4,Citation5 Therefore, there is considerable interest in deciphering the molecular mechanisms involved in leukemia–microenvironment interactions that contribute to leukemia progression. On the other hand, these data evoke innovative approaches to treatment of leukemia, which focus on the microenvironment — the niche — in support of leukemogenesis.
Indeed, the mechanisms of the bone marrow microenvironment-mediated protection are intricate. A complex interplay among stroma-produced cytokines, chemokines, and adhesion molecules is involved in this process. Stromal cell-derived factor-1a (SDF-1a), is constitutively produced by bone marrow stromal cells, and thought to play an important role in both normal and leukemic hematopoietic cell migration, homing in vivo through its receptor CXCR4.Citation6 Binding of SDF-1a to CXCR4 causes CXCR4 to be incorporated into lipid rafts and increases its phosphorylation, and then leads to prolong activation of the extracellular singling-regulated kinase and phosphoinositol-3-kinase pathways, which are key signaling pathways that promote leukemia cells survival.Citation7 In addition, it has also been shown that SDF-1a enhances proliferation in pre-B-ALL via STAT5 activation.Citation8 Owing to a crucial role in survival, some lines of evidence suggest that elevated CXCR4 levels have been described in AML and are found to portend a poor prognosis.Citation9 Furthermore, leukemia cells are able to directly modulate the niche at the expense of normal hematopoietic stem and progenitor cells by down-regulating SDF-1 levels in areas of leukemia infiltration.Citation2
A peculiar aspect of SDF-1a-mediated signaling is epitomized by their interactions with human ether-à-go-go-related gene 1 (hERG1) K+ channels: SDF-1 significantly increased hERG1 K+ tail current to support a hyperpolarization of the membrane potential, and consequently specific hERG1 K+ channels inhibitors significantly blocked SDF-1-induced migration of leukemic cell lines and leukemic blasts.Citation10 Accordingly, hERG channel blockers inhibit cell proliferation, thereby leading to membrane depolarization and an arrest in early G1 phase.Citation11 One of the recent advances in cancer biology is the discovery of the roles of ion channels in regulatory cell proliferation and cell death, especially K+ channels. Among the various types of K+ channels are known thus far, the hERG1 appears of exceptional importance in favoring cancer progression in vivo and in vitro. Strikingly, numerous studies have documented that hERG1 is constantly overexpressed in a variety of primary human cancers and tumor cell lines of different histogenesis including primary leukemia cells and the HL-60 leukemic cell line, but absent from the healthy cells from which the respective tumor cells are derived.Citation12 Based on published studies, three main functions relevant to AML-cell biology can be ascribed to hERG channel activity: (1) the control of cell proliferation;Citation12 (2) the regulation of leukemia cell migration and invasion, possible through interaction with integrin receptors and the vascular endothelial growth factor receptor-1;Citation13 and (3) the control of leukemia cell angiogenesis, through the modulation of vascular endothelial growth factor secretion.Citation13 hERG1 is thus proposed a novel diagnostic and prognostic factor in AML, as well as a target for anti-leukemia therapy.
Based upon these findings, we hypothesized that there is a crosstalk between the hERG1 K+ channels and SDF-1a in leukemic cell proliferation. For the purposes of the present article, we sought to (1) determinate whether hERG1 K+ channels and SDF-1a are associated in leukemic cell proliferation; (2) establish the regulation of hERG1 K+ channels by SDF-1a at the transcriptional levels; and (3) identify the involvement of hERG1 K+ channels in SDF-1a mediated apoptosis. In addition, Wnt signaling plays an important role in cancer. We examined that hERG1 K+ channels appear to be a target of Wnt-dependent pathways activated by SDF-1a.
Materials and Methods
Reagents
E-4031, a specific hERG1 K+ channels inhibitor, was obtained from Sigma. RPMI-1640 and fetal bovine serum (FBS) were obtained from Gibco (Grand Island, NY, USA). Human recombinant SDF-1a was purchased from Upstate Biotechnology (Lake Placid, NY, USA). Annexin-V FITC/PI apoptosis detection kit was obtained from BD Biosciences (Bedford, MA, USA). Cell Counting Kit-8 (CCK-8) was purchased from Beyotime Institute of Biotechnology (Haimen, China). RT-PCR kit was product of Fermentas (Vilnius, Lithuania).
