Abstract
We have used two different probes with distinct detection properties, dichlorodihydrofluorescein diacetate and Amplex Red/horseradish peroxidase, as well as different respiratory substrates and electron transport chain inhibitors, to characterize the reactive oxygen species (ROS) generation by the respiratory chain in calcium-overloaded mitochondria. Regardless of the respiratory substrate, calcium stimulated the mitochondrial generation of ROS, which were released at both the mitochondrial-matrix side and the extra-mitochondrial space, in a way insensitive to the mitochondrial permeability transition pores inhibitor cyclosporine A. In glutamate/malate-energized mitochondria, inhibition at complex I or complex III (ubiquinone cycle) similarly modulated ROS generation at either mitochondrial-matrix side or extra-mitochondrial space; this also occurred when the backflow of electrons to complex I in succinate-energized mitochondria was inhibited. On the other hand, in succinate-energized mitochondria the modulation of ROS generation at mitochondrial-matrix side or extra-mitochondrial space depends on the site of complex III which was inhibited. These results allow a straight comparison between the effects of different respiratory substrates and electron transport chain inhibitors on ROS generation at either mitochondrial-matrix side or extra-mitochondrial space in calcium-overloaded mitochondria.
Introduction
Under physiological conditions, mitochondria play an important role in cellular calcium homeostasis. However, mitochondrial calcium overload in pathological situations is associated with the increase in reactive oxygen species (ROS) generation, oxidation of membrane protein thiols, and induction of the mitochondrial permeability transition (MPT) process. MPT is mediated by the opening of membrane permeability transition pores (PTP) in the presence of calcium and evidenced by cyclosporine A (CsA)-sensitive mitochondrial swelling; it is considered to play an important role in cell death by necrosis and apoptosis.Citation1–Citation5
Mitochondria are the main intracellular sources of ROS. The primary ROS generated by mitochondria is superoxide anion (O2•−), the product of molecular oxygen reduction by the electrons that leak from the electron transport chain. The two main ROS-generating sites in the electron transport chain are complex I (NADH:ubiquinone oxidoreductase) and complex III (ubiquinol:cytochrome c oxidoreductase).Citation6,Citation7 In complex I, O2•− is produced by the oxidation of reduced flavin (FMNH2)/mitochondrial-matrix facing flavin semiquinone (FMNH•)Citation8 or at the iron − sulfur centers.Citation9 In complex III, the source of O2•− is the ubisemiquinone radical intermediate (QH•), which is formed during the Q cycle at the Qo site facing the mitochondrial inter-membrane space.Citation10,Citation11 In vitro, electrons entering at complex II (succinate dehydrogenase) can flow backward through complex I to produce ROS at the FMN site.Citation8 Regardless of the production site, O2•− quickly undergoes dismutation to H2O2, catalyzed by superoxide dismutase in the mitochondrial-matrix (Mn-SOD) or in the inter-membrane space (Cu,Zn-SOD).Citation12 H2O2 can diffuse across the mitochondrial membranes and be detected in the extra-mitochondrial space.Citation13 In the presence of metal ions, H2O2 can be further transformed into hydroxyl radical (OH•) by Fenton chemistry.Citation14 Mitochondria are also believed to be primary source of other reactive species such as nitrogen-centered species, which are derived from nitric oxide and carbonate radical.Citation15,Citation16
Despite the vast literature addressing the respiratory chain sites involved in mitochondrial ROS generation, the process in calcium-overloaded mitochondria has not yet been completely characterized. Here, we have used two different probes with distinct detection properties, as well as different respiratory substrates and electron transport chain inhibitors, to characterize the ROS generation by the respiratory chain in calcium-overloaded mitochondria. This work continues our previous studies on the potential role of calcium on PTP opening.Citation17,Citation18
Materials and methods
Isolation of rat liver mitochondria
Male Wistar rats weighting approximately 200 g were used, according to research protocols approved by the local ethics committee (Comissão de Ética no Uso de Animais do Campus de Ribeirão Preto – USP, CEUA-USP). Mitochondria were isolated by standard differential centrifugation, at 4°C, according to Pedersen et al.Citation19 with minor modifications.Citation17 Mitochondrial protein content was determined by the biuret reaction. Rat liver-isolated mitochondria energized with 5 mM succinate (plus 2 µM rotenone) presented respiratory control ratio and ADP/O in the 4.5–5.5 and 1.3–1.7 ranges, respectively; the mitochondrial membrane potential was ∼160 mV. We have used freshly prepared mitochondria (used within 3 hours) incubated in a medium devoid of inorganic phosphate in order to prevent non-specific sensitization of PTP opening. At 100 µM, but not at 10 µM, calcium significantly increased state-4 respiration rate.
