Introduction
Hereditary hemochromatosis (HH) is an iron overload disease of genetic origin. Its usual form, named HFE-related hemochromatosis (or Type 1 hemochromatosis), affects only white populations where it is one of the most frequent genetic diseases. Other genetic forms of hemochromatosis are rare but present worldwide; they correspond to non-HFE mutations and are named Type 2, 3, or 4 hemochromatosis.
Cause
| •. | Most forms of HH (Types 1, 2, and 3) are due to the decreased production by the liver of the iron hormone hepcidin, leading to an increased absorption of dietary iron. Therefore, iron concentration in the blood increases. | ||||
| •. | One rare form of hemochromatosis (Type 4 hemochromatosis) is due to decreased ferroportin activity. Ferroportin is the protein permitting the iron exit from the cell into the blood. Therefore, iron excess develops within the organs due to decreased iron exit (and not to increased iron entry). Blood iron concentration, in this hemochromatosis form, is diminished, contrasting with elevated body iron stores. Importantly, due to the absence of non-transferrin bound iron, organ damage is less pronounced than in Type 1 hemochromatosis. | ||||
Symptoms and signs
| 1. | For the usual form of hemochromatosis (HFE-related or Type 1 hemochromatosis). | ||||
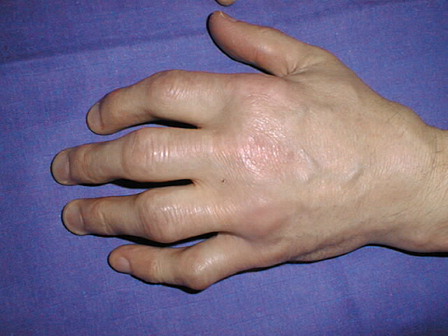
| 2. | a long latent phase, the symptoms appear around 30–40 years of age in men, and 40–50 years in women. The main signs include chronic fatigue (physical, psychological, and sexual, e.g. impotence in men). Joint pains that correspond to inflammatory rheumatism and are often misdiagnosed. One suggestive location concerns the hands (). Osteoporosis may be responsible for backache and vertebral fractures. Skin is abnormally bronzed. Liver symptoms include enlargement, a moderate and chronic increase of serum transaminases, development of hepatic fibrosis with cirrhosis and even hepatic cancer. Diabetes is related to pancreatic iron excess. Cardiac iron overload can be responsible for rhythm disturbances (palpitations) and cardiac failure. | ||||
| 3. | For the non-HFE related forms. The symptoms of Type 2 and Type 3 hemochromatosis resemble Type 1 but Type 2 affects solely younger people (‘juvenile hemochromatosis’) – with predominant cardiac and endocrine symptoms – whereas type 3 can affect both adult and young patients. Type 4 HH (‘ferroportin disease’) is often clinically asymptomatic. | ||||
Figure 1. Typical hemochromatosis rheumatism, affecting the metacarpo-phalangeal and proximal inter-phalangeal joints.
Tests
1. For the usual form of hemochromatosis (HFE-related or type 1 HH)
Blood iron concentration is increased (above 25 µmol/l), together with an increased transferrin saturation: over 60% in men and 50% in men, often reaching 100% (normally <45%), reflecting the increased intestinal absorption of iron.
Blood ferritin concentration (over 300 µg/l in men, and 200 µg/l in women) is present as soon as iron excess starts in the organs.
Increased iron concentration in the liver can be assessed using magnetic resonance imaging (MRI) which has replaced liver biopsy for hepatic iron load evaluation.
After elevated blood iron parameters have been found, genetic testing must be performed on a further blood sample. The HFE mutation is present in duplicate, corresponding to the result: ‘C282Y/C282Y’ (one mutation received from the father, the other one from the mother).
2. For non-HFE-related HH
For Types 2 and 3 HH, blood iron profile is identical to Type 1 (increased iron, transferrin saturation, and ferritin) but genetic testing will search for mutations of the hemojuvelin or hepcidin gene (for Type 2), and for mutations of the Transferrin Receptor 2 gene (for Type 3).
For type 4 HH, blood iron profile is quite different since iron and transferrin saturation are not elevated (and often decreased) whereas ferritin levels are usually much higher than in type1 HH. The genetic test will search for mutations of the ferroportin gene.
For all non-HFE-related HH, genetic testing requires highly specialized laboratories.
Treatment options
1. For the usual form of hemochromatosis (Type 1 HH)
Iron removal by regular (weekly) venesections remains the mainstay of the treatment. A phlebotomy schedule consists of removing 7 ml/kg body weight without exceeding 550 ml per venesection, until ferritin reaches 50 µg/l, and provided hemoglobin levels remains above 11 g/dl.
Iron removal is highly efficient on fatigue and skin hyperpigmentation. It ameliorates hepatic, pancreatic (diabetes), and cardiac status but joint symptoms may not be improved and even worsen.
Once this goal is reached, maintenance therapy consists in one venesection every 1 to 3 months aiming to maintain ferritin close to 50 µg/l.
It is essential to engage a family study, based on the combination of genetic testing with blood iron parameters, in siblings but also in children above 18 years old, and in parents.
Family members without the C282Y mutation, or with C282Y at single dose, are at no risk for developing iron overload.
2. For non-HFE-related HH
For Type 2 HH, the therapeutic measures are identical, except that, due to massive iron excess, in addition to venesection iron chelation may be required (parenteral desferrioxamine or the new oral iron chelators).
For Type 3 HH, treatment is identical to Type 1 HH for adults, and to Type 2 HH when affecting young patients.
For Type 4 HH, venesection therapy is less appropriate (since iron removal is rendered difficult by the limitations of cellular iron exit), so that anemia can develop if the venesection program is too drastic. However, an alleviated schedule (such as one phlebotomy every 2 weeks) is usually correctly tolerated and efficient.
Family studies must also be conducted in these various non-HFE-related HH forms, knowing that Type 4 HH is the only HH with a dominant mode of inheritance.
Outlook for the usual form of hemochromatosis (Type 1 HH)
For the treatment: Iron-removing drugs, such as oral iron chelators, can be discussed in case of contra-indication to venesections. Hepcidin supplementation, when clinically available, will represent the logical therapeutic approach of the disease.
For prevention: HH being clinically silent during the first 30 years of life, and treatment efficacy depending on its early start, it is of utmost importance to check blood iron parameters in every young adult.
Additional resources
Canadian Hemochromatosis Society: http://www.toomuchiron.ca
Iron Disorders Institute: http://www.irondisorders.org/hemochromatosis
Hemochromatosis Information society: http://www.hemoinfo.org/
European Federation of Associations of patients with Haemochromatosis (EFAPH): http://efaph.eu/
Haemochromatosis Society of Australia: [email protected]
Haemochromatosis Society of South Africa: http://www.haemochromatosisza.org
Non-HFE-related HH features
Rare but present worldwide
Can affect young patients (‘juvenile HH’) for Types 2 and 3 HH
Can be relatively asymptomatic for Type 4 HH (‘ferroportin disease’)
High serum ferritin contrasting with low serum iron in Type 4 HH
Venesections can be completed by iron chelation in juvenile HH
Venesections can be poorly tolerated in Type 4 HH
Genetic testing requires highly specialized labs