Cells and cell culture
The HL-60 cell line was provided by Life Science College, Wuhan University (Wuhan, China). Bone marrow specimens from five AML patients and two normal donors were obtained after informed consent. All AML patients’ medical information was listed in . Mononuclear cells were isolated by density-gradient centrifugation and stored in liquid nitrogen until use. Cells were cultured in RPMI-1640 medium supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml), respectively. The culture medium was changed every 2 days. Cells were maintained at 37°C in a humidified atmosphere with 5% CO2.
Table 1. Herg expression in bone marrow from patients
Cell proliferation assay
The proliferative effect of SDF-1a on HL-60 cells was determined by CCK-8 assay. Briefly, cells were cultured in complete RPMI-1640 medium in the presence of SDF-1a (12·5, 25, 50, and 100 ng/ml, final concentrations) for 48 hours in 96-well plates. Thereafter, 10 μl CCK-8 solutions were added to each well. Cells were further incubated at 37°C for 1 hour. The absorbance was detected in a microplate reader at a wavelength of 450 nm using a 96-well multiscanner autoreader (Biotech Instruments μQuant, Piscataway, NY, USA).
RT-PCR
Total RNA was extracted from approximately 1×106 cultured cells using the Trizol method according to the manufacturer’s instructions. For each condition, 1 μg of total RNA was reverse transcribed into cDNA using a RevertAidTM First Strand cDNA Synthesis Kit (MBI Fermentas, Hanover, MD, USA) following the manufacturer’s instructions. cDNA was stored at −20°C. Primers were used as indicated in . A final volume of 20 μl PCR reaction mixture was first incubated at 94°C for 5 minutes, followed by 38 cycles of 94°C for 1 minute, 55°C for 50 seconds, 72°C for 1 minute, with a final extension at 72°C for 10 minutes. After amplification, PCR products were analyzed by electrophoresis in a 1·5% agarose gel. To assess the relative changes in hERG1, beta-catenin, cyclin-D1, and c-myc mRNA contents by SDF-1a treatment, semi-quantitative RT-PCR was performed using beta-actin as an internal standard. The PCR products were quantified using ImageJ software.
Table 2. Primers used in RT-PCR analysis
Cell cycle analysis
HL-60 cells were incubated in RPMI-1640 medium in the presence of SDF-1a with or without E-4031 for 48 hours, and then cell cycle analysis was performed by measuring cellular DNA content using flow cytometry. Cells were collected, washed with PBS and fixed in 70% ethanol. After fixation, cells were pelleted by centrifugation, resuspended in phosphate-buffered saline (PBS), treated with ribonuclease at a final concentration of 10 μg/ml for 30 minutes, and stained with propidium iodide (PI) at a final concentration of 50 μg/ml. The stained cells were analyzed by flow cytometry. The cell cycle distribution patterns (G0/G1, S, and G2/M phases) were quantitated using Cellquest Software. Results were expressed as percentage of the cells in each phase.
Assay for apoptosis
According to the method described in Annexin-V FITC/PI Apoptosis Detection Kit, cells (1×106/ml) were cultured in complete RPMI-1640 medium in the presence of SDF-1a with or without E-4031 for 48 hours. Cells were harvested, and washed with PBS. Afterwards, cells were stained with 5 μl Annexin-V FITC solutions, and PI 20 μg/ml was added for 15 minutes at room temperature in the dark. Samples were then diluted with 300 μl of binding buffer and analyzed by flow cytometry (FACSort; BD Biosciences, San Jose, CA, USA). Early apoptotic cells were stained with Annexin-V alone, whereas necrotic and late apoptotic cells were stained with both Annexin-V and PI.
Statistical analysis
All data were expressed as mean±standard error of the mean. Analysis of statistical significance was performed using Student’s t-test for paired observations. Three or more means were tested using one-way ANOVA and Dunnett’s post hoc tests. Differences between values were considered significant when P value was less than 0·05.