Mitochondrial assays
The standard medium for the mitochondrial assays consisted of 125 mM sucrose, 65 mM KCl, and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-KOH pH 7.4, at 30°C. Mitochondrial ROS were monitored spectrofluorimetrically using as probes 5 µM dichlorodihydrofluorescein diacetate (H2DCFDA)Citation20 or 2 µM Amplex Red plus 1 U/ml horseradish peroxidase (HRP)Citation21 at 503/529 nm (slits 5/10 nm), and 563/587 nm (slits 5/5 nm) excitation/emission wavelength pairs, respectively, in a Model F-4500 Hitachi fluorescence spectrophotometer (Tokyo, Japan). The fluorescence levels of probes assayed in the absence of mitochondria were not altered in the presence of the respiratory substrates or inhibitors used in the study. Mitochondrial calcium uptake was assessed in the standard medium by monitoring the changes in the absorbance of arsenazo III (25 µM) at 675–685 nm. The calcium concentration was determined based on a standard curve under the assay conditions. The concentration of calcium in the medium with mitochondria was ∼2 µM. Mitochondrial membrane potential was assessed spectrofluorimetrically using 10 µM safranine O as a probe at the 495/586 nm excitation/emission wavelength pair. Mitochondrial swelling was estimated spectrophotometrically from the decrease in apparent absorbance at 540 nm, using a Model U-2910 Hitachi spectrophotometer (Japan).
Statistical analysis
Data are represented as average ± standard error of mean of 3–6 repetitions using different mitochondrial preparations. Multiple comparisons were performed using one-way analysis of variance followed by Tukey test. P < 0.05 was considered significant.
Results and discussion
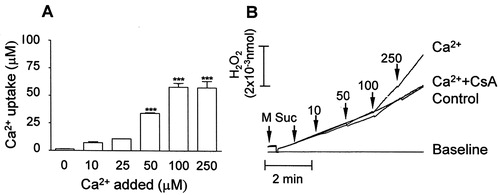
In order to characterize the ROS generation by the respiratory chain in calcium-overloaded mitochondria, we first evaluated the saturation of calcium uptake by the organelles: calcium uptake increased from 10 to 100 µM calcium and no further increase was observed at 250 µM (A). Therefore, we chose to work with 100 µM calcium, a condition which significantly increased the basal rate of mitochondrial H2O2 generation (as demonstrated with the Amplex red probe, B). Then we assessed ROS generation in the presence of calcium plus different respiratory chain substrates and inhibitors by means of two different probes, H2DCFDA and Amplex Red, regarding their specificity for different ROS types and location. It is worth considering that calcium overload as addressed in the present study may be found under specific pathological situations such as excitotoxin-induced neuronal cell death and ischemia/reperfusion injury, as well as during local calcium spikes mediated by IP3 receptors at the endoplasmic reticulum–mitochondrion interface.Citation22 It has been proposed that ROS generation induced by calcium involves either: (1) stimulation of TCA cycle/oxidative phosphorylation enhancing electron transport through the respiratory chain, (2) stimulation of mtNOS generating NO which inhibits complex IV, or (3) displacement of cytochrome c from mitochondrial inner membrane either by competing for cardiolipin binding sites, or through the PTP pore.Citation23
Figure 1. Calcium uptake (A) and calcium-stimulated ROS (H2O2) generation (B) in mitochondria isolated from rat liver. (A) Mitochondria (0.5 mg protein/ml) were energized with 5 mM succinate (+2 µM rotenone) in a standard medium consisting of 125 mM sucrose, 65 mM KCl, and 10 mM HEPES-KOH pH 7.4, at 30°C; calcium (Ca2+) was determined with arsenazo III as described in Materials and methods. (B) Mitochondria were incubated in the standard medium at 30°C and H2O2 was determined with the Amplex Red probe/HRP as described in Materials and methods. Additions: standard medium + Amplex Red probe/HRP (base line); Mitochondria (M) + 2 µM rotenone + 100 µM EGTA + 5 mM succinate (Suc) (Control); M + 2 µM rotenone + Suc + 10–250 µM calcium (Ca2+). Bars are average ± SEM and traces are representative experiments.
