Abstract
The use of low-molecular-weight, non-peptidic molecules that disrupt the interaction between the p53 tumor suppressor and its negative regulator MDM2 has provided a promising alternative for the treatment of different types of cancer. Here, we used small-molecule reactivation of p53 and induction of tumor cell apoptosis (RITA) to sensitize leukemic NALM-6 cells to doxorubicin by upregulating p53 protein. RITA alone effectively inhibited NALM-6 cells viability in dose-dependent manner as measured by 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide assay and induced apoptosis as evaluated by flow cytometry, whereas RITA in combination with doxorubicin enhanced NALM-6 cells to doxorubicin-sensitivity and promoted doxorubicin induced apoptosis. Levels of p53 protein and its proapoptotic target genes, quantified by western blot and real-time PCR respectively, showed that expression of p53 was significantly increased after RITA treatment. Using p53 inhibitors PFT-alpha and PFT-mu it was shown that p53-mediated apoptosis induced by RITA can be regulated by both p53-transcription-dependent and -independent pathways. Moreover, RITA-induced apoptosis was accompanied by the activation of caspase-3 and PARP cleavage. Therefore, exploiting synergistic effects between RITA and chemotherapeutics might be an effective clinical strategy for leukemia chemotherapy.
Introduction
The tumor suppressor gene p53 plays a major role in regulation of the mammalian cellular stress response, in part through the transcriptional activation of genes involved in cell cycle control, DNA repair, and apoptosis. Cells that are insulted by oncogene expression, DNA damage or other forms of stress stabilize the p53 protein by post-translational modifications. Stabilized p53 accumulates in the nucleus to regulate the expression of numerous pro-apoptotic genes.Citation1–Citation3 As a consequence, disruption of p53 function promotes checkpoint defects, cellular immortalization, genomic instability, and inappropriate survival, allowing the continued proliferation and evolution of damaged cells. Given the profound proliferative advantage produced by loss of p53 function, it is not surprising that p53 is the most commonly inactivated tumor suppressor gene in human cancer.Citation4–Citation6 Inactivation of p53, by mutation or by overexpression of its negative regulators occurs in the majority, if not all tumors. The findings that inactivation of p53 is crucial for the survival of established tumors provide strong support for the strategies aimed at reactivation of p53 function in tumors.Citation7–Citation9 In vivo studies show that established tumors of different types remain vulnerable to p53-mediated suppression, supporting the notion that the restoration of p53 function may be an attractive strategy for treating cancer.Citation10 In normal unstressed cells, p53 is regulated by a feedback loop with the negative regulator protein MDM2 (murine double-minute clone 2, referred to as human double-minute clone 2, HDM2, in humans).Citation11 Recent studies have shown that rescue of p53 function by disruption of the p53-MDM2 interaction may be a promising strategy for developing new anti-cancer drugs.Citation12Citation12,13 It has been previously demonstrated that small molecule RITA which binds with high affinity to p53 and reduces p53–MDM2 interaction triggers apoptosis in wild-type p53–expressing colon, breast, lung carcinoma, osteosarcoma, and renal cell carcinoma cells. Notably, activation of p53 by RITA is much more potent in tumor cells than in normal cells.Citation14–Citation16 Using the p53-reactivating molecule RITA, we addressed the questions of whether and how p53 can promote apoptotic signals in pre-B acute lymphoblastic leukemia (ALL) cells. We showed that RITA is able to induce p53 accumulation and acetylation. In addition, we demonstrate that p53 activated by RITA potentiates the set of proapoptotic genes that play a critical role in p53-induced apoptosis.
Material and Methods
Reagents
Antibodies were purchased from the following manufacturers: acetyl-p53 (Lys382) (Cell Signaling Technology, UK); p53 (DO-1), procaspase-3 and secondary antibodies conjugated to horseradish peroxidase (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA); Anti-PARP-1 from Epitomics; anti-beta-actin, Pifithrin-alpha, Pifithrin-mu, doxorubicin and 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma. Annexin V-FLOUS-Staining Kit was from Roche. RITA was prepared from Alexis Biochemicals.