Results
SDF-1a increased hERG1 mRNA levels
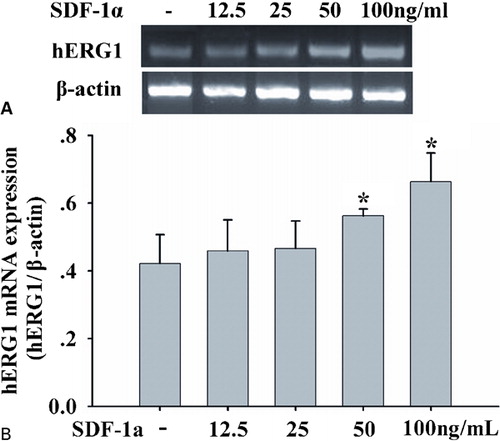
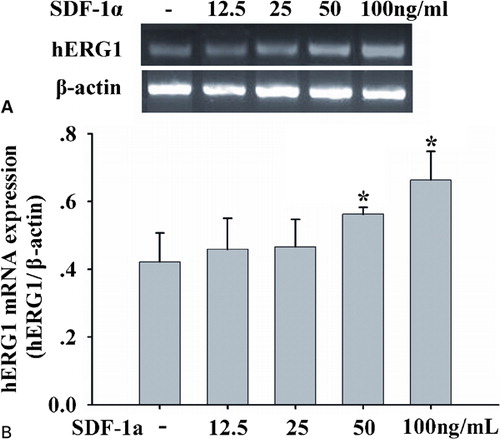
Since our hypothesis was that hERG1 might be a potential target for SDF-1a, cells were incubated in the presence of different SDF-1a concentrations, and hERG1 gene expression was studied. RT-PCR data showed that SDF-1a stimulated mRNA levels of hERG1 in a dose-dependent manner (). After exposures to 100 ng/ml SDF-1a, hERG1 mRNA levels increased significantly, by approximately 1·5-fold above control levels. It indicates that SDF-1a modulated herg1 gene transcription.
Figure 1. SDF-1alpha enhances hERG1 mRNA expression. (A) RT-PCR of hERG1 gene( upper panel) and β-actin gene (lower panel) in HL-60 cells treated with SDF-1a at 12.5, 25, 50, and 100 ng/ml for 48 hours; (B) SDF-1a up-regulated mRNA levels of hERG1 in a dose-dependent manner. hERG1 mRNA levels were quantified and normalized to the amplified beta-actin products. Each column represents mean±SE, and each assay was repeated there times, *P<0·05.
hERG1 K+ channels involved in SDF-1a-induced leukemic cell proliferation
Since SDF-1a is constitutively secreted by stromal cells in the bone marrow and this chemokine contributes to the migration and survival of leukemic blasts. It has previously been shown that recombinant SDF-1a enhances pre-B leukemic cell proliferation in vitro.Citation8 Therefore, we explored whether recombinant SDF-1a had any biological role for AML-cell proliferation. To evaluate the mitogenic effect of SDF-1a, we used a serum medium as control condition. HL-60 cells were treated with SDF-1a at various concentrations (12·5, 25 50, and 100 ng/ml) for 48 hours and evaluated the cell viability by CCK-8 kit. As shown in , SDF-1a enhanced cell proliferation in a dose-dependent manner. The maximal increase by 1·6 times was obtained for 100 ng/ml. We consequently chose to use this concentration in all subsequent experiments.
Figure 2. The hERG1 K+ blocker impaired leukemic cell proliferation induced by SDF-1a and accumulated cells in the G0/G1 phase. (A) SDF-1a increased cell proliferation in a dose-dependent manner. Cells were cultured in 96-well plates with SDF-1a at 12·5, 25, 50, and 100 ng/ml for 48 hours. Cell viability was determined by CCK-8 assay. Values indicated were expressed as mean ±SE. Each assay was repeated three times. *P<0·05, **P<0·001. (B) The hERG1 K+ blocker inhibited proliferation induced SDF-1a in a dose-dependent manner. (C) The hERG1 K+ channel blocker and SDF-1a accumulated cells in the G0/G1 phase. HL-60 cells were treated with SDF-1a (100 ng/ml) in the absence or presence of E-4031at 1 μM for 48 hours. Thereafter, cells were collected and stained with propidium iodide. The DNA content was measured by flow cytometry.