The H2O2 in the extra-mitochondrial (inter-membrane) space can be measured by the reaction with the non-fluorescent probe Amplex Red in conjunction with HRP, with formation of the fluorescent product resorufin.Citation21 In ROS assays with H2DCFDA, the probe crosses the mitochondrial membranes and is enzymatically hydrolyzed by the matrix esterase forming the non-fluorescent H2DCF product.Citation24 Once in the mitochondrial matrix and in the presence of hydroxyl, carbonate, or nitrogen dioxide radicals, H2DCF is rapidly oxidized to the highly fluorescent dichlorofluorescein (DCF) product.Citation25 In virtue of the wide spectrum of detection, data from DCF indicate the overall ROS status rather than any particular species. These different characteristics of detection allowed us to assess both overall ROS status in the matrix side and H2O2 in the extra-mitochondrial space of calcium-overloaded organelles.
Under the control conditions (presence of ethylene glycol tetra-acetate, EGTA), only the Amplex Red/HRP assay showed a significant difference between ROS generated in mitochondria energized with glutamate/malate (A) or succinate (B), being higher for succinate. Since mitochondria energized with succinate, in the absence of rotenone, generates ROS by the backflow of electrons to complex I,Citation8 this result suggests that H2O2 was released at the extra-mitochondrial space (detection by Amplex Red/HRP), without significant ROS release at the mitochondrial-matrix side (lack of detection by H2DCFDA). Calcium, in its turn, stimulated ROS release in mitochondria energized with either glutamate/malate, or succinate, which was detected by both Amplex Red () and H2DCFDA (), indicating ROS release at both the mitochondrial-matrix side and extra-mitochondrial space.
Figure 2. H2O2 generation assessed with the Amplex Red probe/HRP in mitochondria energized with glutamate/malate (A) or succinate (B). Mitochondria were incubated in the standard medium described in , in the presence of 5 mM glutamate/malate (A) or 5 mM succinate (B) as described in Materials and methods; 100 µM EGTA (control) or 100 µM calcium (Ca2+) were added at the initial time (0 seconds). The inhibitors rotenone (Rot, 2 µM), antimycin A (AA, 1 µM), or myxothiazol (Myx, 2 µM) were added after 300 seconds. Bars (A, B) are average ± SEM of the relative fluorescence at 600 seconds from the traces represented in A-1 and B-1. *P < 0.05, succinate vs. glutamate/malate; #P < 0.05 vs. Control; ‡P < 0.05 vs. Ca2+.
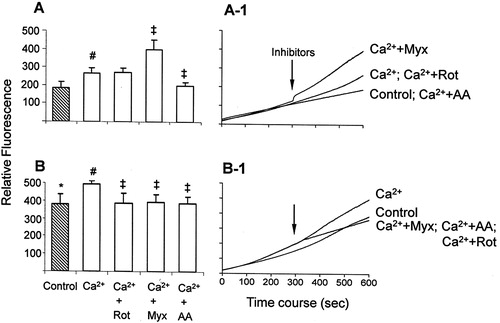
Figure 3. ROS generation assessed with the H2DCFDA probe in mitochondria energized with glutamate/malate (A) or succinate (B). The assays were performed as described in . #P < 0.05 vs. control; ‡P < 0.05 vs. Ca2+.