Cell culture
NALM-6 human pre-B ALL cells were grown in suspension in RPMI medium supplemented with 2 mM L-glutamine, 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin in a humidified 5% CO2 incubator at 37°C under standard cell culture conditions.
Cell viability measured by MTT assay
The effect of various concentrations of RITA on cell viability was assayed by the MTT colorimetric method. Briefly, exponentially growing cancer cells were seeded into a 96-well culture plate at a density of 5×103 cells/well and incubated with various concentrations of RITA for 24 and 48 hours. After removing the medium, cells were incubated with MTT solution (5 mg/ml in PBS) for 4 hours and the resulting formazan was solubilized with DMSO (100 μl). The absorbance of each well was measured at 570 nm in an ELISA reader.
Sub-G1 DNA content analysis
Apoptotic cells were detected using PI staining of RITA-treated cells followed by flow cytometry to detect the so-called sub-G1 peak. Briefly, NALM-6 cells were seeded into six well plates at the concentration of 0·8×106 cells/ml and incubated with different concentrations of RITA for 48 hours. NALM-6 cells were then harvested and washed twice with PBS and fixed with 70% ethanol for 24 hours. Then cells were treated with 100 μg/ml RNase in PBS, and incubated at 37°C for 30 minutes before staining with 50 μg/ml PI for 30 minutes. The cells were analyzed using a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Phosphatidylserine externalization (annexin-V assay)
NALM-6 cells were treated with compounds studied and were washed with PBS after the incubation time. A total of 5×105 cells per sample were resuspended in a total volume of 100 μl of the incubation buffer. Annexin-V-Flous (0·5 μl per sample) and propidium iodide (final concentration 20 μg/ml) were added, and cell suspensions were incubated for 20 minutes in the dark. Fluorescence was then measured using flow cytometry. The data were evaluated using the CellQuest Software (Becton Dickinson) and expressed as a percentage of the cells positive for annexin-V and negative for propidium iodide (early-apoptotic phase).
Western blot analysis
Cells were centrifuged at different time points after various treatments, and cellular pellets were washed with cold PBS and lysed (5×106 cells/aliquots) in 0·2 ml of RIPA buffer (10 mM Tris-HCl, pH 7·4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0·1% sodium dodecyl sulfate, and 0·5% sodium deoxycholate) containing protease and phosphatase inhibitor cocktails (Sigma). After centrifugation at 13 000×g for 20 minutes at 4°C, the supernatant was collected. Protein concentrations were determined by Bradford protein assay, and equivalent amounts of total cellular protein were separated by 10% SDS-PAGE, according to the method of Laemmli. The gels were then electroblotted onto nitrocellulose membranes (Hybond-ECL, Amersham Corp., Louisville, CO, USA). Subsequently, membranes were blocked with 5% nonfat dry milk in TBS containing 0·1% (v/v) Tween-20, 1 hour at room temperature or overnight at 4°C, and probed with specific primary antibodies overnight at 4°C. After 5 washes in TBS-T, membranes were incubated with horseradish-peroxidase-conjugated secondary antibodies. Proteins were then visualized with a chemiluminescence detection system (Amersham ECL Advance Kit, GE Healthcare)
RNA isolation and preparation of cDNA
RNA from NALM-6 cells was isolated at different times after treatment with doxorubicin by using TriPure Isolation Reagent according to the instructions of the manufacturer (Roche Applied Science). RT-reaction was performed using the RevertAid First Strand cDNA Synthesis kit from Fermentas. A 20 μl reaction contained 4 μl 5×PCR buffer, 1 μl DEPC-treated water, 2 μl dNTP (10 mM), 1 μ RNase inhibitor (20 U/μl), 1 μl random hexamers and 1 μl M-MuLV reverse transcriptase (200 U/μl) as a master mix, and 2 μl of total RNA was added prior to reaction start. Based on photometric measurement the total RNA template concentration was below the reaction capacity of ⩽1 μg RNA per reaction. Adapted times and temperature profiles for the reverse transcription were used: incubation for 5 minutes at 65°C, 5 minutes at 25°C followed by 60 minutes at 42°C. The reaction was terminated by heating at 70°C for 5 minutes.