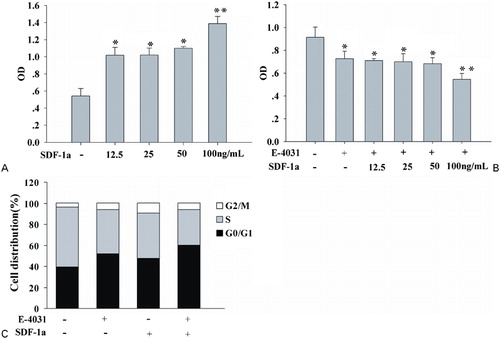
In previous studies, we used the patch-clamp technique to test the relationship between SDF-1a and hERG1 K+ channels.Citation10 In this context, we investigated whether the inhibition of hERG1 K+ channels activity would suppress leukemic cell proliferation induced by SDF-1a. It has been demonstrated that E-4031 blocked the proliferation of leukemic cells at a concentration of 1 μM.Citation12 So 1 μM E-4031 was therefore applied in all subsequent experiments. E-4031 and SDF-1a were added to the cell culture medium. We confirmed that E-4031 inhibited leukemic cell proliferation as previous stated. Simultaneously, E-4031 inhibited leukemic cell proliferation induced by SDF-1a. Not as expected, when the concentrations of SDF-1a were less than 50 ng/ml, there was no significant antagonistic effect upon proliferation by the hERG1 K+ channels inhibitor and SDF-1a. However, E-4031 significantly impaired cell proliferation induced by SDF-1a with a concentration of 100 ng/ml by 40·3±8·4% ().
Furthermore, to determine whether the inhibition of SDF-1a proliferation induced by E-4031 was accompanied by cell arrest in a specific cell cycle phase, we used flow cytometry to analyze the cell cycle status of HL-60 cells incubated in SDF-1a alone or with E-4031. Cells were incubated in SDF-1a (100 ng/ml) in the presence or absence of E-4031 for 48 hours. Control cells in G0/G1, S, and G2/M phases of the cell cycle accounted for 39·35±1·15%, 57·02±0·52%, and 3·63±0·79%, respectively. However, E-4031-treated cells induced an accumulation of cells in G0/G1 (52·25±2·68%) and the G2/M phase (5·89±0·86%), and a reduction in the number of cells in the S phase (41·86±3·5%). The same effects were observed when cells were incubated in SDF-1a with or without E-4031 (). Taken together, these results showed that suppression of hERG1 K+ channels activity inhibited SDF-1a-stimulated HL-60 proliferation by inducing G0/G1 arrest. Collectively, we concluded that hERG1 K+ channels are strongly involved in HL-60 cell proliferation induced by SDF-1a.
hERG1 K+ channels mediated SDF-1a-induced apoptosis in leukemic cell line
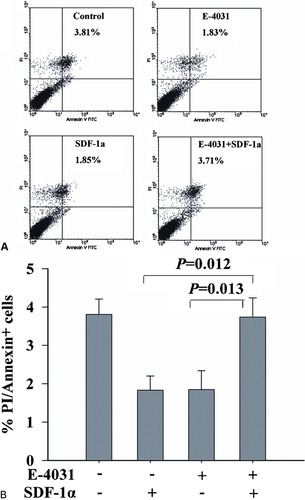
Cell proliferation and apoptosis are two counterparts that share the responsibility for maintaining cell homeostasis. To explore whether hERG1 K+ channels activity involved in apoptosis is regulated by SDF-1a. We used Annexin-V FITC/PI double-labeled flow cytometry to elucidate the role of SDF-1a alone or with E-4031 in conferring sensitivity to apoptosis (). Cells were incubated in E-4031 1 μM for 48 hours, which showed that the percentage of apoptotic cells was decreased from (3·81±0·41 to 1·83±0·35% (P<0·05). SDF-1a (100 ng/ml) suppressed apoptosis rate to 1·85±0·38% (P<0·05). The results indicate that hERG1 K+ channels promote apoptotic cell death, while SDF-1a can protect leukemic cell apoptosis. Interestingly, when SDF-1a is combined with E-4031, 3·71±0·40% of cells were apoptotic.