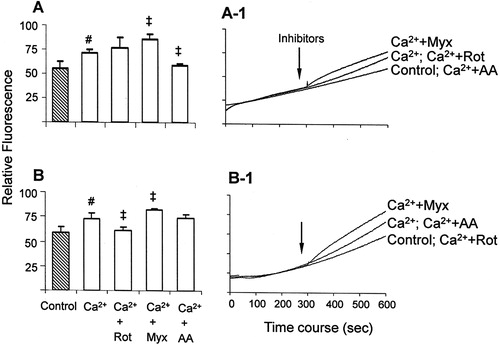
In mitochondria energized with glutamate/malate and in the presence of respiratory chain inhibitors, Amplex Red (A) and H2DCFDA (A) assays showed similar results. Rotenone, which blocks electron transfer from Fe–S center to ubiquinone and increases ROS generation at complex I,Citation9,Citation26 had no significant effect on calcium-stimulated ROS generation. Under this condition, ROS generation is supported only by complex I, since the blockage of electrons flow to ubiquinone renders it in an oxidized state unable to donate electrons to oxygen, thus reducing ROS generation at complex III.Citation27 This suggests an interference of rotenone at the site where calcium-stimulated ROS is generated, i.e. it decreases at complex III and increases at complex I. On the other hand, the complex III inhibitors myxothiazol and antimycin A had opposite effects as compared with each other: myxothiazol increased, while antimycin A decreased, calcium-stimulated ROS generation (A and A). The effects of complex III inhibitors on mitochondrial ROS generation, in the absence of calcium, have already been described: antimycin A inhibits the transfer of an electron from cytochrome b to ubiquinone at the Qi site, thus accumulating semiquinone at the Qo site, which favors O2•− generation.Citation28 However, our results with both probes showed that in mitochondria energized with glutamate/malate, antimycin A prevents calcium-stimulated ROS generation. Myxothiazol inhibits O2•− generation by preventing the transfer of an electron from ubiquinol to the Rieske iron–sulfur protein in the respiratory chain, thereby inhibiting semiquinone formation.Citation29 Its effect has been reported either aloneCitation30 or in associationCitation31 with antimycin A, which enhances ROS generation. An explanation is that (1) myxothiazol-induced ROS originate from an electropositive site of complex III, and (2) an increase in O2•− production occurs from the accumulation of QH2, resulting in auto-oxidation of ubiquinol with leakage of electrons to oxygen.Citation32 Since our study focused on the inhibitors' effects on calcium-stimulated ROS generation, these results suggest that in mitochondria energized with glutamate/malate, complex I, and ubiquinone cycle at complex III are the main sources of ROS, which are released both at the mitochondrial-matrix side and the extra-mitochondrial space. Moreover, the modulation of complex III at Qo and Qi produces opposite effects on calcium-stimulated ROS generation.
In succinate/rotenone-energized mitochondria both probes Amplex Red (B) and H2DCFDA (B) showed a decrease in calcium-stimulated ROS generation. Since rotenone prevents the generation of ROS via backflow of electrons to complex I,Citation8 this is a probable site of calcium-stimulated ROS generation in succinate-energized mitochondria. On the other hand, the effects of complex III inhibitors myxothiazol and antimycin A in succinate-energized mitochondria, similar to the observations with glutamate/malate, were detected differently by Amplex Red (B) and H2DCFDA (B). In fact, in succinate-energized mitochondria in the absence of calcium, complex III inhibitors lead to the entry of electrons through complex II, generating ROS at complex III.Citation32 In the presence of calcium, the increase of ROS generation induced by myxothiazol in succinate-energized mitochondria was significantly detected only by H2DCFDA. This suggests that when the Qo site of complex III is inhibited, higher ROS levels are released at the mitochondrial-matrix side, but H2O2 is not generated/released at the extra-mitochondrial space. Moreover, in succinate-energized mitochondria, the effect of antimycin A was also detected differently by Amplex Red and H2DCFDA. The decrease of calcium-stimulated ROS generation promoted by antimycin A was only detected by Amplex Red (not by H2DCFDA); this result suggests that blockage at the Qi site of complex III attenuates the calcium-stimulated generation of H2O2 at the extra-mitochondrial space. Therefore, in succinate-energized mitochondria both backflow of electrons to complex I and the ubiquinone cycle at complex III may be sources of calcium-stimulated ROS generation. Moreover, the complex III inhibition at Qo or Qi sites promotes distinct effects on ROS release concerning mitochondrial-matrix side or extra-mitochondrial space.
