
Introduction
Haemophilia A is a rare inherited bleeding disorder resulting from the deficiency of factor VIII (FVIII), an essential component of the intrinsic pathway of blood coagulation. The incidence of haemophilia A is 1 in 5000 male live births and affected individuals have severe, moderate, and mild forms of the disease, defined by FVIII plasma levels of less than 1% of normal, 1 to 5%, and 6 to 30%, respectively. The reduction of this plasma protein causes persons with haemophilia to have a lifelong bleeding tendency, of clinical severity proportional to the degree of FVIII reduction.
Cause
Haemophilia A is caused by various mutations in the FVIII gene, which is located on the X-chromosome. Since this disorder is transmitted as an X-linked recessive trait, it occurs mainly in males. Usually, the affected boy has inherited the abnormal gene from his carrier mother, although about 30% of cases arise from a new mutation. Some types of FVIII gene mutations (i.e. inversions, large deletions) are also involved in the risk of development of FVIII inhibitory alloantibodies, which occur in up to 30% of severely affected patients. They are the most important complication of haemophilia treatment, because they render replacement therapy ineffective and expose patients to an increased risk of morbidity and mortality.
Symptoms and signs
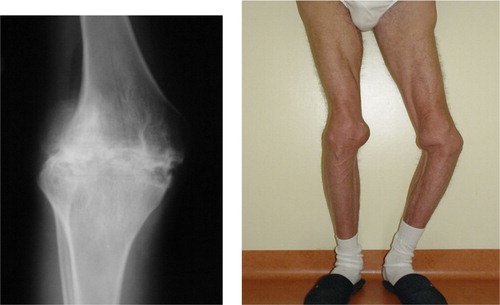
Patients with mild forms of haemophilia seldom have unprovoked haemorrhages and usually bleed only after trauma or surgery. By contrast, patients with moderate disease only occasionally have spontaneous haemorrhages, while those with severe disease develop spontaneous haemorrhages from early infancy, usually at the time of beginning ambulation. Carrier females with low FVIII levels in the range of mild haemophilia may experience bleeding symptoms such as oral, surgical, and trauma-related bleeding. The clinical hallmarks of haemophilia A are joint and muscle haemorrhages, and prolonged and potentially fatal haemorrhage after trauma or surgery. The joints more frequently involved are the knees, elbows, and ankles. Joint bleeds usually begin with mild discomfort and a slight limitation of joint motion, accompanied by pain, swelling, and cutaneous warmth. If untreated, joint haemorrhage usually leads to severe motion limitation. The pathologic processes continue even after bleeding stops, because inflammation causes damage of the blood-filled joints leading to synovitis, which in turn increases the likelihood of more frequent bleeds in target joints. The final step of this vicious circle is haemophilic arthropathy, characterized by narrowing of the joint space due to loss of cartilage, development of bone cysts, and severe limitation of motion resulting in permanent disability (). Intramuscular haematomas can cause severe problems by compressing vital structures. Closed-space bleeding is especially dangerous, since it can lead to nerve paralysis or to vascular or airway obstruction. Bleeding into the iliopsoas muscle is a typical symptom in severe haemophilia, and may lead to muscle contracture, atrophy, and nerve palsies. Haemorrhage into the central nervous system is the most dangerous hemorrhagic event requiring urgent management. Intracranial bleeding may be unprovoked but in approximately half of the cases a previous relatively minor trauma can be recognized. Symptoms often occur soon after trauma, but sometimes they are delayed, as for instance in the presence of a subdural hematoma. Thus, haemophilic patients with unexplained and persistent headache should always be suspected of having haemorrhage in the brain parenchyma, a subdural or epidural hematoma.
| • | Mild haemophilia A (FVIII 6–30%)
| ||||||||||||||||
| • | Moderate haemophilia A (FVIII 1–5%)
| ||||||||||||||||
| • | Severe haemophilia A (FVIII <1%)
| ||||||||||||||||
Figure 1. Severe arthropathy in a 65-year-old severe haemophilia A patient born before the advent of modern FVIII replacement therapy.
Tests
A diagnosis of haemophilia A should be suspected whenever unusual bleeding occurs in a male and is supported by the results of simple laboratory tests, including a prolonged partial thromboplastin time (APTT) contrasting with a normal platelet count, prothrombin time (PT), and bleeding time. The definitive diagnosis relies on the assay for FVIII coagulant activity in plasma. The analysis of FVIII gene mutation is useful to further characterize the FVIII defect and the risk of inhibitor development, which must be measured with a specific test.
| • | Prolonged APTT | ||||
| • | Normal PT, platelet count, bleeding time | ||||
| • | Reduced FVIII activity | ||||
| • | FVIII mutation analysis | ||||
Treatment options
Replacement therapy with intravenously delivered products containing FVIII in concentrated form corrects the coagulation defect and hence is the cornerstone of haemophilia A therapy. In addition, the administration of desmopressin (DDAVP) may be useful in milder forms of the disease.
DDAVP
This synthetic analogue of the antidiuretic hormone vasopressin, given intravenously or subcutaneously, can be used in patients with mild hemophilia A for the treatment of minor bleeding or minor surgery, such as dental extractions. In order to test the patient degree of FVIII increase after DDAVP, a test infusion prior to its therapeutic use is recommended.
FVIII concentrates
Since the 1970s, the production of lyophilized plasma-derived FVIII concentrates allowed the modern management of hemophilia, leading to the widespread adoption of home replacement therapy, the early control of bleeding and the reduction of the musculoskeletal damage typical until then of untreated or poorly treated patients. At the same time the implementation of primary prophylaxis (i.e. the regular infusion of FVIII concentrate), applied first in Sweden and then in other countries, achieved the goal of preventing the majority of bleeding episodes and further reduced the impact of arthropathy. Several plasma-derived and recombinant FVIII concentrates are available, all with a high degree of safety and hemostatic efficacy. As a result of the recent progress made in the field of therapy, the quality of life of hemophilia A patients has dramatically improved and their life-expectancy has progressively become similar to that of males in the general population, at least in high-income countries. The treatment of patients with inhibitors is more problematic and the therapeutic strategies are mainly focused on the treatment or prevention of bleeding episodes using agents that bypass the inhibitor (i.e. plasma-derived activated prothrombin complex concentrates and recombinant activated factor VII). Eradication of the inhibitor through long-term intensive treatment with large doses of FVIII (immune tolerance induction) is effective in approximately two thirds of cases.
Resources
There are many resources online but the most important are: the World Federation of Hemophilia (http://www.WFH.org), European Association for Haemophilia and Allied Disorders (http://www.eahad.org), and European Haemophilia Safety Surveillance (http://www.euhass.org).
A recent free full-text review on haemophilia can be found in the journal Orphanet Journal of Rare Diseases, volume 7, page 24 (2012).