Figure 3. hERG1 K+ channels mediated SDF-1a-induced apoptosis in leukemic cell line. To explore whether the hERG1 K+ channels were involved in SDF-1a mediated apoptosis, HL-60 cells were stimulated by RPMI 1640 medium supplemented with 10% FBS, E-4031, and SDF-1a in the absence or presence of E-4031 for 48 hours. Cells were collected and stained with Annexin V FITC/PI. The induction of apoptosis was determined by Annexin-V FITC/PI double-staining assay (A and B). The upper-right quadrant indicated cell apoptosis.
hERG1 K+ channels mediated SDF-1a-induced apoptosis in AML patients’ derived mononuclear cells
After 48 hours of incubation with E-4031 and SDF-1a, mononuclear bone marrow cells were assayed for the expression of PI/Annexin V by flow cytometry. Two normal bone marrow samples were assayed as controls. As reported in , the exposure to the combination E-4031 and SDF-1a is associated with a marked apoptosis induction. Obviously, by blockage with the hERG1 K+ channels, E-4031 impaired the protection effect of SDF-1a in AML.
Figure 4. hERG1 K+ channels mediated SDF-1a-induced apoptosis in AML patients’ derived mononuclear cells. Cells were stimulated by RPMI 1640 medium supplemented with 10% FBS, E-4031, and SDF-1a in the absence or presence of E-4031 for 48 hours. At the end of the incubation, apoptosis was determined by Annexin-V FITC/PI double staining assay (A and B). The upper-right quadrant indicated cell apoptosis.
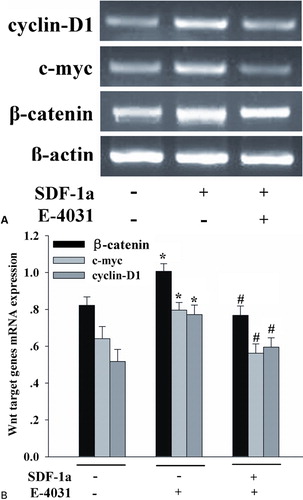
hERG 1 K+ channels mediated the effects of SDF-1a on Wnt/beta-catenin signaling
Recently, it has been reported that the Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML, and aberrant beta-catenin expression predicts an adverse prognosis in AML patients.Citation14,Citation15 To determine whether SDF-1a-induced proliferation is related with Wnt/beta-catenin signaling pathway, RT-PCR was employed to detect the expression of transcription for some Wnt target genes. Beta-catenin, c-myc, and cyclin-D1 are established target genes of the Wnt/beta-catenin pathway.Citation16 To assess the ability of SDF-1a to alter beta-catenin, c-myc, and cyclin-D1 transcript expression, HL-60 cells were incubated in SDF-1a for 48 h, and then analyzed by RT-PCR. As shown in , SDF-1a significantly increased beta-catenin, c-myc, and cyclin-D1 mRNA expression. Interestingly, hERG1 K+ channels had a negative effect on the Wnt/beta-catenin signaling pathways. Treatment with SDF-1a in the presence of E-4031 notably depressed the expression of these Wnt target genes. These data demonstrate that SDF-1a activates the Wnt/beta-catenin pathway, and hERG1 K+ channels modulate this process.
Figure 5. The hERG1 K+ blocker decreased the expression of Wnt target genes induced by SDF-1a. A synergistic effect of hERG1 K+ channels and SDF-1a enhanced the Wnt/beta-catenin target genes expression. HL-60 cells were incubated with SDF-1a with or without E-4031 for 48 hours. Cells were then harvested to detect the expression of Wnt/beta-catenin target genes, including beta-catenin, cyclin-D1, and c-myc. SDF-1a significantly increased beta-catenin, c-myc, and cyclin D1 mRNA expression, whereas E-4031 notably depressed the expression of these Wnt target genes. Each column represents mean±SE (n = 3). *P<0·05, compared with control; #P<0·05, compared with treatment of SDF-1a.