Quantitative real-time PCR
Real-time PCR was performed using the 12·5 μl of Maxima SYBR green master mix (Fermentas), 2 μl of the cDNA product in a total volume of 25 μl. DNA was amplified in a 40-cycle PCR reaction, using the following conditions: denaturation at 95°C for 15 seconds, annealing and elongation at 60°C for 60 seconds. The reaction took place in the Rotor Gene 6000 Real Time PCR System (Corbett Research, Hilden, Germany). A melting curve analysis was performed to verify the specificity of the products, and the fold induction or repression was measured relative to control and calculated after adjusting for reference gene GAPDH using the comparative CT (2-▵▵CT) method. The list of primer used and their respective sequences are given in .
Table 1. Nucleotide sequences of the primers used for real-time PCR
Statistical analyses
The significance of differences between experimental variables was determined by the use of the Student’s t-test. A probability level of P<0·05 was considered statistically significant.
Results
RITA induced cell death in a time and dose-dependent manner in pre-B ALL NALM-6 cells
Viability of NALM-6 cells was assessed by MTT assay after treatment of cells with various concentrations of RITA. The cytotoxic effect of RITA in NALM-6 cells was dose- and time-dependent. After 24 hours incubation, it was not seen cell death induced by RITA (data not shown), but after 48 hours incubation RITA-induced cell death was significantly observed in a dose-dependent fashion (). To further confirm our data, flow cytometry analysis was performed to detect apoptotic cells in sub-G1 region. NALM-6 cells were exposed to different concentrations of RITA for 48 hours and the percentages of apoptotic cells was measured by sub-G1 DNA content analysis. As shown in , by increasing concentrations of RITA the cell population in sub-G1 region was increased which confirms the apoptotic effect of RITA on NALM-6 cells. In addition, it was shown that co-incubation of RITA 2 μM with doxorubicin 25 nM resulted in a synergistic increase of apoptosis in the pre-B ALL NALM-6 cells ().
Figure 1. Effects of RITA on human acute lymphoblastic leukemia cells. (A) NALM-6 cells were cultured in the presence of 0·5, 1, 2, 4, 8 or 10 μM RITA; 48 hours later viability of cells and the percentage of apoptotic cells was measured by MTT assay (A) and flow cytometry (B) respectively. The percentage of apoptotic cells with PS externalization after 30 hours (C). The results are expressed as mean±standard deviations (SD) of at least three independent experiments. The symbol denotes significant difference (P<0·05): * relative to cells treated with doxorubicin or RITA alone.
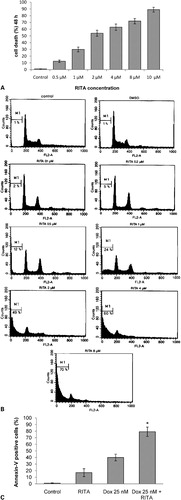
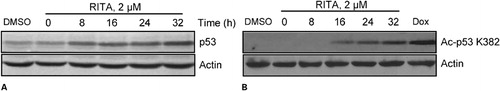
Figure 2. RITA stimulates p53 protein expression in a time-dependent fashion. NALM-6 cells were treated with RITA 2 μM and cells were harvested at different times. Total cell lysates were prepared and protein was quantified by Bradford’s assay. Equal amount of solubilized protein (40 μg) was electrophoresed on a 10% SDS-PAGE gel and transferred electrophoretically onto a nitrocellulose membrane. The blots were incubated with the anti-p53 DO-I (A) and anti-acetylated p53 (B) antibodies separately.
RITA induced p53 accumulation and post-translational acetylation in pre-B ALL cells
Since RITA inhibits interaction of p53-MDM2, we sought to examine the effect of RITA on p53 protein levels in NALM-6 cells. Therefore, we incubated NALM-6 with RITA 2 μM and cells were harvested at different times for examination of p53 protein levels. shows that exposure of these cells to RITA led to accumulation of p53 within 8 hours. Previous studies revealed that p300/CBP-mediated p53 acetylation is negatively regulated by MDM2 through recruiting a complex containing class I histone deacetylase (HDAC1). Interestingly, recruitment of HDAC1 by MDM2 promotes p53 degradation by removing these acetyl groups.Citation17 Since the acetylated p53 lysine residues overlap with those that are ubiquitylated, we wished to show the effect of RITA on acetylation of p53. To do so, we treated NALM-6 cells with 2 μM concentrations of RITA at different times. In addition, we used doxorubicin which is a potent inducer of p53 acetylation. As presented in , p53 acetylation levels is progressively increased by the passage of time which indicates that RITA prevented p53 ubiquitination by MDM2.