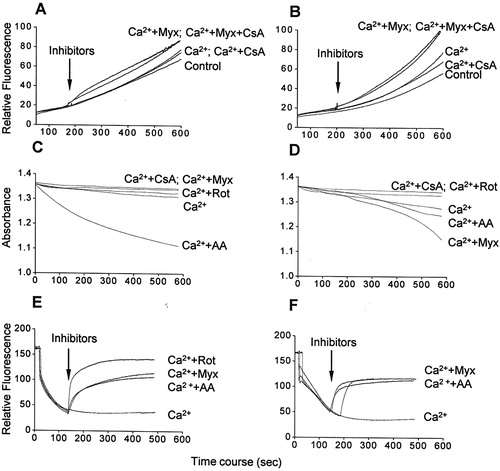
Mitochondria-generated ROS favor PTP-opening, which in turn generates more ROS.Citation33 In order to verify whether ROS release in calcium-overloaded mitochondria results from PTP opening, we assessed ROS in the presence of the classical PTP opening inhibitor CsA. H2DCFDA detection showed that ROS release in the presence of calcium and/or myxothiazol either in glutamate/malate- (A) or succinate-energized mitochondria (B) was only weakly inhibited by CsA, indicating no significant contribution of PTP opening. In this context, we also examined whether ROS release in the presence of calcium and/or myxothiazol favors calcium-induced PTP opening as evidenced by mitochondrial swelling, as well as a possible involvement of changes in mitochondrial membrane potential. As expected, calcium induced a slight swelling in either glutamate/malate- (C) or succinate-energized (D) mitochondria, both inhibited by CsA. The rate and extent of swelling were higher in succinate-energized mitochondria compared to glutamate/malate-energized organelles; in the former case, the rate was higher in the absence of rotenone (D). Antimycin A induced mitochondrial swelling regardless of the respiratory substrate, being larger with glutamate/malate, whereas myxothiazol only induced swelling in succinate-energized mitochondria. Under all conditions, swelling was significantly prevented by the antioxidant Trolox (50 µM, data not shown), suggesting that oxidative stress provided by the presence of calcium is required. Remarkably, the patterns of swelling and ROS generation in the presence of calcium and/or respiratory chain inhibitors were quite similar. Concerning mitochondrial membrane potential, antimycin A and myxothiazol promoted its disruption in calcium-overloaded mitochondria energized with either glutamate/malate (E), or succinate (F), apparent consequence of the respiratory chain inhibition. These results show that PTP opening and mitochondrial membrane potential dissipation do not have any significant role on calcium-stimulated ROS generation and suggest a calcium action at the respiratory chain level.
Figure 4. ROS generation (A and B), mitochondrial swelling (C and D), and membrane potential dissipation (E and F) in the presence of calcium/respiratory chain inhibitors in glutamate/malate-energized (A, C, and E) or succinate-energized (B, D, and F) mitochondria. Mitochondria were incubated in the standard medium with 100 µM calcium, as described in and Materials and methods. Additions: only Ca2+ (Ca2+), 1 µM cyclosporine A (CsA), 2 µM rotenone (Rot), 1 µM antimycin A (AA), and 2 µM myxothiazol (Myx). Traces are representative of three independent preparations.
Conclusion
Regardless of the respiratory substrate, calcium stimulates the mitochondrial generation of ROS, which are released at both the mitochondrial-matrix side and extra-mitochondrial space. In glutamate/malate-energized mitochondria, inhibition at complex I or complex III (ubiquinone cycle) similarly modulates ROS generation at the either the mitochondrial-matrix side or the extra-mitochondrial space; this also occurs when the backflow of electrons to complex I in succinate-energized mitochondria is inhibited. In succinate-energized mitochondria, the modulation of ROS generation at the mitochondrial-matrix side or extra-mitochondrial space relies on the site of complex III which is inhibited. These results allow a straight comparison between the effects of different respiratory substrates and electron transport chain inhibitors on ROS generation at either the mitochondrial-matrix side or extra-mitochondrial space in calcium-overloaded mitochondria.
Acknowledgements
This work was supported by grants from FAPESP, CAPES and CNPq, Brazil. The authors thank Neife Aparecida Guinaim dos Santos for the careful revision of the manuscript.
References
- Hunter DR, Haworth RA. The calcium-induced membrane transition in mitochondria: III. Transitional calcium release. Arch Biochem Biophys 1979;195:468–77.
- Kowaltowski AJ, Castilho RF, Vercesi AE. Opening of the mitochondrial permeability transition pore by uncoupling or inorganic phosphate in the presence of Ca2+ is dependent on mitochondrial-generated reactive oxygen species. FEBS Lett 1996;378:150–2.
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007;87:99–163.
- Skulachev VP. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis 2006;11:473–85.
- Zoratti M, Szabò I. The mitochondrial permeability transition. Biochim Biophys Acta 1995;1241:139–76.
- Chance B, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev 1979;59:527–605.
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol 2003;552:335–44.
- Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem 2002;80:780–7.