Discussion
The importance of bone marrow microenvironment for leukemia progression is becoming widely recognized in recent years. SDF-1 is the main chemokine produced by bone marrow stromal cells and has been found to mediate migration and homing of leukemic cells via its receptor CXCR4.Citation1,Citation17 Indeed, SDF-1 can regulate cell proliferation and also act as a survival factor for human AML progenitor cells.Citation7 Recently, we found that SDF-1 significantly increased hERG1 K+ tail current and hERG1 K+ channels inhibitor significantly blocked SDF-1-induced migration of leukemic blasts, which led us to hypothesize that SDF-1 promoted AML-cell proliferation may involve in regulation of hERG1 K+ channels.
As previously reported,Citation17–Citation20 we confirmed that SDF-1 and hERG1 K+ channels can regulate cell proliferation in vitro respectively. Furthermore, the effect of SDF-1 was dose-dependent. To evaluate whether hERG1 K+ channels were directly involved in SDF-1- induced proliferation of leukemic blasts, we evaluated the effect of hERG1 K+ channel inhibitor in the present of SDF-1 in proliferation assay. Surprisingly, inhibition of hERG1 K+ channel significantly impaired SDF-1-induced proliferation of AML cells. Our present results suggest that proliferation of SDF-1 on AML cells could be mediated through activation of hERG1 K+ channels. Previous results reported that the herg gene can be included in the set of genes activated early on during the G0/G1 to S transition of hematopoietic progenitor cells.Citation21 Indeed, we demonstrated that the hERG1 K+ channel inhibitor suppresses AML-cell proliferation in the absence or presence of SDF-1 in the G0/G1 phase, and the latter is more seriously impaired, which indicates that this type of K+ channels plays a critical regulatory role in SDF-1-dependent cellular proliferation of human AML cells. It had been showed that inhibition of CXCR4/SDF-1 axis in AML cells led to reduce proliferation/survival,Citation17 and SDF-1 promoted AML-cell proliferation also by regulating the expression of elastase.Citation19 In light of the data presented here, it may be possible that SDF-1 promotes AML-cell proliferation also by regulating the hERG1 K+ channel, which we found to be involved in regulation of AML-cell proliferation.
To explain how hERG1 K+ channels are linked to cell proliferation induced by SDF-1, we recently explored a transcriptional effect of SDF-1 on the hERG1 K+ channel. Indeed, our molecular biology results clearly showed that SDF-1 was able to increase hERG1 mRNA levels after 48-hour incubation. More importantly, SDF-1 enhanced herg1 gene expression in a dose-dependent manner. The clinical data demonstrated that herg1 overexpression in AML patients had a higher probability of relapse, a shorter time to relapse, and their overall survival was significantly shorter compared with that of herg1 patients.Citation13 Herein, these results may provide a novel mechanism that bone marrow microenvironments regulate leukemic cell proliferation, survival, and metastasis via the interaction of SDF-1 with hERG1 K+ channels.
Since apoptosis frequently parallels abnormalities in cell proliferation and differentiation. The role of K+ channels in apoptosis was proposed initially on the basis of observations demonstrating involvement of K+ channels in regulating cell cycles.Citation22 The present results found that both the hERG1 K+ channel inhibitor and SDF-1 regulated AML-cell apoptosis. Of interest, the hERG1 K+ channel inhibitor enhanced SDF-1-induced apoptosis. It is inconceivable that K+ channels favor tumour cell proliferation and also promote apoptotic cell death. From this, it seems that hERG1 K+ is involved in SDF-1-induced apoptosis and hERG1 K+ channels promote AML-cell apoptosis activating different potential signaling pathways; further work is required to confirm the mechanism.
Aberrant cell signaling is thought to play a role in maintenance of the leukemic clone in AML. The Wnt pathway is constitutively active in AML and a significant proportion of AML cases show aberrant expression of components of the Wnt pathway including Wnt-1, Wnt-2b, and LEF-1.Citation23 In addition, expression of beta-catenin predicts enhanced clonogenic capacities and poor prognosis.Citation24 What’s more, Wnt/beta-catenin signaling pathway is required for self-renewal of leukemia stem cells, while beta-catenin is not absolutely required for self-renewal of adult hematopoietic stem cells; thus, targeting the Wnt/beta-catenin pathway may represent a new therapeutic opportunity in AML.Citation15 In this content, we assume that SDF-1-induced proliferation activates Wnt/beta-catenin signaling and hERG1 K+ channel activity is involved in modulating this event.
