
Introduction
Sickle cell disease (SCD) is a group of disorders affecting the haemoglobin protein leading to red blood cell sickling. It is the most common form of inherited blood disorders. Patients are born with the disease and can experience complications of anaemia and organ damage to any part of the body starting in infancy. The complications of SCD are caused by abnormal red cells that take on a crescent or sickled shape in low oxygen conditions. These sickled red cells break apart more easily than normal shaped red blood cells and result in anaemia. The abnormal and rigid sickled cells occlude small blood vessels impairing blood flow and oxygen delivery, leading to progressive organ damage. SCD is more common in certain ethnic groups whose ancestors lived in malaria prone regions of the world, which includes people of African descent, Hispanics from South and Central America, and people from the Middle East and Indian subcontinent.
Cause
The oxygen-carrying protein in red blood cells is called haemoglobin. A single amino acid mutation in the beta globin gene, a component of the haemoglobin protein, causes the sickle haemoglobin (HbS) to have an abnormal property whereby it can be triggered to form protein chains causing the red cells to change shape under certain conditions. The trigger for red cell sickling is not fully understood. One generally accepted hypothesis is that red cell sickling is triggered by low oxygen environment, a condition found in small blood vessels as the red cells release oxygen to tissues. Sickle cell anaemia, the most common and severe form of the disease, results from the inheritance of two copies of the HbS mutation (HbSS). Inheritance of one copy of the HbS gene without any other beta globin gene mutation is known as HbS trait and is a benign condition affecting about 1 in 12 African Americans. Variations of SCD occur when patients carry the HbS mutation in addition to another mutation in their other beta globin gene, such as haemoglobin SC and haemoglobin S/beta thalassemia. These other types of SCD can be associated with a mild-to-severe course depending on various genetic and environmental factors.
Symptoms and signs
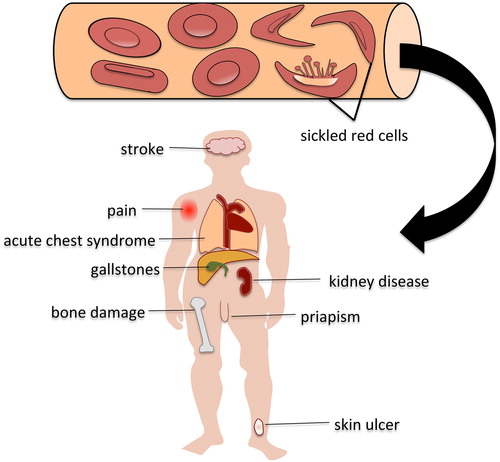
Patients with SCD have marked differences in severity of disease and complications. Symptoms can range from mild and self-limiting to severe and life threatening. Progressive organ damage can occur anywhere in the body as a result of recurrent episodes of blood flow blockage and impaired oxygen delivery. The most common complication is a vaso-occlusive crisis (VOC), which is a recurring pain event triggered by tissue damage and inflammation caused by red cell sickling and obstruction of blood flow. Patients experience pain most often in the lower spine and joints of the arms and legs. Fatigue, shortness of breath, and dizziness occur as a result of anaemia. Patients with sickle cell are at risk of infections due to damage to the spleen and compromise in the immune system due to chronic inflammation from red cell destruction and tissue damage. Increased red cell destruction leads to the appearance of yellow skin and eyes. Children can have pain and swelling in their hands and feet during a VOC event, a condition known as hand-foot syndrome or dactylitis. Acute chest syndrome, marked by fever, shortness of breath, chest pain, and an infiltrate on a chest x-ray similar to pneumonia can be a potentially fatal complication of SCD. Impaired blood flow in the blood vessels of the brain in the form of a stroke or transient ischaemic attack can occur in children or adults. Males with SCD may develop unwanted and painful erections call priapism that can lead to erectile dysfunction and impotence.
Other complications of SCD include:
| • | leg ulcers | ||||
| • | damage or engorgement of the spleen | ||||
| • | avascular necrosis (damage to bone) | ||||
| • | gallstones | ||||
| • | pulmonary hypertension (elevated blood pressure in the lung blood vessels). | ||||
Tests
Testing for SCD can be done with blood tests, called haemoglobin electrophoresis or high performance liquid chromatography, which can detect the abnormal HbS. If there is a need to clarify the diagnosis or define the genetic mutations further, DNA-based mutation testing can be ordered. The United Kingdom and United States have testing for sickle cell anaemia as part of their newborn screening programmes. If the initial screening test shows that a newborn has HbS, the parents will be notified and referred to a physician who can order tests to confirm the type of SCD.
Treatment options
Currently, the mainstay treatment for SCD is aimed at reducing complications and treating symptoms. For patients with severe disease, including those with sickle cell anaemia (HbSS), treatment with a medication called hydroxyurea can decrease complications of sickle cell by increasing the amount of foetal haemoglobin in red cells. Foetal haemoglobin decreases red cell sickling and destruction. Patients experience less pain events and have improved anaemia, which reduces the need for red blood cell transfusions. In children, treatment with daily penicillin is aimed at preventing infections, which in the past was the leading cause of death in children with SCD. All patients are encouraged to have timely immunizations such as yearly influenza vaccine but also vaccines against pneumococcal, meningococcal, and haemophilus influenza for those without a functional spleen. Blood transfusions are used to treat severe anaemia and reduce the risk of stroke in children but can cause iron overload and immunologic complications.
Figure 1. Complications of sickle cell
Stem cell transplantation offers a potential cure for sickle cell anaemia but comes with significant risks during and after transplant. Traditionally, this procedure is reserved for patients who have significant complications from SCD and have a full sibling who can be a matched donor of stem cells. With continued improvements in stem cell transplant technique and therapy to control its complications, transplant may become a more available and accepted treatment option for a broader group of patients with sickle cell anaemia.
Outlook
There are marked differences in disease severity and complications among patients with SCD even with the same genetic mutations. In general, the most severe form of the disease is sickle cell anaemia (HbSS) or haemoglobin S coupled with a severe beta thalassemia mutation. Genetic and environmental factors that affect severity of the disease are not understood fully. Current treatment is aimed mainly at preventing and treating complications. Although stem cell transplant can offer a cure, it is associated with significant risks and limited to those patients who have a full matched sibling. Further development of safe and effective treatments for SCD is needed.
Additional resources
There is abundant information available online with the more reliable resources at the following sites:
| • | From the Centers for Disease Control and Prevention (http://www.cdc.gov/ncbddd/sicklecell/index.html) | ||||
| • | From the National Institute of Health (http://www.nhlbi.nih.gov/health/health-topics/topics/sca/) | ||||
| • | From the National Health Services (http://www.nhs.uk/conditions/sickle-cell-anaemia/Pages/Introduction.aspx) | ||||