Inhibition of p53-HDM2 interaction by RITA up-regulated expression of p53 target genes
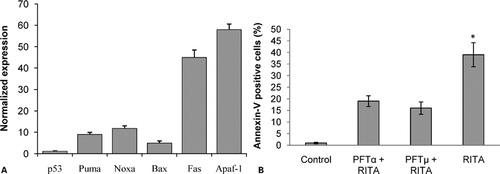
Like IR therapy, most clinically used standard cytotoxic chemotherapeutic agents induce DNA damage signaling cascades which are involved in p53 activation. Activated p53 binds to transcriptional regulatory elements in DNA and induces expression of several target genes mediating apoptosis, cell cycle arrest and DNA repair.Citation18 To this end, we examined the effect of RITA on p53 target genes in NALM-6 cells. After 28 hours incubation with RITA 2 μM, cells were harvested, RNA was isolated and cDNA synthesis was performed. As presented in , we performed real-time qPCR analysis of several known p53 target genes and found that RITA treatment resulted in the induction of endogenous p53 target genes PMAIP1 (Noxa), BBC3 (PUMA), Fas, Bax and APAF1. We also detected p53 gene expression level and we found that RITA had no effect on p53 mRNA steady-state levels. To further examine the p53-dependence of RITA effects, we applied two chemical inhibitors of p53. Pifithrin-alpha (PFTalpha) which is known to prevent transcriptional activity of p53 and pifithrin-mu (PFTmu) which has been reported to prevent the induction of transcriptional-independent branch of p53-mediated apoptosis.Citation19Citation19,20 To this end, NALM-6 cells were pretreated with PFTalpha (10 μM) or PFTmu (10 μM) three hours prior to RITA (2 μM) treatment. The incubation was continued for a further 36 hours. Cells were then harvested and subjected to annexin-V assay. As indicated in , both PFTalpha and PFTmu abolished apoptosis (annexin-V positive cells) induced by RITA in these cells ().
Figure 3. RITA induces p53-dependent apoptosis in NALM-6 cells. (A) NALM-6 cells were cultured with RITA 2 μM for 28 hours, and then assessed for p53 and p53 target genes mRNA expression as detailed in the Materials and Methods section. (B) The percentages of apoptotic cells with PS externalization after incubation of cells with RITA alone, RITA plus Pifithrin-alpha or RITA plus Pifithrin-mu. The results are expressed as mean±standard deviations (SD) of at least three independent experiments. The symbol denotes significant difference (P<0·05): * relative to cells treated with RITA plus Pifithrin-alpha or RITA plus Pifithrin-mu.
Effect of RITA on procaspase-3 and PARP-1
Stress-induced mitochondrial outer-membrane permeabilization allows for the release of cyctochrome c from the mitochondrial intermembrane space into cytosol where it contributes in the formation of the apoptosome, which mediates autocleavage and activation of caspase 9. Once activated, caspase 9 cleaves and activates various effector caspases, including caspase 3, which in turn carries out the proteolysis of a number of substrates, such as Poly(ADP-ribose) polymerase (PARP), during the execution phase of apoptosis.Citation21
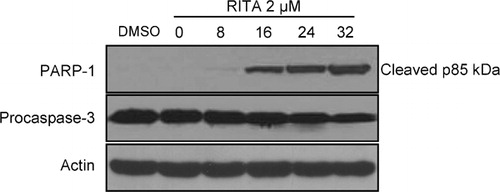
Therefore we next examined the effect of RITA on cleavage of procaspase 3 and PARP. NALM-6 cells were treated with RITA 2 μM and examined for the expression of cleaved PARP-1 and procaspase 3 by Western blot analysis. As seen in , cleavage of procaspase 3 and PARP were detected clearly within 16 hours and increased further by 32 hours post-treatment. In addition, it was shown that RITA induced expression level of APAF-1 gene () which acts as a coactivator of caspase-9 and helps initiate the caspase cascade. These data show that in accordance with its stimulatory effect on p53 target genes, RITA induced activation of the proteins known to be involved in execution of apoptosis.