The Wnt signaling is activated by binding Wnt ligands to receptors on the cell surface and results in beta-catenin accumulation in the cytoplasm.Citation25,Citation26 The accumulated beta-catenin translocates to the nucleus and activates transcription of Wnt target genes, including c-myc and cyclin-D1.Citation27,Citation28 Increasing evidences demonstrate that overexpression of cyclin-D1 and c-myc is highly associated with accumulation of beta-catenin in many types of tumors.Citation14,Citation29 In this study, we showed that stimulation of AML cells with SDF-1a enhanced nuclear translocation of beta-catenin. Furthermore, SDF-1a increased expression of Wnt target genes: cyclin-D1 and c-myc. These data suggest that SDF-1a may activate the Wnt pathway to regulate proliferation. Recent work has shown that SDF1a/CXCR4 axis can lead to beta-catenin stabilization and participate in the transcriptional activation of Wnt target genes.Citation30 However, the crosstalk among SDF-1a, the hERG1 K+ channel, and β-catenin has rarely been evaluated. The present study showed that the specific hERG1 K+ inhibitor suppressed the expression of beta-catenin, cyclin-D1, and c-myc induced by SDF-1a. This result suggests that hERG1 K+ channels involve in regulating SDF-1 through activation of Wnt/beta-catenin pathway. C-myc and cyclin-D1 are clearly relevant in proliferation, apoptosis, and cell-cycle regulation.Citation31 SDF-1a enhanced the expression of cyclin-D1 and c-myc, while this effect was down-regulated by the hERG1 K+ channel inhibitor. Based on these results, it may be confirmed that the hERG1 K+ channel is involved in AML-cell proliferation mediated by SDF-1a.
In conclusion, our data indicate that SDF-1a stimulates cell proliferation by up-regulating the hERG1 K+ channel mRNA level. In particular, this process is possibly through Wnt/beta-catenin pathway. Indeed, the hERG1 K+ channel participates in regulating the Wnt/beta-catenin pathway activated by SDF-1a. Evidence suggests that SDF-1a enhances cell proliferation by modulating membrane hERG1 K+ channels on AML cells in vitro; this mechanism may elaborate the crosstalk between bone marrow microenvironment and AML cells. A better understanding of the mechanism of bone marrow stromal cells in AML-cell biology will possibly lead to new therapeutic approaches for AML.
The financial support of this work is received from the National Nature Sciences Foundation of China (no. 30971112).
References
- Konopleva M, Tabe Y, Zeng Z, Andreeff M. Therapeutic targeting of microenvironmental interactions in leukemia: mechanisms and approaches. Drug Resist Updat 2009;12:103–13.
- Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 2008;322:1861–5.
- Fiegl M, Samudio I, Clise-Dwyer K, Burks JK, Mnjoyan Z, Andreeff M. CXCR4 expression and biologic activity in acute myeloid leukemia are dependent on oxygen partial pressure. Blood 2009;113:1504–12.
- Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood 2005;106:1154–63.
- Estey E, Dohner H. Acute myeloid leukaemia. Lancet 2006;368:1894–907.
- Kryczek I, Wei S, Keller E, Liu R, Zou W. Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. Am J Physiol Cell Physiol 2007;292:C987–95.
- Zeng Z, Shi YX, Samudio IJ, Wang RY, Ling X, Frolova O, et al.. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood 2009;113:6215–24.
- Mowafi F, Cagigi A, Matskova L, Björk O, Chiodi F, Nilsson A. Chemokine CXCL12 enhances proliferation in pre-B-ALL via STAT5 activation. Pediatr Blood Cancer 2008;50:812–7.
- Spoo AC, Lubbert M, Wierda WG, Burger JA. CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood 2007109786–91.
- Li H, Du YM, Guo L, Jie S, Zhang S, Du W, et al.. The role of hERG1 K+ channels and a functional link between hERG1 K+ channels and SDF-1 in acute leukemic cell migration. Exp Cell Res 2009;315:2256–64.