- Herrero A, Barja G. Localization of the site of oxygen radical generation inside the complex I of heart and nonsynaptic brain mammalian mitochondria. J Bioenerg Biomemb 2000;32:609–15.
- Muller FL, Roberts AG, Bowman MK, Kramer DM. Architecture of the Qo site of the cytochrome bc1 complex probed by superoxide production. Biochemistry 2003;42:6493–9.
- St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem 2002;277:44784–90.
- Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu, Zn-SOD in mitochondria. J Biol Chem 2001;276:38388–93.
- Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem 2003;278:5557–63.
- Halliwell B, Gutteridge JM. Role of free radicals and catalytic metal ions in human disease: an overview. Meth Enzymol 1990;186:1–85.
- Bonini MG, Radi R, Ferrer-Sueta G, Ferreira AM, Augusto O. Direct EPR detection of the carbonate radical anion produced from peroxynitrite and carbon dioxide. J Biol Chem 1999;274:10802–6.
- Medinas DB, Cerchiaro G, Trindade DF, Augusto O. The carbonate radical and related oxidants derived from bicarbonate buffer. IUBMB Life 2007;59:255–62.
- Pestana CR, Silva CH, Pardo-Andreu GL, Rodrigues FP, Santos AC, Uyemura SA, et al. Ca(2+) binding to c-state of adenine nucleotide translocase (ANT)-surrounding cardiolipins enhances (ANT)-Cys(56) relative mobility: a computational-based mitochondrial permeability transition study. Biochim Biophys Acta 2009;1787:176–82.
- Pestana CR, Silva CH, Uyemura SA, Santos AC, Curti C. Impact of adenosine nucleotide translocase (ANT) proline isomerization on Ca2+-induced cysteine relative mobility/mitochondrial permeability transition pore. J Bioenerg Biomembr 2010;42:329–35.
- Pedersen PL, Greenawalt JW, Reynafarje B, Hullihen J, Decker GL, Soper JW, et al. Preparation and characterization of mitochondria and submitochondrial particles of rat liver and liver-derived tissues. Meth Cell Biol 1978;20:411–81.
- Wrona M, Patel KB, Wardman P. Reactivity of 2′,7′-dichlorodihydrofluorescein and dihydrorhodamine 123 and their oxidized forms towards carbonate, nitrogen dioxide, and hydroxyl radicals. Free Rad Biol Med 2005;38:262–70.
- Zhou M, Panchuk-Voloshina N, Haugland RP, Diwu Z. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal Biochem 1997;253:162–8.
- Brenner C, Grimm S. The permeability transition pore complex in cancer cell death. Oncogene 2006;25:4744–56.
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 2004;287:817–33.
- LeBel C, Ischiropoulos H, Bondy S. Evaluation of the probe 2 V,7 V-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol 1992;5:227–31.
- Wardman P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Rad Biol Med 2007;43:995–1022.
- Shiryaeva A, Arkadyeva A, Emelyanova L, Sakuta G, Morozov V. Superoxide anion production by the mitochondrial respiratory chain of hepatocytes of rats with experimental toxic hepatitis. J Bioenerg Biomembr 2009;41:379–85.
- Hoffman DL, Brookes PS. Oxygen sensitivity of mitochondrial reactive oxygen species generation depends on metabolic conditions. J Biol Chem 2009;284:16236–45.
- Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys 1985;237:408–14.
- Young TA, Cunningham CC, Bailey SM. Reactive oxygen species production by the mitochondrial respiratory chain in isolated rat hepatocytes and liver mitochondria: studies using myxothiazol. Arch Biochem Biophys 2002;405:65–72.
- Sousa SC, Maciel EN, Vercesi AE, Castilho RF. Ca2+-induced oxidative stress in brain mitochondria treated with the respiratory chain inhibitor rotenone. FEBS Lett 2003;543:179–83.
- Cadenas E, Boveris A. Enhancement of hydrogen peroxide formation by protophores and ionophores in antimycin-supplemented mitochondria. Biochem J 1980;188:31–7.
- Starkov AA, Fiskum G. Myxothiazol induces H2O2 production from mitochondrial respiratory chain. Biochem Biophys Res Commun 2001;281:645–50.
- Peng TI, Jou MJ. Oxidative stress caused by mitochondrial calcium overload. Ann NY Acad Sci 2010;1201:183–8.