Figure 4. PARP-1 and procaspase-3 cleavage was induced in NALM-6 cells by RITA. Cells were treated as described in the legend to , harvested at the indicated times, and examined by immunoblotting with the indicated antibodies. Equal loading was verified using anti-actin antibody. One representative experiment of at least three performed is presented.
Discussion
Somatic missense mutations in the TP53 gene are present in 50% of human cancers. However, the observation that p53 function is lost in most wild-type p53 (wtp53)-carrying cancers, due to amplification or overexpression of its negative regulator MDM2, has rendered the strategy of releasing p53 from MDM2 an attractive therapeutic target in oncology.Citation22Citation22,23 The use of conventional chemotherapy or radiotherapy in cancer treatment is limited by serious side effects that arise due to toxicity in normal cells. Therefore, modern drug design focuses on the development of targeted therapies. Indeed, in the past decade, induction of p53-mediated apoptosis in tumor cells by exquisitely selective MDM2 inactivation without stimulation of genotoxic damage has been one of the leading ideas to avoid unwanted side effects and limit the induction of secondary cancers resulting from genomic damage.Citation24Citation24,25 In this study, we showed that small molecule RITA can lead to a dose-dependent apoptosis in pre-B ALL cells harboring wild type p53. Interestingly, synergistic effect was seen by combination of the non-genotoxic molecule, RITA, with a conventional chemotherapeutic drug doxorubicin in NALM-6 cells. Our results are consistent with the study by Nahi et al.Citation26 reporting that co-incubation of RITA with fludarabine acts synergistically in chronic lymphocytic leukemia cells. Mutations in TP53 constitute a clinical problem in hematological malignancies, as they are associated with resistance to conventional chemotherapeutic drugs, and poor survival.Citation27Citation27,28 In contrast to most solid tumors, the p53 gene is mutated in approximately 10–15% of both myeloid and lymphoid leukemias at diagnosisCitation29 and is found mainly in aggressive non-Hodgkin’s lymphoma,Citation30 progressive B-cell chronic lymphocytic leukemia,Citation31 and chronic prolymphocytic leukemia.Citation32 This data implie that activation of the p53 pathway by non-genotoxic target-specific drugs in human ALL cells could be a promising strategy to improve chemotherapy. However, in a recent study it was demonstrated that RITA is even capable of reactivating mutant p53 in different tumor cells.Citation19 Our findings that RITA induced p53 accumulation and its transcriptional activity are in accordance with previous studies.Citation33 In addition, we found that in NALM-6 cells, along with transcription-dependent pathway, a transcription-independent mechanism of p53-mediated cell death was involved, as manifested by the ability of PFT-mu to prevent cell death induced by RITA (). In conclusion, although RITA can induce apoptosis alone, it can act in concert with conventional chemotherapeutics to activate p53, with the potential of improving efficacy or lowering the genotoxic burden by enabling dose reduction. However, the effective combination of the conventional or novel chemotherapeutic drugs with the small molecule RITA should be further tested in primary ALL cells.
This study was supported by the grant P-800 from Iran University of Medical Sciences.
References
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000;408:307–10.
- Amundson SA, Myers TG, Fornace AJ. Roles for p53 in growth arrest and apoptosis: putting on the brakes after genotoxic stress. Oncogene 1998;17:3287–99.
- Xu Y. Regulation of p53 responses by post-translational modifications. Cell Death Differ 2003;10:400–3.
- Beroud C, Soussi T. The UMD-p53 database: new mutations and analysis tools. Hum Mutat 2003;21:176–81.
- Soussi T, Wiman KG. Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell 2007;12:303–12.
- Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov 2008;7:979–87.
- Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer 2009;9:862–73.
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al.. Restoration of p53 function leads to tumour regression in vivo. Nature 2007;445:661–5.
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al..: Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007;445:656–60.