- Wang J, Wang H, Zhang Y, Gao H, Nattel S, Wang Z. Impairment of HERG K+ channel function by tumor necrosis factor-alpha: role of reactive oxygen species as a mediator. J Biol Chem 2004;279:13289–92.
- Pillozzi S, Brizzi MF, Balzi M, Crociani O, Cherubini A, Guasti L, et al.. HERG potassium channels are constitutively expressed in primary human acute myeloid leukemias and regulate cell proliferation of normal and leukemic hemopoietic progenitors. Leukemia 2002;16:1791–8.
- Pillozzi S, Brizzi MF, Bernabei PA, Bartolozzi B, Caporale R, Basile V, et al.. VEGFR-1 (FLT-1), beta1 integrin, and hERG K+ channel for a macromolecular signaling complex in acute myeloid leukemia: role in cell migration and clinical outcome. Blood 2007;110:1238–50.
- Chen CC, Gau JP, You JY, Lee KD, Yu YB, Lu CH, et al.. Prognostic significance of beta-catenin and topoisomerase IIalpha in de novo acute myeloid leukemia. Am J Hematol 2009;84:87–92.
- Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, et al.. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010;327:1650–3.
- Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis 2008;4:68–75.
- Tavor S, Petit I, Porozov S, Avigdor A, Dar A, Leider-Trejo L, et al.. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res 2004;64:2817–24.
- Smith GA, Tsui HW, Newell EW, Jiang X, Zhu XP, Tsui FW, et al.. Functional up-regulation of HERG K+ channels in neoplastic hematopoietic cells. J Biol Chem 2002;277:18528–34.
- Tavor S, Petit I, Porozov S, Goichberg P, Avigdor A, Sagiv S, et al.. Motility, proliferation, and egress to the circulation of human AML cells are elastase dependent in NOD/SCID chimeric mice. Blood 2005;106:2120–7.
- Scotton CJ, Wilson JL, Scott K, Stamp G, Wilbanks GD, Fricker S, et al.. Multiple actions of the chemokine CXCL12 on epithelial tumor cells in human ovarian cancer. Cancer Res 2002;62:5930–8.
- Crociani O, Guasti L, Balzi M, Becchetti A, Wanke E, Olivotto M, et al.. Cell cycle-dependent expression of HERG1 and HERG1B isoforms in tumor cells. J Biol Chem 2003;278:2947–55.
- Wang Z. Roles of K+ channels in regulating tumour cell proliferation and apoptosis. Pflugers Arch 2004;448:274–86.
- Simon M, Grandage VL, Linch DC, Khwaja A. Constitutive activation of the Wnt/beta-catenin signalling pathway in acute myeloid leukaemia. Oncogene 2005;24:2410–20.
- Ysebaert L, Chicanne G, Demur C, de Toni F, Prade-Houdellier N, Ruidavets JB, et al.. Expression of beta-catenin by acute myeloid leukemia cells predicts enhanced clonogenic capacities and poor prognosis. Leukemia 2006;20:1211–6.
- Wilusz M, Majka M. Role of the Wnt/beta-catenin network in regulating hematopoiesis. Arch Immunol Ther Exp (Warsz) 2008;56:257–66.
- Macheda ML, Stacker SA. Importance of Wnt signaling in the tumor stroma microenvironment. Curr Cancer Drug Targets 2008;8:454–65.
- Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, et al.. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA 1999;96:5522–7.
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al.. Identification of c-MYC as a target of the APC pathway. Science 1998;281:1509–12.
- Li YJ, Wei ZM, Meng YX, Ji XR. Beta-catenin up-regulates the expression of cyclinD1, c-myc and MMP-7 in human pancreatic cancer: relationships with carcinogenesis and metastasis. World J Gastroenterol 2005;11:2117–23.
- Luo Y, Cai J, Xue H, Mattson MP, Rao MS. SDF1alpha/CXCR4 signaling stimulates beta-catenin transcriptional activity in rat neural progenitors. Neurosci Lett 2006;398:291–5.
- Kikuchi A. Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci 200394225–9.