- Kastan MB. Wild-type p53: tumors can’t stand it. Cell 2007;128:837–40.
- Marine JC, Lozano G. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ 2010;17:93–102.
- Chene P. Inhibition of the p53-hdm2 interaction with low molecular weight compounds. Cell Cycle 2004;3:460–1.
- Klein C, Vassilev LT. Targeting the p53-MDM2 interaction to treat cancer. Br J Cancer 2004;91:1415–9.
- Doggrell SA. RITA–a small-molecule anticancer drug that targets p53. Expert Opin Investig Drugs 2005;14:739–42.
- Zhao CY, Szekely L, Bao W, Selivanova G. Rescue of p53 function by smallmolecule RITA in cervical carcinoma by blocking E6-mediated degradation. Cancer Res 2010;70:3372–81.
- Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, et al.. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med 2004;10:1321–8.
- Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E, et al.. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J 2002;21:6236–45.
- Lu C, El-Deiry WS. Targeting p53 for enhanced radio- and chemo-sensitivity. Apoptosis 2009;14:597–606.
- Zhao CY, Grinkevich VV, Nikulenkov F, Bao W, Selivanova G. Rescue of the apoptotic-inducing function of mutant p53 by small molecule RITA. Cell Cycle 2010;9:1847–55.
- Strom E, Sathe S, Komarov PG, Chernova OB, Pavlovska I, Shyshynova I, et al.. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat Chem Biol 2006;2:474–9.
- Chipuk JE, Green DR. Dissecting p53-dependent apoptosis. Cell Death Differ 2006;13:994–1002.
- Selivanova G. Therapeutic targeting of p53 by small molecules. Semin Cancer Biol 2010;20:46–56.
- Leach FS, Tokino T, Meltzer P, Burrell M, Oliner JD, Smith S, et al.. p53 Mutation and MDM2 amplification in human soft tissue sarcomas. Cancer Res 1993;53:2231–4.
- Saha MN, Micallef J, Qiu L, Chang H. Pharmacological activation of the p53 pathway in haematological malignancies. J Clin Pathol 2010;63:204–9.
- Fojo T. Commentary: novel therapies for cancer: why dirty might be better. Oncologist 2008;13:277–83.
- Nahi H, Selivanova G, Lehmann S, Mollgard L, Bengtzen S, Concha H, et al.. Mutated and non-mutated TP53 as targets in the treatment of leukaemia. Br J Haematol 2008;141:445–53.
- Dohner H, Fischer K, Bentz M, Hansen K, Benner A, Cabot G, et al.. p53 gene deletion predicts for poor survival and non-response to therapy with purine analogs in chronic B-cell leukemias. Blood 1995;85:1580–9.
- Hawkins JM, Moorman AV, Hoffbrand AV, Martineau M, Wright FS, Mehta AB, et al.. Association of 17p loss with late-stage or refractory disease in hematologic malignancy. Cancer Genet Cytogenet 1994;77:134–43.
- Mitani N, Niwa Y, Okamoto Y. Surveyor nuclease-based detection of p53 gene mutations in haematological malignancy. Ann Clin Biochem 2007;44:557–9.
- Wilson WH, Teruya-Feldstein J, Fest T, Harris C, Steinberg SM, Jaffe ES, et al.. Relationship of p53, bcl-2, and tumor proliferation to clinical drug resistance in non-Hodgkin’s lymphomas. Blood 1997;89:601–9.
- Gaidano G, Ballerini P, Gong JZ, Inghirami G, Neri A, Newcomb EW, et al.. p53 mutations in human lymphoid malignancies: association with Burkitt lymphoma and chronic lymphocytic leukemia. Proc Natl Acad Sci USA 1991;88:5413–7.
- Lens D, De Schouwer PJ, Hamoudi RA, Abdul-Rauf M, Farahat N, Matutes E, et al.. p53 abnormalities in B-cell prolymphocytic leukemia. Blood 1997;89:2015–23.
- Enge M, Bao W, Hedstrom E, Jackson SP, Moumen A, Selivanova G. MDM2-dependent downregulation of p21 and hnRNP K provides a switch between apoptosis and growth arrest induced by pharmacologically activated p53. Cancer Cell 2009;15:171–83.